Introduction
Asthma is one of the most common chronic
inflammatory airway diseases along with airway epithelial cells
injury, which seriously threat to the health of human worldwide
(1,2). It is surveyed by World Health
Organization (WHO) that there are approximately 235,000,000 people
suffering from this disease and causes about 255,000 deaths
worldwide annually (3). The airway
epithelium contacts physical stimuli, inhaled allergens, pollution,
bacteria, viruses, and respiratory drugs firstly, and plays the
part of an obstruction between the interior of living bodies and
exterior environment (4). Previous
studies have demonstrated that the repair process of airway
epithelium in asthma was defective fundamentally, and abnormal
airway epithelial shedding was observed in asthmatic patients
(5–7). The proposed therapeutic approach of
asthma was the combination of glucocorticoids (GCs) and
β2 receptor agonist. However, the treatment was unable
to prevent the recurrence of asthma and the rising of incidence
year by year. Hence, to investigate new exact mechanisms and
molecular pathways activated in asthma is necessary in developing
novel treatment measures and therapeutic drugs in asthma.
Dexamethasone (DEX) represents one of the common GCs
drugs, which have been widely used in the treatment of asthma,
chronic obstructive pulmonary disease, and rheumatoid arthritis
(8,9). GCs play a role in regulating the target
gene transcription of glucocorticoid receptors (GR) (10). In the meantime, GCs also possess
anti-inflammatory effect through inhibiting the transcription of
inflammatory transcription factors, including nuclear factor-kappa
B (NF-κB) and activator protein 1 (AP-1) (11). It has been reported that GCs could
significantly reduce the proliferation of airway epithelial cells
in asthmatic patients and inhibit the airway epithelium repair
through restraining the early proliferation and migration of airway
epithelial cells (12,13). Nevertheless, the accurate mechanism
in the process of airway epithelium repair inhibited by GCs is not
clear.
The repair of airway epithelium is achieved
primarily through the interaction of epidermal growth factor (EGF)
with its epidermal growth factor receptor (EGFR) to activate the
mitogen-activated protein kinase (MAPK)/extracellular regulated
kinase (ERK) signaling pathway. MAPK/ERK pathway plays a critical
role in the process of cell survival, proliferation, migration and
differentiation (14). Increasing
evidences have shown that the activation of MAPK/ERK pathway
promoted the proliferation, migration and differentiation of
peripheral cells around the damage (15,16).
Recent studies also have revealed that mitogen-activated protein
kinase (Mek1/2) and Erk1/2 specific inhibitors in the MAPK/ERK
pathway markedly inhibited the repair of airway epithelium injury
(13,17). Furthermore, GCs significantly
inhibited the phosphorylation of v-raf-1 murine leukemia viral
oncogene homolog 1 (Raf-1) and activation of downstream MAPK/ERK
pathway activated by growth factors (18,19).
However, Raf-1 phosphorylation is widely considered to be the
up-stream signal molecule that plays a key role in the activation
of MAPK/ERK pathway. Therefore, we conjectured that GCs inhibited
human airway epithelial cells repair might through inhibiting the
phosphorylation of Raf-1 and the activation of MAPK/ERK
pathway.
Indoleamine 2, 3-dioxygenase (IDO) is a
rate-limiting enzyme, which has the function of catalyzing the
oxidative decomposition of pyrrole ring in tryptophan molecule
(20). IDO widely distributed in the
tissues of humans and other mammals (21), including lung, neuroglia, endothelial
cells, and visceral epithelial cells (22–25). And
the secretion of IDO is impacted by lipopolysaccharide, cytokines,
tumor necrosis factor-α (TNF-α), and interferon γ (IFN-γ) (26). It has been demonstrated that the IDO
transcriptional induction is the mechanism of neuroendocrine
disorders triggered by chronic inflammation (27). Studies also have found that IDO
played an essential role in depressive disorder and Alzheimer's
disease (28,29). Nevertheless, as far as we known, the
effects and mechanisms of IDO in human airway epithelial cells
repair inhibited by DEX have not been reported yet.
Collectively, in this study, we explored the
biological roles of IDO on the repair of human airway epithelium
inhibited by DEX. Furthermore, mechanisms analysis indicated that
IDO positively regulated the expression levels of Raf-1, Mek1/2,
and Erk1/2. Together, our study elucidated that IDO promoted the
repair of human airway epithelial cells inhibited by DEX through
affecting the MAPK/ERK pathway.
Materials and methods
Reagents
All of the products utilized in cell-culture were
obtained from Gibco; Thermo Fisher Scientific, Inc., (Waltham, MA,
USA). Antibodies were obtained from Abcam (Cambridge, UK). The
inhibitor of ERK (PD98059; Item: ML4829; CAS: 167869-21-8) was
gained from YuduoBio (Shanghai, China). And the inhibitor of HMGB1
(R, S-Sulforaphane; Item: S6317; CAS: 142825-10-3) was gained from
HengfeiBio (Shanghai, China).
Cell culture
The human lung bronchial epithelial cell line
(16HBE) was obtained from AATCC (American Type Culture Collection,
Manassas, VA, USA). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) mixed with 10% fetal bovine serum (FBS) in a
5% CO2 atmosphere at 37°C. Afterwards, cells were
observed under an inverted microscope for the growth status.
IDO gene recombinant adenovirus
transfection
Cultured 16HBE cells were transfected with objective
gene adenovirus (CMV-IDO1-EGFP; FUBIO, Shanghai, China) and sowed
into 12-well plates at a density of 1×105 cells per
well. After the cells were complete adherence, transfection could
be carried out. Before transfection, 500 µl fresh 16HBE cells
medium were added to each well to replace the quondam cells. 15 µl
adenovirus was added to the wells, and then cells were cultured in
the incubator after shaking (37°C, 5% CO2, 95% relative
humidity). 8 h later, the medium with adenovirus was removed and
cells were washed by PBS twice. Fresh medium were added to the
cells and cells were then cultured in the incubator (37°C, 5%
CO2, 95% relative humidity). After 30 h, 16HBE cells
transfected with IDO gene were harvested.
Western blot analysis
Proteins from 16HBE cells were obtained and
bicinchoninic acid assay was performed to detect the protein
concentration. After that, equal quantity of proteins (50 µg) were
solubilized in 5× sodium dodecyl sulfate (SDS)-sample buffer and
separated on the SDS polyacrylamide gels (Thermo Fisher Scientific,
Inc.). Separated proteins were then transferred onto a
polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA,
USA). After blocking, the membranes were incubated with anti-IDO
(dilution, 1:500; ab55305); anti-p-Raf-1 (dilution, 1:500;
ab208449); anti-Raf-1 (dilution, 1:1,000; ab50858); anti-p-Mek1/2
(dilution, 1:1,000; ab194754); anti-Mek1/2 (dilution, 1:1,000;
ab215263); anti-p-Erk1/2 (dilution, 1:1,000; ab201015); anti-Erk1/2
(dilution, 1:1,000; ab17942); anti-GAPDH (dilution, 1:1,000,
ab8245; all from Abcam) antibodies overnight at 4°C. After that,
horseradish peroxidase-conjugated secondary antibodies (bs-0293M;
BIOSS, Beijing, China) were added and incubated at room temperature
for 1 h. The results of all the assessments were evaluated by
enhanced chemiluminescent reagents (EMD Millipore).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
About 5×105/well 16HBE cells were cultured in 6-well
plates and treated with control, NC [Adenovirus empty vector;
FUBIO, Shanghai, China], DEX (D1756; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), or IDO. And then the total RNA was extracted
from 16HBE cells by TRIzol (DP405-02; Tiangen Biotech Co., Ltd.,
Beijing, China) as suggested by the manufacturers. Afterwards, two
microliters of RNA was used for cDNA synthesis with a first strand
cDNA kit (Sigma-Aldrich; Merck KGaA) following the specification.
RT-qPCR was performed on the SYBR Green Premix Reagent (Takara Bio,
Inc., Otsu, Japan) by ABI 7500 Thermocycler (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The PCR cycles were under the
following conditions: 10 min pretreatment at 94°C, 95°C for 5 sec,
65°C for 30 sec (35 cycles), 95°C for 15 sec, 60°C for 1 min, 95°C
for 15 sec, a final extension at 72°C for 10 min and held at 4°C.
The primers were designed by Shanghai Sangong Pharmaceutical Co.,
Ltd., (Shanghai, China) and were as followed: IDO, forward:
5′-ATGCAGACTGTGTCTTGGCA-3′ and reverse: 5′-GCGCCTTTAGCAAAGTGTCC-3′
(product: 222 bp); Ki67, forward: 5′-CGTCCCAGTGGAAGAGTTGT-3′ and
reverse: 5′-GCCATTACGTCCAGCATGTT-3′ (product: 673 bp); HMGB1,
forward: 5′-CGGAGGGATTACGCTGACGA-3′ and reverse:
5′-CTTTGGGAGAGCGGACTACG-3′ (product: 212 bp); glyceraldehyde
3-phosphate dehydrogenase (GAPDH), forward:
5′-AATGGGCAGCCGTTAGGAAA-3′ and reverse: 5′-GCGCCCAATACGACCAAATC-3′
(product: 168 bp). And GAPDH was used as the control for the input
RNA level.
Cell viability analysis
Cell Counting Kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Shanghai, China) assay was performed to evaluate the
cell viability. About 6×104 cells/ml of 16HBE cells in
the logarithmic phase were sowed into the wells of 96-well plates
and cultured in the incubator (37°C, 5% CO2) for 12 h.
Cells were treated with control, NC, DEX, or IDO, respectively.
Cells were then maintained in the incubator (37°C, 5%
CO2) for 12 h. The absorbance at 450 nm was read by
enzyme labeling instrument. Cell viability was evaluated by the
percentage of cell survival compared with control.
ELISA
Cultured 16HBE cells were treated with control, NC,
DEX, or IDO, respectively. Then wells were sealed up by adhesive
tape, and incubated for 90 min at 37°C. 100 µl biotinylated
antibody fluids were added to each well, except for the blank
wells. Then wells were sealed up by adhesive tape and maintained at
37°C for 60 min. Enzyme solution was prepared in advance for 30
min, and placed away from the light at room temperature. After
washing, 100 µl enzyme solutions were added to each well. Wells
were sealed with adhesive tape and maintained at 37°C for 30 min.
After washing, chromogenic substrate was added to each well, except
for the blank wells. Then plates were incubated in the dark at 37°C
for 10–15 min. The stop solution was added to each well, and mixed
in 10 min immediately. Finally, the OD450 value was measured.
5(6)-carboxyfluorescein diacetate
succinimidyl ester (CFSE) labeling
Cultured 16HBE cells were suspended in PBS at a
final concentration of 1×106 cells/ml. After that, 5 µM
CFSE (Thermo Fisher Scientific, Inc.) was added into the cells.
After maintaining at 37°C for 10 min, cells were treated with 10
volumes of ice-cold culture medium (10% FBS). Then cells were
maintained on ice for 5 min. After centrifugation, cells were
washed by medium twice and subsequently seeded in 24-well plates
(about 1×105 cells per well). Afterwards, flow cytometry
was performed to evaluate the CFSE fluorescence intensity.
Wound-healing assay
Cultured 16HBE cells were seeded in 24-well plates
(about 1×105 cells per well) and treated with control,
NC, DEX, or IDO, respectively. Cells were incubated for 15 days
until the fusion rate to 100%. Horizontal lines were drawn by a 10
µl pipette tip at the bottom of the 24-well plates through the
culture hole. Cells were washed by Hanks liquor three times to
remove the crossed cells. The serum-free medium (1% B27, 2 mmol/l
glutamine, and 10 µl/ml penicillin-streptonmycin) was added to
cells. Cells were maintained in the incubator (37°C, 5%
CO2) and pictures were taken using a fluorescence
microscope after culturing for 24 or 0 h and 24 h,
respectively.
Statistical analysis
Our data were expressed as mean ± SEM of at least
three independent experiments. Statistical analysis was performed
using IBM SPSS statistical software (v19; IBM Corp., Armonk, NY,
USA). The differences in characteristics among the experimental
groups in western blot analysis, RT-qPCR analysis, cell viability
analysis, ELISA, CFSE labeling, and wound-healing assay were
examined by one-way ANOVA and Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of IDO mRNA and IDO protein
in 16HBE cells
We detected the effect of IDO on the expression of
IDO mRNA and IDO protein in 16HBE cells. We found that IDO
significantly enhanced mRNA levels of IDO after treatment compared
with the untreated control cells and treated with NC cells
(P<0.05; Fig. 1A). According to
the western blot results (Fig. 1B),
the protein levels of IDO in 16HBE cells treated with IDO were
markedly higher than control group and NC group (P<0.05). Thus,
it was confirmed that the expression levels of IDO were enhanced by
IDO gene in airway epithelial cells in vitro.
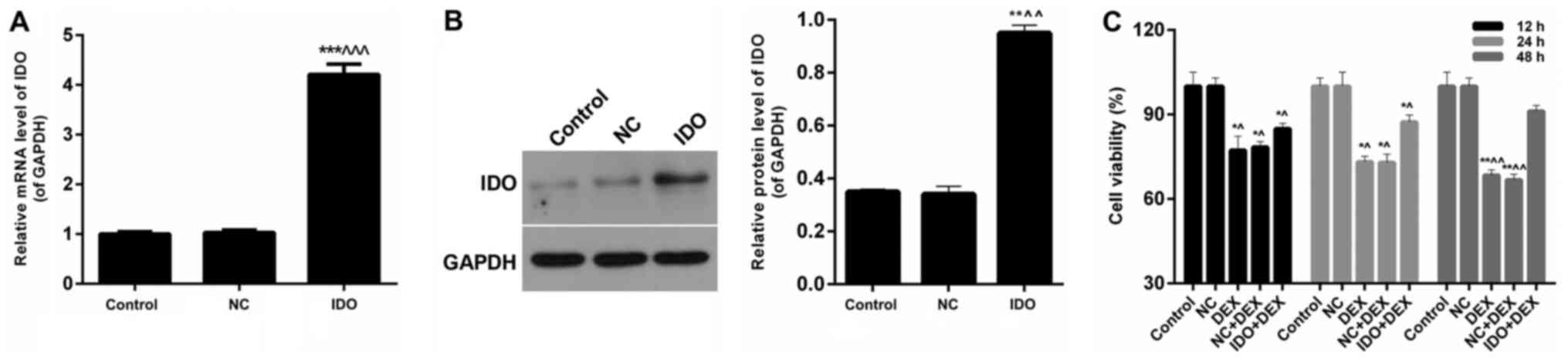 | Figure 1.Overexpression of IDO enhanced cell
viability of 16HBE cells. (A) RT-qPCR and (B) western blot assays
were performed to assess the IDO expression levels in 16HBE cells
treated with control, NC, and IDO. 16HBE cells were treated with
control, NC, DEX (10 µM), NC+DEX, and IDO+DEX. (C) CCK-8 was
carried out to evaluate the cell viability of 16HBE cells.
*P<0.05, **P<0.01, ***P<0.001 vs. control,
^P<0.05, ^^P<0.01,
^^^P<0.001 vs. NC. IDO, indoleamine 2, 3-dioxygenase;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; CCK-8, Cell Counting Kit-8; NC, negative control; DEX,
dexamethasone. |
Overexpression of IDO enhanced the
cell viability of 16HBE cells inhibited by DEX
The results of CCK-8 assays showed that the cell
viability of 16HBE cells treated with DEX and NC+DEX were
significantly lower than untreated control cells, while a sharp
increase was observed in 16HBE cells treated with IDO+DEX
(P<0.05; Fig. 1C). According to
the processing time, we found that the trends of cell viability
with 48 h treatment were more obvious than those with 12 and 24 h.
These results indicated that overexpression of IDO enhanced the
cell viability of 16HBE cells inhibited by DEX.
Overexpression of IDO promoted
proliferation and migration of 16HBE cells inhibited by DEX
As CFSE labeling results shown in Fig. 2, the cell proliferation rates of
16HBE cells treated with DEX were markedly lower than control
(P<0.05). However, after treating with IDO, the cell
proliferation rate was distinctly increased (P<0.05; Fig. 2), which indicated that overexpression
of IDO enhanced the cell proliferation capacity of 16HBE cells
inhibited by DEX. Moreover, we found that DEX obviously restrained
the 16HBE cells migration, while IDO evidently remitted the
suppression and promoted the cell migration, as assessed by
wound-healing assay (P<0.05; Fig.
3). Furthermore, we also evaluated the expression levels of
related proliferation- and migration-associated factors in 16 HBE
cells in each group. We found that the EGF expression in five
treatment groups had no significant differences, which indicated
that DEX and overexpression of IDO had little effect on the EGF
expression (Fig. 4A). The mRNA
expression levels of Ki67 and high-mobility group box 1 protein
(HMGB1) were significantly reduced by treating with DEX, while
sharp increases were observed in IDO+DEX groups (P<0.05;
Fig. 4B). These results revealed
that DEX significantly down-regulated the expression levels of Ki67
and HMGB1, while IDO markedly up-regulated the Ki67 and HMGB1
expression. Therefore, we could draw the conclusion that
overexpression of IDO promoted the proliferation and migration
abilities of 16HBE cells inhibited by DEX through regulating the
expression levels of Ki67 and HMGB1, respectively.
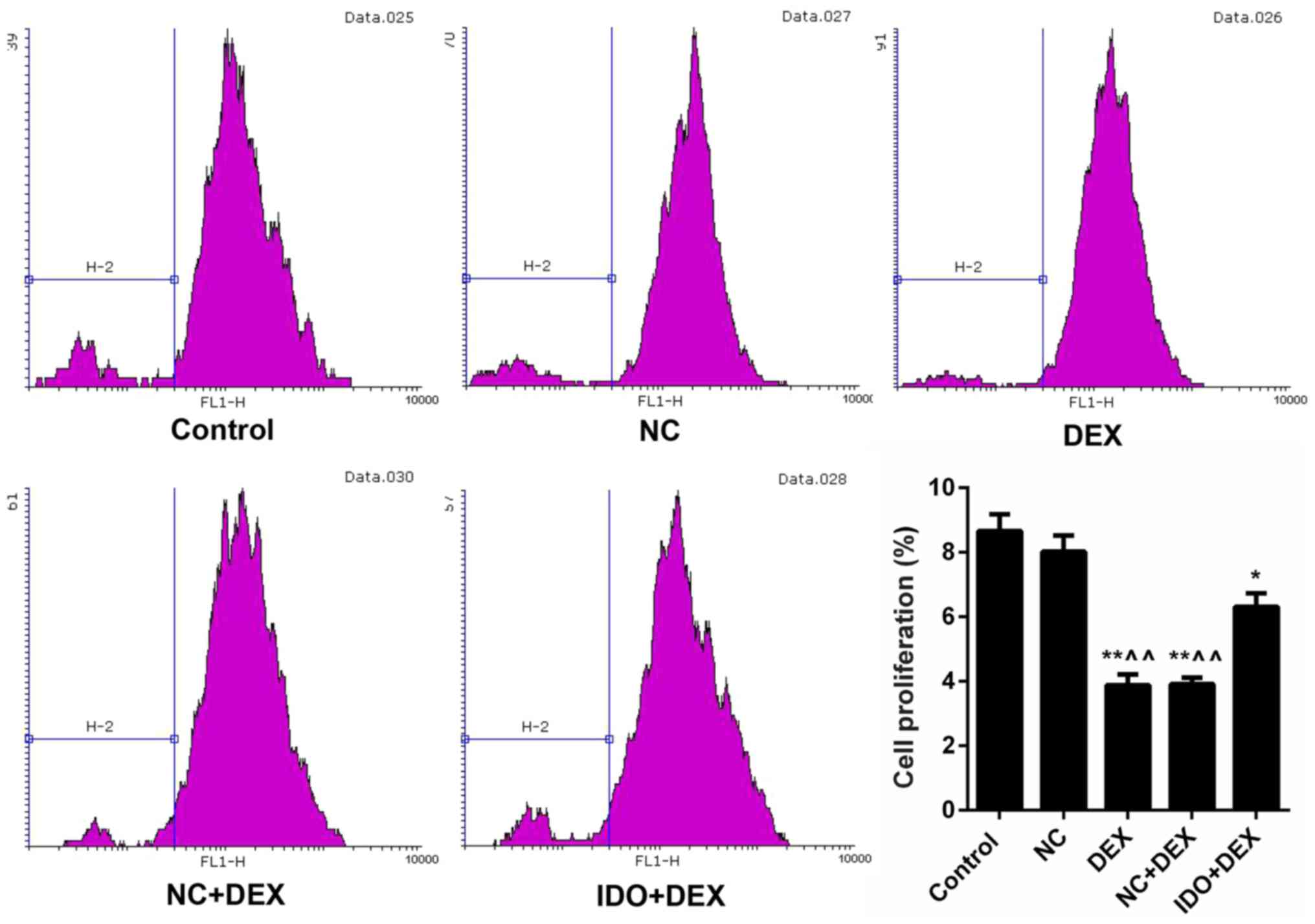 | Figure 2.Overexpression of IDO strengthened
cell proliferation of 16HBE cells. CFSE labeling was performed to
assess the cell proliferation of 16HBE cells treated with control,
NC, DEX, NC+DEX, and IDO+DEX. *P<0.05, **P<0.01 vs. control,
^^P<0.01 vs. NC. IDO, indoleamine 2, 3-dioxygenase;
CFSE, 5(6)-carboxyfluorescein diacetate succinimidyl ester; NC,
negative control; DEX, dexamethasone. |
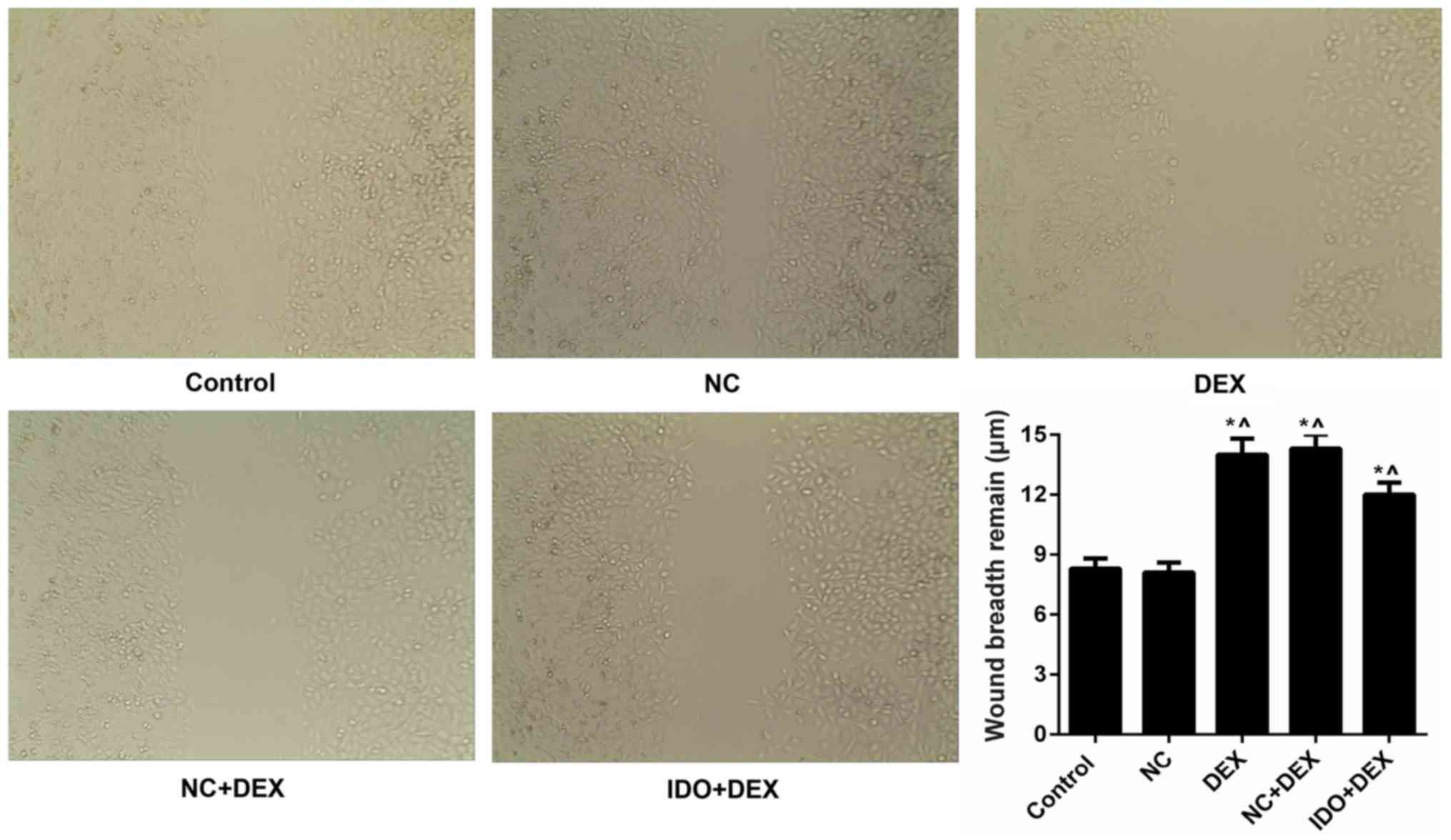 | Figure 3.Overexpression of IDO promoted the
cell migration of 16HBE cells. Wound-healing assay was carried out
to evaluate th cell migration of 16HBE cells treated with control,
NC, DEX, NC+DEX, and IDO+DEX. *P<0.05 vs. control,
^P<0.05 vs. NC. IDO, indoleamine 2, 3-dioxygenase;
16HBE, human lung bronchial epithelial cell line; NC, negative
control; DEX, dexamethasone. |
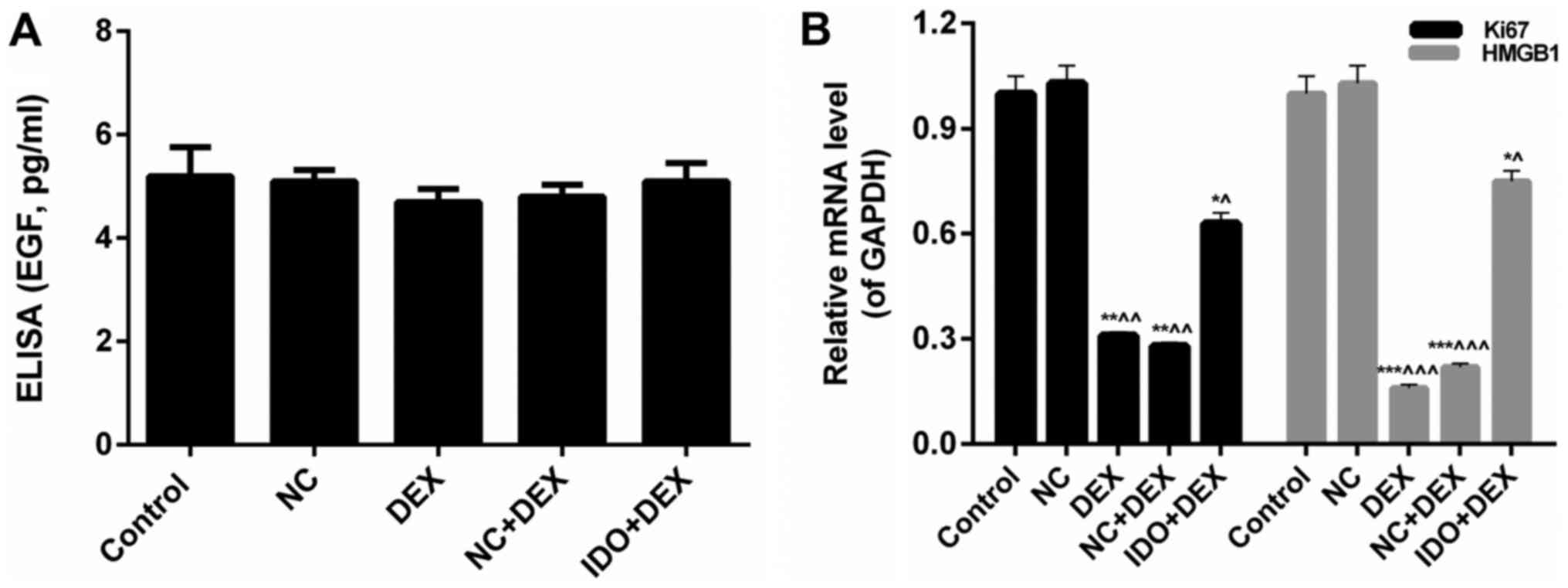 | Figure 4.Overexpression of IDO regulated the
expression of related factors. 16HBE cells were treated with
control, NC, DEX (10 µM), NC+DEX, and IDO+DEX. (A) ELISA was
performed on the EGF expression in the supernatant of 16HBE cells.
(B) RT-qPCR assay was performed to distinct the mRNA expression
levels of Ki67 and HMGB1 in 16HBE cells. *P<0.05, **P<0.01,
***P<0.001 vs. control, ^P<0.05,
^^P<0.01, ^^^P<0.001 vs. NC. IDO,
indoleamine 2, 3-dioxygenase; 16HBE, human lung bronchial
epithelial cell line; NC, negative control; DEX, dexamethasone;
EGF, epidermal growth factor; HMGB1, high-mobility group box 1
protein. |
Overexpression of IDO affected the
MAPK/ERK pathway
Furthermore, we detected the expression levels of
p-Raf-1, Raf-1, p-Mek1/2, Mek1/2, p-Erk1/2, and Erk1/2 in 16 HBE
cells from each group. Western blot results indicated that the
expression levels of p-Raf-1 in 16HBE cells treated with DEX were
significantly lower than control; in the meantime, distinct
increases of p-Raf-1 levels were observed in the 16HBE cells
treated with IDO (P<0.05; Fig.
5A). However, the expression levels of Raf-1 in five groups had
no significant difference (Fig. 5A).
The expression levels of p-Mek1/2 in DEX groups were markedly lower
than control, while the p-Mek1/2 levels were evidently increased by
treating with IDO (P<0.05; Fig.
5B). Nevertheless, the expression levels of Mek1/2 in five
groups had no significant difference (Fig. 5B). And compared with control group,
the expression levels of p-Erk1/2 were markedly reduced by DEX,
while the p-Erk1/2 expression in 16HBE cells treated with IDO was
significantly enhanced (P<0.05; Fig.
5C). Besides, the expression levels of Erk1/2 in all of the
groups had no significant difference (Fig. 5C). Therefore, it was affirmed that
overexpression of IDO affected the MAPK/ERK pathway in the repair
of human airway epithelial cells inhibited by DEX.
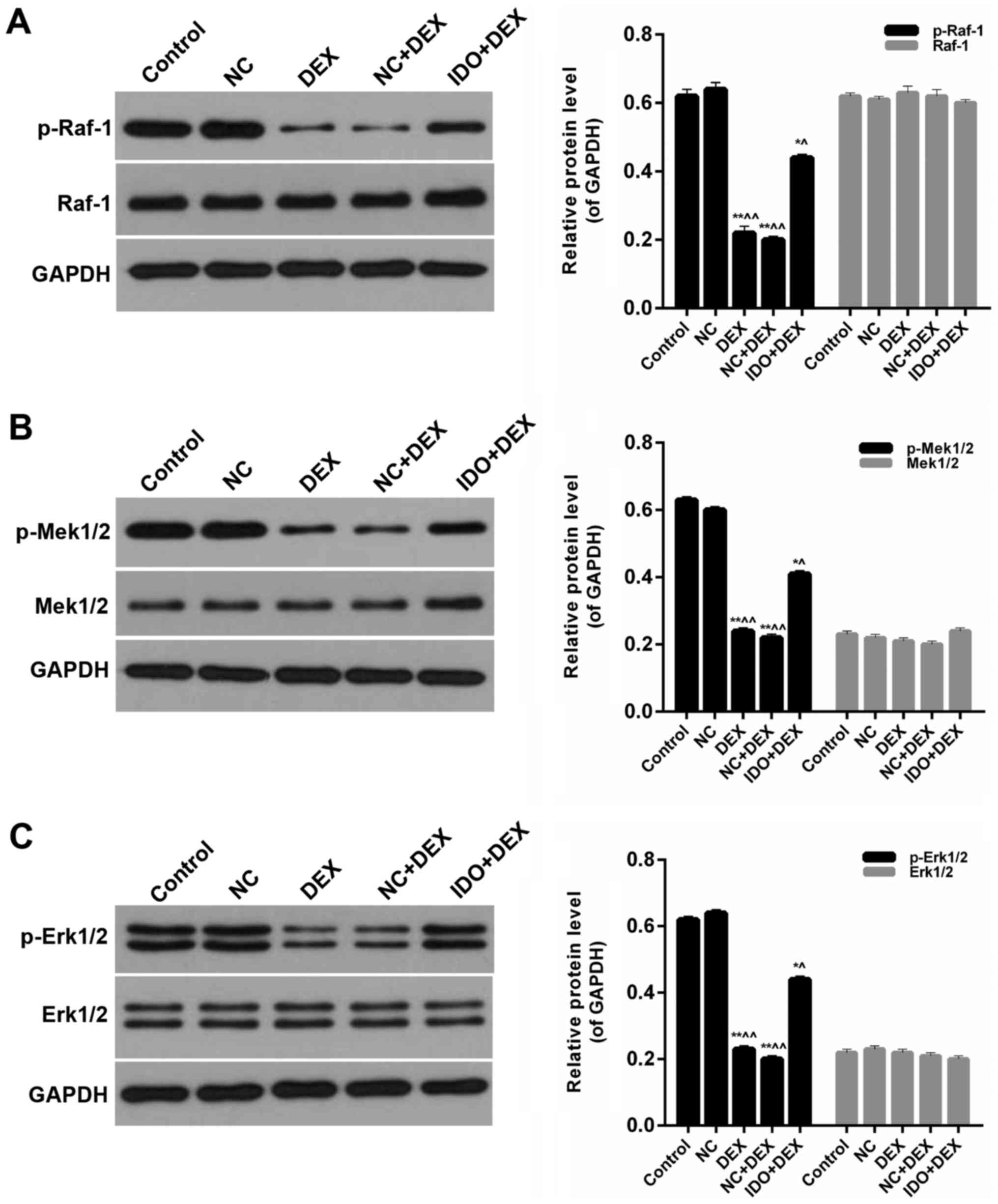 | Figure 5.Overexpression of IDO affected the
MAPK/ERK pathway. Western blot assay was performed on the protein
expression levels of (A) p-Raf-1, Raf-1, p-Mek1/2 Mek1/2 (B)
p-Erk1/2 and (C) Erk1/2 in 16HBE cells treated with control, NC,
DEX, NC+DEX, and IDO+DEX. *P<0.05, **P<0.01 vs. control,
^P<0.05, ^^P<0.01 vs. NC. IDO,
indoleamine 2, 3-dioxygenase; MAPK, mitogen-activated protein
kinase; ERK, extracellular regulated kinase; p-Raf-1,
phosphorylated-v-raf-1 murine leukemia viral oncogene homolog 1;
p-Mek1/2, phosphorylated-mitogen-activated protein kinase; NC,
negative control; DEX, dexamethasone. |
In order to further verify the impact of ERK and
HMGB1 on the cell proliferation and migration modulated by
overexpression of IDO, the ERK inhibitor (PD98059) and HMGB1
inhibitor (R, S-Sulforaphane) were used in our current study.
According to the CFSE labeling results, we found that after
inhibiting the expression of ERK, the cell proliferation of 16HBE
cells from each treatment group was evidently reduced (P<0.05;
Fig. 6A). The wound-healing assay
data also indicated that inhibition of ERK obviously lessened the
migration ability of 16HBE cells (P<0.05; Fig. 6B). Moreover, it was proved that the
migration ability of 16HBE cells was distinctly decreased by
treating with HMGB1 inhibitor (P<0.05; Fig. 6C). Hence, we could testify the
conclusion above that overexpression of IDO modulated the cell
proliferation and migration of 16HBE cells through regulating the
expression levels of ERK and HMGB1.
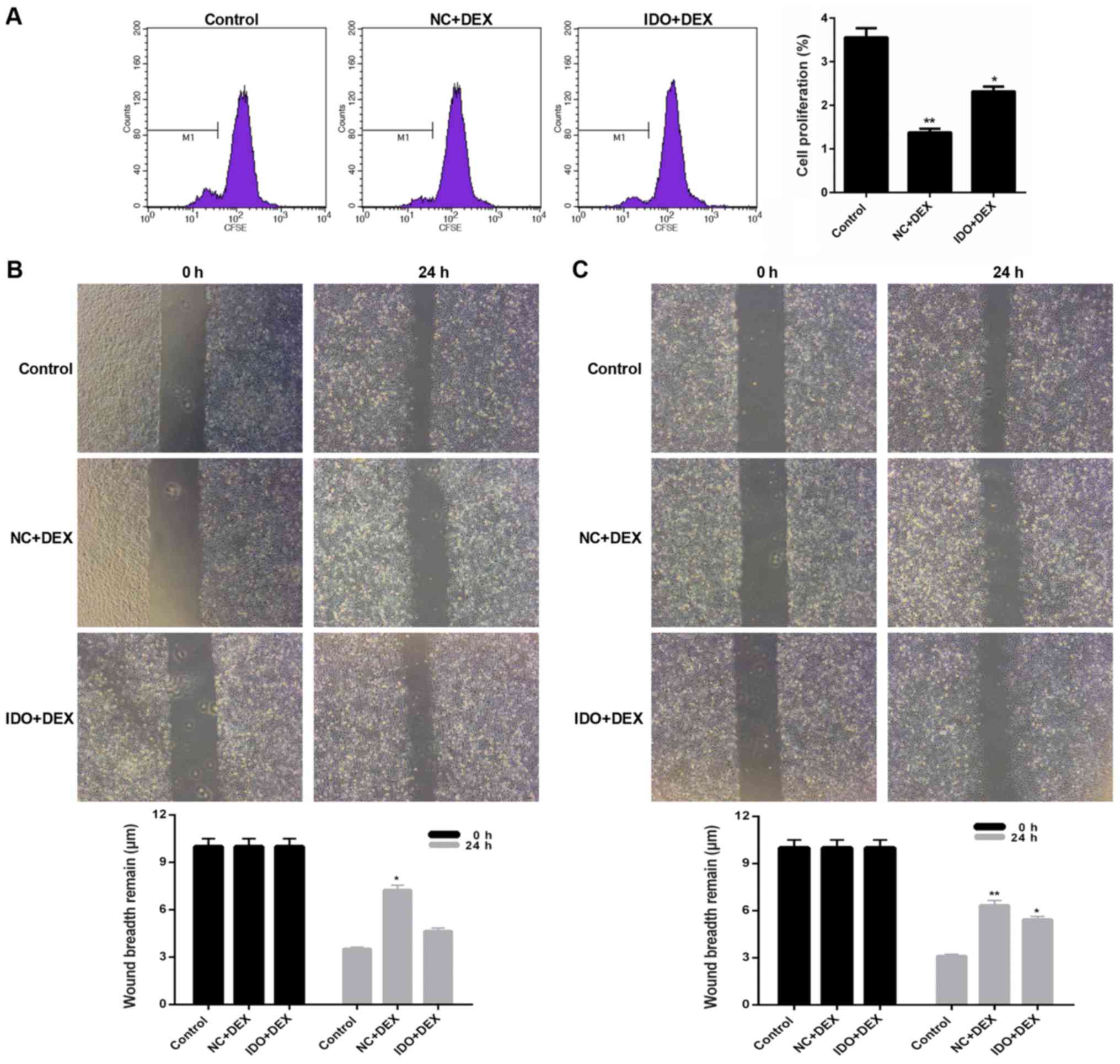 | Figure 6.Inhibition of ERK or HMGB1 suppressed
the cell proliferation and migration of 16HBE cells. 16HBE cells
were treated with ERK inhibitor (PD98059) in advance, and then
treated with control, NC+DEX, and IDO+DEX. (A) CFSE labeling was
carried out to evaluate the cell proliferation of 16 HBE cells. (B)
Wounding-healing assay was performed on the cell migration of 16HBE
cells. 16HBE cells were treated with HMGB1 inhibitor (R,
S-Sulforaphane) in advance, and then treated with control, NC+DEX,
and IDO+DEX. (C) Wounding-healing assay was performed to measure
the cell migration of 16HBE cells. *P<0.05, **P<0.01 vs.
control. ERK, extracellular regulated kinase; 16HBE, human lung
bronchial epithelial cell line; NC, negative control; DEX,
dexamethasone; IDO, indoleamine 2, 3-dioxygenase. |
Discussion
Asthma is mainly characterized by chronic airway
inflammation, and airway epithelial injury represents a critical
pathological basic of asthma. The dysfunctional epithelium in
asthmatic patients reveals abnormal structures, biochemical and
immune functions, which together lead to barriers of the airway
epithelium defence. It has been proved that the desquamation parts
of airway epithelium had significant structural changes (30). Ovalbumin (OVA) sensitization is a
common method used for establishing asthma models, which was
typical of airway epithelium abscission together with cell
apoptosis and could not been inhibited by regular hormone therapy
(6). Studies have demonstrated that
the related proteins maintaining integrity of airway epithelial
cells in asthmatic patients were changed, including tight junction
protein, adhesion junction protein, E-cadherin, basal cell marker,
and cytokeratin-5 (31–33). In the meantime, abnormal release of
growth factors and production of extracelluar matrix from airway
epithelial cells in asthmatic patients were observed (34,35).
These reasons above are important factors that affect the repair of
airway epithelial cells.
IDO is known as an immunosuppressive enzyme, which
plays an important role in inducing autoimmune diseases, maternal
fetal immune tolerance, tumor escape, and transplantation immune
tolerance (36). Previous studies
have proved that IDO was expressed by many tumors as well as by
infiltrating leucocytes presented within the tumor microenvironment
(37–39). It has been demonstrated recently that
airway epithelial cells produced IDO, and IDO contributed to the
epithelial cells-mediated inhibition of T cell activation (40). Therefore, in the present study, we
investigated the mRNA and protein expression levels of IDO in human
airway epithelial cells treated with IDO. It was suggested that
compared with NC, IDO significantly up-regulated the expression of
IDO in 16HBE cells. There is evidence that long-term inhalation of
GCs might give rise to lung damage in asthmatic patients (41), and DEX has been proved that inhibited
the repair of airway epithelium (42). However, the effects of IDO on the
cell viability and proliferation capacity of airway epithelial
cells are not clear. In our study, we found that the 16HBE cell
proliferation capacity suppressed by DEX was obviously enhanced by
overexpression of IDO. Hence, to the best of our knowledge, we
confirmed that IDO expression affected the 16HBE cells growth and
proliferation. Furthermore, we evaluated the expression levels of
EGF and Ki67 in 16HBE cells treated with IDO. EGF and Ki67 act as
growth factors, and play a critical role in the airway epithelial
cell growth and proliferation. According to the previous studies,
the expression levels of EGF in human airway epithelial cells could
be regulated by retinol and all transretinoic acid (43,44).
However, our results revealed that the EGF expression in 16HBE
cells was not affected by IDO. Additionally, overexpression of IDO
markedly enhanced the expression level of Ki67 in 16 HBE cells. All
together, it was demonstrated that overexpression of IDO
strengthened the cell proliferation capacity of airway epithelial
cells suppressed by DEX through regulating the expression level of
Ki67. Besides, we also found that overexpression of IDO
significantly enhanced the migration ability of 16HBE cells
inhibited by DEX. The abilities of migration and wound repair of
airway epithelial cells were demonstrated that could be
strengthened by ATP-mediated activation of dual oxidase 1 (45). Nevertheless, the effective role of
IDO played in promoting airway epithelial cell migration was first
studied in our research. We also investigated the exact mechanisms
of IDO affecting the airway epithelial cell migration. On the basis
of reported literature (46), we
chose HMGB1 as the object of study. It was indicated that
overexpression of IDO significantly enhanced the mRNA expression
level of HMGB1 in 16HBE cells suppressed by DEX. These results
suggested that overexpression of IDO enhanced the migration ability
of 16HBE cells inhibited by DEX via up-regulating the expression
level of HMGB1. It has been demonstrated that the expression of
MAPK/ERK pathway in human airway epithelial cells could be impacted
by 15-Lipoxygenase 1 interacts with
phosphatidylethanolamine-binding protein. Nevertheless, the
relationship between IDO and the regulation of MAPK/ERK pathway in
human airway epithelial cells is not clear. Therefore, we assessed
the expression levels of phosphorylated Raf-1, Mek1/2, and Erk1/2
in 16HBE cells treated with DEX and IDO. The results showed that
overexpression of IDO enhanced the phosphorylation of Raf-1,
Mek1/2, and Erk1/2 in 16HBE cells suppressed by DEX. These
consequences indicated that overexpression of IDO might be an
important regulator in the control of MAPK/ERK pathway in human
airway epithelium.
In our study, we proved that overexpression of IDO
contributed to the repair of 16HBE human airway epithelial cells
inhibited by DEX via affecting the MAPK/ERK pathway. However, there
also had some limitations in our works. We have only assessed one
human lung bronchial epithelial cell line in our investigation,
which might have limitations about the current conclusion. Hence,
in the future, the in vivo DEX-induced rat models will be
further used to investigate the roles and mechanisms of IDO in the
therapy of asthma. Moreover, we noted that Akt/mTOR signaling
pathway might be involved in mediating the effects of IDO treatment
on cell migration and proliferation, which will also be considered
in our subsequent research.
In the current study, it is suggested that the
overexpression of IDO is conducive to the repair of human airway
epithelial cells inhibited by DEX. According to the previous
article, functional IDO expression is high in the lung (47). It has been proved that immature
dendritic cells expressing IDO inhibit OVA-induced allergic airway
inflammation in vivo (48).
In addition, another study has confirmed that knocking down IDO
activity markedly decreased the ability of human airway epithelial
cells (40). However, the IDO-only
treated control group has not been designed in this study. Of
course, the addition of the IDO-only treated control group will
make the experiment more convincing.
Studies have reported that the asthmatic airway
epithelium revealed injury and shedding after GCs treatment. Our
results found that overexpression of IDO enhanced the proliferation
and migration of 16HBE cells inhibited by DEX. Besides, IDO
overexpressing up-regulated MAPK/ERK pathway in 16HBE cells treated
with DEX. Altogether, our findings first suggested that
overexpression of IDO contributed to the repair of human airway
epithelium suppressed by DEX through up-regulating MAPK/ERK
pathway. Our work provided an understanding that IDO might be a
potential genetic therapeutic agent and supported the utilization
of IDO in asthma.
In conclusion, the inhibition of epithelial injury
repair by DEX was ameliorated in part by the overexpression of IDO,
which enhanced airway epithelial cell viability, proliferation, and
migration through up-regulating the MAPK/ERK pathway.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SJ wrote the article. SJ, PG and XG performed the
experiments. SJ and PG designed the study. SJ, HW, JL and XF
performed data analysis. HW, JL and XF contributed to manuscript
revisions and all authors reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nakawah MO, Hawkins C and Barbandi F:
Asthma, chronic obstructive pulmonary disease (COPD), and the
overlap syndrome. J Am Board Fam Med. 26:470–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Papaiwannou A, Zarogoulidis P, Porpodis K,
Spyratos D, Kioumis I, Pitsiou G, Pataka A, Tsakiridis K, Arikas S,
Mpakas A, et al: Asthma-chronic obstructive pulmonary disease
overlap syndrome (ACOS): Current literature review. J Thorac Dis. 6
Suppl 1:S146–S151. 2014.PubMed/NCBI
|
|
3
|
Zalewska M, Furmańczyk K, Jaworski S,
Niemiro W and Samoliński B: The prevalence of asthma and declared
asthma in Poland on the basis of ECAP survey using correspondence
analysis. Comput Math Methods Med. 2013:5978452013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tam A, Wadsworth S, Dorscheid D, Man SF
and Sin DD: The airway epithelium: More than just a structural
barrier. Ther Adv Respir Dis. 5:255–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barbato A, Turato G, Baraldo S, Bazzan E,
Calabrese F, Panizzolo C, Zanin ME, Zuin R, Maestrelli P, Fabbri LM
and Saetta M: Epithelial damage and angiogenesis in the airways of
children with asthma. Am J Respir Crit Care Med. 174:975–981. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dorscheid DR, Wojcik KR, Sun S, Marroquin
B and White SR: Apoptosis of airway epithelial cells induced by
corticosteroids. Am J Respir Crit Care Med. 164:1939–1947. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Holgate ST: The airway epithelium is
central to the pathogenesis of asthma. Allergol Int. 57:1–10. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kardos Z: Treatment of chronic obstructive
pulmonary disease with inhaled pharmacotherapy: Role of
corticosteroids. Acta Pharm Hung. 82:33–41. 2012.(In Hungarian).
PubMed/NCBI
|
|
9
|
Spangler DL: The role of inhaled
corticosteroids in asthma treatment: A health economic perspective.
Am J Manag Care. 18 2 Suppl:S35–S39. 2012.PubMed/NCBI
|
|
10
|
De Bosscher K, Vanden Berghe W and
Haegeman G: Mechanisms of anti-inflammatory action and of
immunosuppression by glucocorticoids: Negative interference of
activated glucocorticoid receptor with transcription factors. J
Neuroimmunol. 109:16–22. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Davies L, Karthikeyan N, Lynch JT, Sial
EA, Gkourtsa A, Demonacos C and Krstic-Demonacos M: Cross talk of
signaling pathways in the regulation of the glucocorticoid receptor
function. Mol Endocrinol. 22:1331–1344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benayoun L, Letuve S, Druilhe A,
Boczkowski J, Dombret MC, Mechighel P, Megret J, Leseche G, Aubier
M and Pretolani M: Regulation of peroxisome proliferator-activated
receptor gamma expression in human asthmatic airways: Relationship
with proliferation, apoptosis, and airway remodeling. Am J Respir
Crit Care Med. 164:1487–1494. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wadsworth SJ, Nijmeh HS and Hall IP:
Glucocorticoids increase repair potential in a novel in vitro human
airway epithelial wounding model. J Clin Immunol. 26:376–387. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pearson G, Robinson F, Gibson Beers T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montiel M, Quesada J and Jiménez E:
Activation of calcium-dependent kinases and epidermal growth factor
receptor regulate muscarinic acetylcholine receptor-mediated
MAPK/ERK activation in thyroid epithelial cells. Cell Signal.
19:2138–2146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lind CR, Gray CW, Pearson AG, Cameron RE,
O'Carroll SJ, Narayan PJ, Lim J and Dragunow M: The
mitogen-activated/extracellular signal-regulated kinase kinase 1/2
inhibitor U0126 induces glial fibrillary acidic protein expression
and reduces the proliferation and migration of C6 glioma cells.
Neuroscience. 141:1925–1933. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ayroldi E, Zollo O, Macchiarulo A, Di
Marco B, Marchetti C and Riccardi C: Glucocorticoid-induced leucine
zipper inhibits the Raf-extracellular signal-regulated kinase
pathway by binding to Raf-1. Mol Cell Biol. 22:7929–7941. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rhen T and Cidlowski JA: Antiinflammatory
action of glucocorticoids-new mechanisms for old drugs. N Engl J
Med. 353:1711–1723. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dolušić E and Frédérick R: Indoleamine
2,3-dioxygenase inhibitors: A patent review (2008–2012). Expert
Opin Ther Pat. 23:1367–1381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X, Newton RC, Friedman SM and Scherle
PA: Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer
therapy. Curr Cancer Drug Targets. 9:938–952. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herbert A, Ng H, Jessup W, Kockx M,
Cartland S, Thomas SR, Hogg PJ and Wargon O: Hypoxia regulates the
production and activity of glucose transporter-1 and indoleamine
2,3-dioxygenase in monocyte-derived endothelial-like cells:
possible relevance to infantile haemangioma pathogenesis. Br J
Dermatol. 164:308–315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jasperson LK, Bucher C,
Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH and Blazar
BR: Indoleamine 2,3-dioxygenase is a critical regulator of acute
graft-versus-host disease lethality. Blood. 111:3257–3265. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnson TS, Munn DH and Maria BL:
Modulation of tumor tolerance in primary central nervous system
malignancies. Clin Dev Immunol. 2012:9372532012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Romani L, Fallarino F, De Luca A,
Montagnoli C, D'Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti
MC, Grohmann U, et al: Defective tryptophan catabolism underlies
inflammation in mouse chronic granulomatous disease. Nature.
451:211–215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watcharanurak K, Zang L, Nishikawa M,
Yoshinaga K, Yamamoto Y, Takahashi Y, Ando M, Saito K, Watanabe Y
and Takakura Y: Effects of upregulated indoleamine 2, 3-dioxygenase
1 by interferon γ gene transfer on interferon γ-mediated antitumor
activity. Gene Ther. 21:794–801. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oxenkrug GF: Metabolic syndrome,
age-associated neuroendocrine disorders, and dysregulation of
tryptophan-kynurenine metabolism. Ann N Y Acad Sci. 1199:1–14.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Myint AM and Kim YK: Network beyond IDO in
psychiatric disorders: Revisiting neurodegeneration hypothesis.
Prog Neuropsychopharmacol Biol Psychiatry. 48:304–313. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zoga M, Oulis P, Chatzipanagiotou S,
Masdrakis VG, Pliatsika P, Boufidou F, Foteli S, Soldatos CR,
Nikolaou C and Papageorgiou C: Indoleamine 2,3-dioxygenase and
immune changes under antidepressive treatment in major depression
in females. In Vivo. 28:633–638. 2014.PubMed/NCBI
|
|
30
|
Dunnill MS, Massarella GR and Anderson JA:
A comparison of the quantitative anatomy of the bronchi in normal
subjects, in status asthmaticus, in chronic bronchitis, and in
emphysema. Thorax. 24:176–179. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Boer WI, Sharma HS, Baelemans SM,
Hoogsteden HC, Lambrecht BN and Braunstahl GJ: Altered expression
of epithelial junctional proteins in atopic asthma: Possible role
in inflammation. Can J Physiol Pharmacol. 86:105–112. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kicic A, Sutanto EN, Stevens PT, Knight DA
and Stick SM: Intrinsic biochemical and functional differences in
bronchial epithelial cells of children with asthma. Am J Respir
Crit Care Med. 174:1110–1118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trautmann A, Kruger K, Akdis M,
Muller-Wening D, Akkaya A, Brocker EB, Blaser K and Akdis CA:
Apoptosis and loss of adhesion of bronchial epithelial cells in
asthma. Int Arch Allergy Immunol. 138:142–150. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hackett TL, Warner SM, Stefanowicz D,
Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick
SM, Bai TR and Knight DA: Induction of epithelial-mesenchymal
transition in primary airway epithelial cells from patients with
asthma by transforming growth factor-beta1. Am J Respir Crit Care
Med. 180:122–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stevens PT, Kicic A, Sutanto EN, Knight DA
and Stick SM: Dysregulated repair in asthmatic paediatric airway
epithelial cells: The role of plasminogen activator inhibitor-1.
Clin Exp Allergy. 38:1901–1910. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
MacKenzie CR, Heseler K, Müller A and
Däubener W: Role of indoleamine 2,3-dioxygenase in antimicrobial
defence and immuno-regulation: Tryptophan depletion versus
production of toxic kynurenines. Curr Drug Metab. 8:237–244. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Litzenburger UM, Opitz CA, Sahm F,
Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X,
et al: Constitutive IDO expression in human cancer is sustained by
an autocrine signaling loop involving IL-6, STAT3 and the AHR.
Oncotarget. 5:1038–1051. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Munn DH, Sharma MD, Hou D, Baban B, Lee
JR, Antonia SJ, Messina JL, Chandler P, Koni PA and Mellor AL:
Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic
cells in tumor-draining lymph nodes. J Clin Invest. 114:280–290.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yeung AW, Terentis AC, King NJ and Thomas
SR: Role of indoleamine 2,3-dioxygenase in health and disease. Clin
Sci (Lond). 129:601–672. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aldajani WA, Salazar F, Sewell HF, Knox A
and Ghaemmaghami AM: Expression and regulation of immune-modulatory
enzyme indoleamine 2,3-dioxygenase (IDO) by human airway epithelial
cells and its effect on T cell activation. Oncotarget.
7:57606–57617. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guilbert TW, Morgan WJ, Zeiger RS, Mauger
DT, Boehmer SJ, Szefler SJ, Bacharier LB, Lemanske RF Jr, Strunk
RC, Allen DB, et al: Long-term inhaled corticosteroids in preschool
children at high risk for asthma. N Engl J Med. 354:1985–1997.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu J, Zhang M, Niu C, Luo Z, Dai J, Wang
L, Liu E and Fu Z: Dexamethasone inhibits repair of human airway
epithelial cells mediated by glucocorticoid-induced leucine zipper
(GILZ). PLoS One. 8:e607052013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Baybutt RC, Smith BW, Donskaya EV, Hu L,
Li T and Wang W: The proliferative effects of retinoic acid on
primary cultures of adult rat type II pneumocytes depend upon cell
density. In Vitro Cell Dev Biol Anim. 46:20–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miller LA, Cheng LZ and Wu R: Inhibition
of epidermal growth factor-like growth factor secretion in
tracheobronchial epithelial cells by vitamin A. Cancer Res.
53:2527–2533. 1993.PubMed/NCBI
|
|
45
|
Wesley UV, Bove PF, Hristova M, McCarthy S
and van der Vliet A: Airway epithelial cell migration and wound
repair by ATP-mediated activation of dual oxidase 1. J Biol Chem.
282:3213–3220. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shim EJ, Chun E, Lee HS, Bang BR, Kim TW,
Cho SH, Min KU and Park HW: The role of high-mobility group box-1
(HMGB1) in the pathogenesis of asthma. Clin Exp Allergy.
42:958–965. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Takikawa O, Yoshida R, Kido R and Hayaishi
O: Tryptophan degradation in mice initiated by indoleamine
2,3-dioxygenase. J Biol Chem. 261:3648–3653. 1986.PubMed/NCBI
|
|
48
|
An XJ, Bai CX, Xia JB, Dang T, Qian P,
Qian GS and Liao W: Immature dendritic cells expressing indoleamine
2,3-dioxygenase suppress ovalbumin-induced allergic airway
inflammation in mice. J Investig Allergol Clin Immunol. 21:185–192.
2011.PubMed/NCBI
|














