Article
Open Access
Report and literature review on two cases with different kinds of Creutzfeldt-Jakob disease
- Authors:
- Chunhua Tang
- Changyue Gao
-
View Affiliations / Copyright
Affiliations:
Department of Neurology, Daping Hospital, The Third Military Medical University, Yuzhong, Chongqing 400042, P.R. China
-
Pages:
854-858
|
Published online on:
November 10, 2017
https://doi.org/10.3892/etm.2017.5481
- Expand metrics +
Metrics:
Total
Views: 0
(Spandidos Publications: | PMC Statistics:
)
Metrics:
Total PDF Downloads: 0
(Spandidos Publications: | PMC Statistics:
)
This article is mentioned in:
Abstract
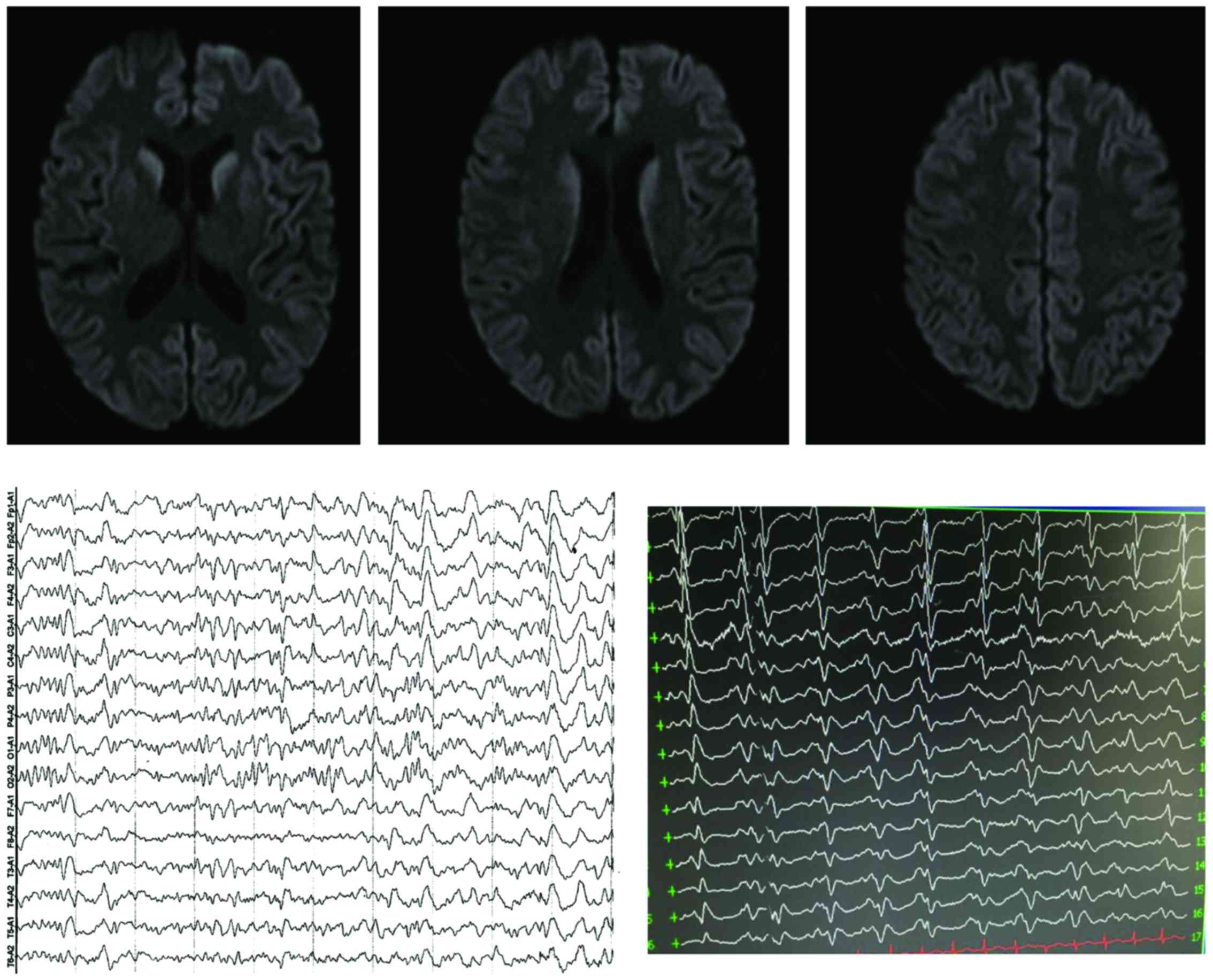
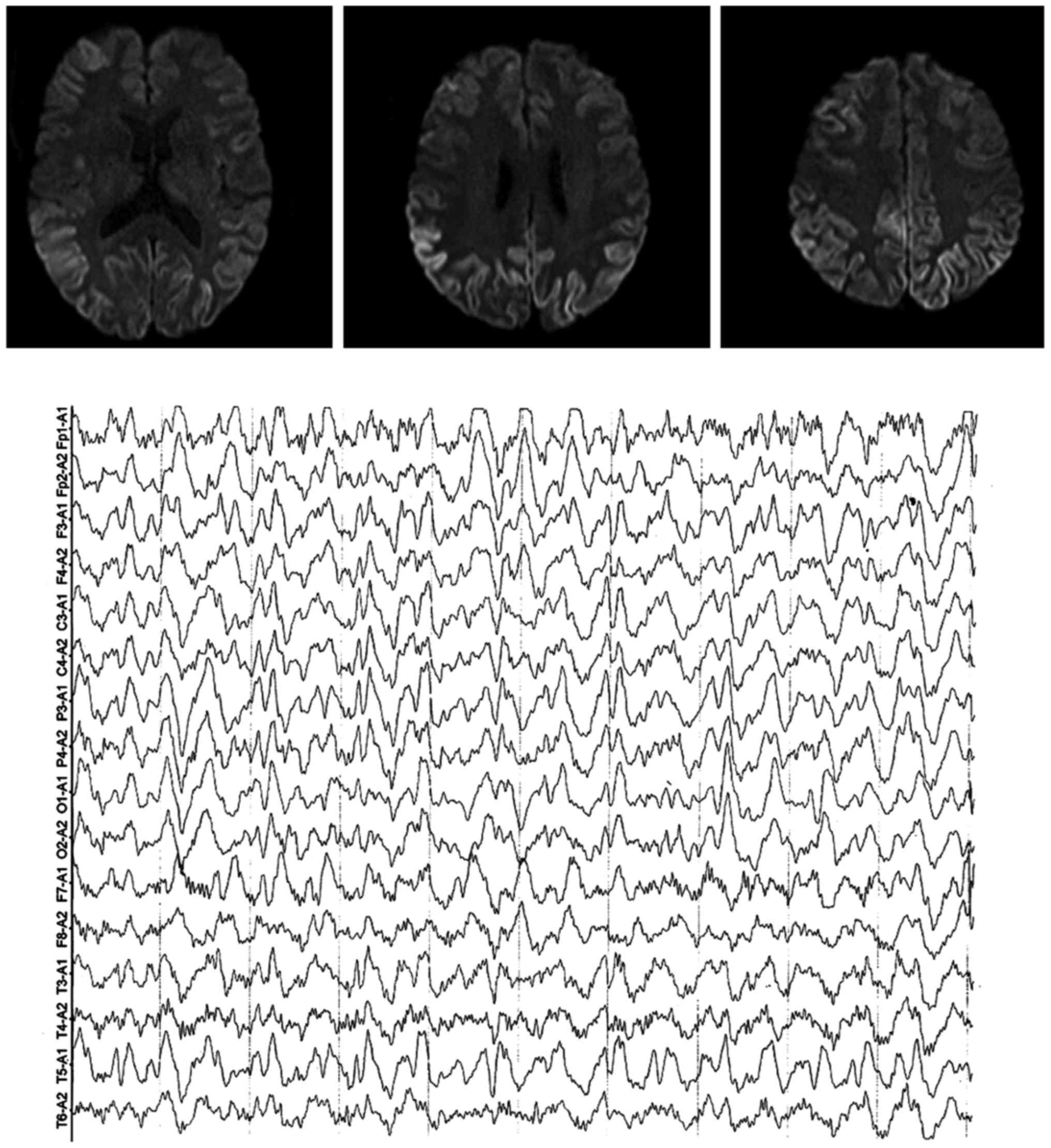
Creutzfeldt-Jakob disease (CJD), also known as corticostriate spinal degeneration, subacute spongiform encephalopathy or infectious spongiform encephalopathy, is a type of degenerative disease of the central nervous system caused by prion protein (PrP) infection, which is the most common type of human PrP disease. CJD is genetic and infectious, and is one of the most common causes of rapid progressive dementia with rare clinical occurrence. Herein, we report the clinical conditions of 2 cases of patients with different type of CJD we treated and followed up recently, and a review of relevant literature. The patient in case 1 was admitted due to ‘dizziness with hypomnesis, and mental and behavior disorder’. He was considered to suffer from a central nervous system infection - a viral encephalitis, but one month later, a repeated cranial MRI showed lace sign of bilateral frontotemporal parietal lobe in DWI sequence, an AEEG indicated periodic synchronous discharge, and the detection of cerebrospinal fluid 14-3-3 protein was positive. It was suggested to be diagnosed as the sporadic CJD. The patient in case 2 was admitted because of ‘progressive hypomnesis’. Cerebrospinal fluid 14-3-3 protein detection was negative, but the V203I-related mutation was found in the PRNP gene detection. The patient was suggested to be diagnosed as genetic CJD. Both patients died in a short time. An earlier diagnosis can provide a time window for treatment, and avoid unnecessary transmission in hospital, as well as doctor-patient dispute.
View References
|
1
|
Sikorska B, Knight R, Ironside JW and
Liberski PP: Creutzfeldt-Jakob disease. Adv Exp Med Biol.
724:76–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosenbloom MH and Atri A: The evaluation
of rapidly progressive dementia. Neurologist. 17:67–74. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ironside JW: Variant Creutzfeldt-Jakob
disease: An update. Folia Neuropathol. 50–56. 2012.PubMed/NCBI
|
|
4
|
Zanusso G, Fiorini M, Ferrari S, Gajofatto
A, Cagnin A, Galassi A, Richelli S and Monaco S: Cerebrospinal
fluid markers in sporadic Creutzfeldt-Jakob disease. Int J Mol Sci.
12:6281–6292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao C, Shi Q, Tian C, Chen C, Han J, Zhou
W, Zhang BY, Jiang HY, Zhang J and Dong XP: The epidemiological,
clinical, and laboratory features of sporadic Creutzfeldt-Jakob
disease patients in China: Surveillance data from 2006 to 2010.
PLoS One. 6:e242312011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geschwind MD, Shu H, Haman A, Sejvar JJ
and Miller BL: Rapidly progressive dementia. Ann Neuro. l64:1–108.
2008.
|
|
7
|
WHO, . Global surveillance, diagnosis and
therapy of human Transmissible Spongiform Encephalopathies: Report
of a WHO consultation. Geneva, Switzerland: 9-11–February. 1998,
World Health Organization; Geneva, Switzerland: 1998.
|
|
8
|
Zerr I, Kallenberg K, Summers DM, Romero
C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B,
Ladogana A, et al: Updated clinical diagnostic criteria for
sporadic Creutzfeldt-Jakob disease. Brain. 132:2659–2668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marandi Y, Farahi N, Sadeghi A and
Sadeghi-Hashjin G: Prion diseases - current theories and potential
therapies: A brief review. Folia Neuropathol. 50:46–49.
2012.PubMed/NCBI
|