Introduction
Gemfibrozil (GEM) is a member of the fibrate class
of lipid-lowering pharmaceuticals and has been widely used in the
therapy of different forms of hyperlipidemia and
hypercholesterolemia since the early 1970s (1). Fibrates act as agonists of the nuclear
receptor peroxisome proliferator-activated receptor α (PPARα),
which regulates gene expression for lipid catabolism and
lipoprotein metabolism (2). Fibrates
cause a moderate decrease in the content of plasma triglycerides
(TG) and increase cholesterol level in high density lipoproteins
(3). Clinical trials have
demonstrated that fibrates have a benignant effect on vascular
remodeling, inflammation, cardiovascular and coronary events
(4,5). However, there is currently little
understanding about the effects on fatty liver disease after drug
treatment, particularly in hepatocytes.
Non-alcoholic fatty liver disease (NAFLD) includes a
broad spectrum of liver injury, which is characterized by fat
infiltration (steatosis) with a TG content >5% liver weight with
no alcohol consumption, which is different from alcoholic fatty
liver disease (6). NAFLD has been
correlated with obesity, diabetes, insulin resistance,
hypertriglyceridemia and cardiovascular diseases, and represents
the hepatic manifestation of the metabolic syndrome (7–9).
Furthermore, the development process of liver disease includes
several stages, ranging from regional steatosis to nonalcoholic
steatohepatitis, and even to serious liver disease, such as
cirrhosis and hepatocellular carcinoma (10). It was estimated that the prevalence
of NAFLD ranges from 17–33% in the general population of Western
countries in 2003 (11).
Unfortunately, current effective therapies for NAFLD are limited
and therefore there is a critical requirement to identify the
mechanisms of NAFLD. According to the two-hit theory, the hallmark
of NAFLD is triacylglycerol accumulation in lipid droplets within
hepatocytes (12,13). In the present study, oleate-treated
human hepatoma SMMC-7721 cells were utilized as a model of
steatosis (14,15) to investigate the role of GEM in
regulating hepatic lipid metabolism.
Materials and methods
Antibodies and reagents
Cluster of differentiation (CD)36, sterol regulatory
element-binding protein 1 (SREBP1) and PPARα antibodies were
separately purchased from Santa Cruz Biotechnology, Inc., (Dallas,
TX, USA). β-actin rabbit monoclonal antibody was purchased from
Abcam (Cambridge, MA, USA). Secondary horseradish
peroxidase-labeled goat anti-rabbit immunoglobulin G (H+L) was
purchased from Novoprotein Scientific, Inc. (Summit, NJ, USA). GEM
was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Oleic acid (OA) and bovine serum albumin (BSA; fatty acid free)
were obtained from Sangon Biotech Co., Ltd., (Shanghai, China).
Cell culture and treatment
Human hepatoma SMMC-7721 cells were supplied by the
Institute of Cell Biology (Shanghai, China). The cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(Biological Industry, Kibbutz Beit Haemek, Israel), 1%
penicillin-streptomycin (10,000 U/ml penicillin and 10 mg/ml
streptomycin; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) and maintained at 37°C with humidified air in 5%
CO2. SMMC-7721 cells were first exposed to GEM when they
reached 75% confluence. Different dilutions (10–200 µM) of GEM were
made with DMSO and 10 µl of GEM solutions were then added to 1 ml
culture medium, respectively. To investigate the effect of OA on
fat-overloading, cultures were exposed to different concentrations
of OA, diluted in 10% BSA, ranging from 0.5–2 mM.
Cell viability assay
To determine the effect of GEM or OA on SMMC-7721
cell viability, the cells were treated with GEM at different
concentrations (0, 50, 100 and 200 µM) for 24 and 48 h at 37°C.
Following this, 1×106 cells in 96-well plates were
treated with OA at different concentrations (0, 0.5, 1, 1.5 and 2
mM) with 10% BSA overnight at 37°C, respectively. Cell viability
was determined using Cell Counting kit-8 dye (Beyotime Institute of
Biotechnology, Beijing, China), according to manufacturer's
instructions. Absorbance was measured at 450 nm with a GENios
multifunction-reader (Tecan GENios Pro; Tecan Group, Ltd.,
Männedorf, Switzerland).
Evaluation and quantification of lipid
accumulation
Oil red O (Sangon Biotech Co. Ltd., Shanghai, China)
was used to monitor the content of lipids in SMMC-7721 cells,
according to the manufacturer's instructions. Cells were seeded in
a 6-well plate at a density of 1.0×105 cells/well.
Following adherence, cells were treated with GEM at different
concentrations (0, 10, 25, 50 and 100 µM) together with 1 mM OA for
24 h at 37°C, respectively. Subsequently, cells were fixed
overnight at 37°C with 4% paraformaldehyde and stained with Oil red
O at 37°C for 30 min. Images were photographed with an inverted
fluorescent microscope (Nikon Eclipse TI; Nikon Corp., Tokyo,
Japan). Following this, Oil red O was extracted using isopropanol
(100 µl) for 1 h at 37°C. Then the extracted sample was moved to
another 96-well plate. Absorbance was measured at 510 nm in a
spectrophotometer for quantitative analysis (16).
Extraction and quantification of
TG
For quantitative estimation of TG, lipids were
extracted from cells using Triton X-100 (2%) for 30 min at 37°C. An
enzymatic assay was then performed using an EnzyChromTM
Triglyceride Assay kit (Bioassay Systems LLC, Hayward, CA, USA),
according to the manufacturer's protocols. Total lipid extraction
and separation was conducted by thin layer chromatography (TLC),
according to previous methods (16).
The cell pellets were harvested by centrifugation at 1,000 × g for
5 min at 37°C, washed twice with phosphate-buffered saline (PBS),
snap-frozen and smashed in 1 ml methanol chloroform mix (v/v, 2/1).
These components were mixed well and then centrifuged at 12,000 × g
for 5 min at room temperature to allow phase separation. The
chloroform phase was transferred to a new tube and blow-dried. The
lipid fractions were separated by TLC using a developing solvent
(hexane/diethyl ether/acetic acid; 40:80:2, v/v/v). To visualize
different fractions of total lipids extracted from cells, the TLC
plate (Sinopharm Chemical Reagent Co., Ltd., Beijing, China) was
stained with iodine vapor at 60°C for 30 min and photographed using
a DNR Bio-Imaging System, Ltd. (Neve Yamin, Israel).
RNA isolation, reverse
transcription-polymerase chain reaction (RT-PCR) and
RT-quantitative PCR (RT-qPCR) analyses
Total RNA was extracted using RNAiso Plus (Takara
Biotechnology Co. Ltd., Dalian, China), according to the
manufacturer's instructions, and quantified using a NanoDrop 2000c
(Thermo Fisher Scientific Inc., Waltham, MA, USA). First-strand
cDNA synthesis (1 µg) and PCR reactions were performed using the
PrimeScriptTM RT reagent kit with gDNA Eraser (Takara Biotechnology
Co. Ltd., Dalian, China), according to the manufacturer's
instructions. Following this, mRNA levels were determined by PCR
(EmeraldAmp PCR MasterMix; Catalogue no. RR300A; Takara
Biotechnology Co. Ltd., Dalian, China), as described in the study
by Bergman et al (17). The
primers were designed using Primer 5.0 software (Premier Biosoft
International, Palo Alto, CA, USA) and are listed in Table I. 18S ribosomal (r)RNA was selected
as an internal control. The PCR reaction conditions were as
follows: 98°C for 10 sec, 55°C for 30 sec and 72°C for 1 min for a
total 30 cycles. qPCR was performed in triplicate assays using SYBR
Premix Ex Taq (Tli RNaseH Plus; catalogue no. RR420; Takara
Biotechnology Co. Ltd., Dalian, China) in a CFX96 Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The qPCR reaction conditions were as follows: Activation of the Taq
DNA polymerase at 95°C for 30 sec, followed by 40 cycles of 95°C
for 10 sec and 60°C for 32 sec. mRNA expression levels were
analyzed by the 2−ΔΔCq method (18), relative to 18S rRNA expression.
 | Table I.Sequences of primers used in the
present study. |
Table I.
Sequences of primers used in the
present study.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Gene | Forward | Reverse |
|---|
| CD36 |
GAGAACTGTTATGGGGCTAT |
TTCAACTGGAGAGGCAAAGG |
| PPARα |
GCGATCTAGAGAGCCCGTTATC |
GCCAAAGCTTCCAGAACTATCC |
| SREBP1 |
CTGGTCGTAGATGCGGAGAA |
CATTGATGGAGGAGCGGTAG |
| LIPIN1 |
GACCTCACAGACATGGATCCTGAAG |
ACCGGGCTCCGTTGTCGCTTGCATG |
| LIPIN2 |
AACAAGTCATCGTATCACAGG |
CTCGCCAGTAGCAGAAGG |
| DGAT1 |
GCAGCCTCTTTCCTTCACTT |
GACCTCCCGCTACCATCAA |
| DGAT2 |
CGAAAGCCACTTCTCATACA |
TGCCTACTACTGCCCTCAC |
| CPT1 |
AAATTACGTGAGCGACTGG |
CTGCCTGAATGTGAGTTGGA |
| CPT2 |
CTGGTCAATGCGTATCCC |
GCCCAGATGTCTCGGTTC |
| ACOX1 |
GAAACCGCTGAGTAACAA |
ACAAACTGGAAGGCATAG |
| HADHA |
GGGATGTGGCAGTTGTTC |
GGACGGCACTTCTGATTT |
| 18S rRNA |
CGGCTACCACATCCAAGGAAG |
AGCTGGAATTACCGCGGCT |
Western blotting
Cells were incubated with either OA (1 mM) or OA
together with GEM (50 µM) for 24 h at 37°C and lysed with
pre-chilled radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology) for 30 min on ice. Following this,
lysates were centrifuged at 13,000 × g for 20 min at 4°C and
quantified by a Bradford protein assay (Bio-Rad Laboratories, Inc.)
(19). Proteins (50 µg) were
separated by 8% SDS-PAGE and transferred to Amersham Hybond-P
polyvinylidene fluoride membranes (GE Healthcare Life Sciences,
Little Chalfont, UK). Membranes were blocked with 5% milk powder in
PBS for 2 h at 37°C and then incubated with primary specific
antibodies overnight at 4°C, including CD36 (catalogue no. sc-9154;
1:2,000), PPARα (catalogue no. sc-9000; 1:1,000), SREBP1 (catalogue
no. sc-8984; 1:1,000) and β-actin (catalogue no. ab8226; 1:5,000)
rabbit monoclonal antibodies. The membrane was placed in PBS-Tween
20 (PBST) and cleaned 3 times for 10 min. Then samples were
incubated with secondary horseradish peroxidase (HRP)-labeled goat
anti-rabbit immunoglobulin G (H+L) (catalogue no. L153B; 1:1,000)
at room temperature for 1.5 h. After completion of secondary
antibody incubation, wash 3 times in PBST for 10 min/time Target
proteins on the membranes were visualized using Pro-light HRP
Chemiluminescent kit (cat. no. PA112; Tiangen Biotech Co., Ltd.,
Beijing, China) following the manufacturers protocol. The protein
bands were analyzed by densitometry using ImageJ software, version
1.37 (National Institutes of Health, Bethesda, MD, USA) after
exposure of the membranes to gel capture software, version 2.0 (DNR
Bio-Imaging System, Ltd., Jerusalem, Israel).
Statistical analysis
All experiments were performed at least three times.
Values were expressed as the mean ± standard deviation. Statistical
analysis was performed using the Student's t-test using SPSS
version 19.0 (IBM Corp., Armonk, NY, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
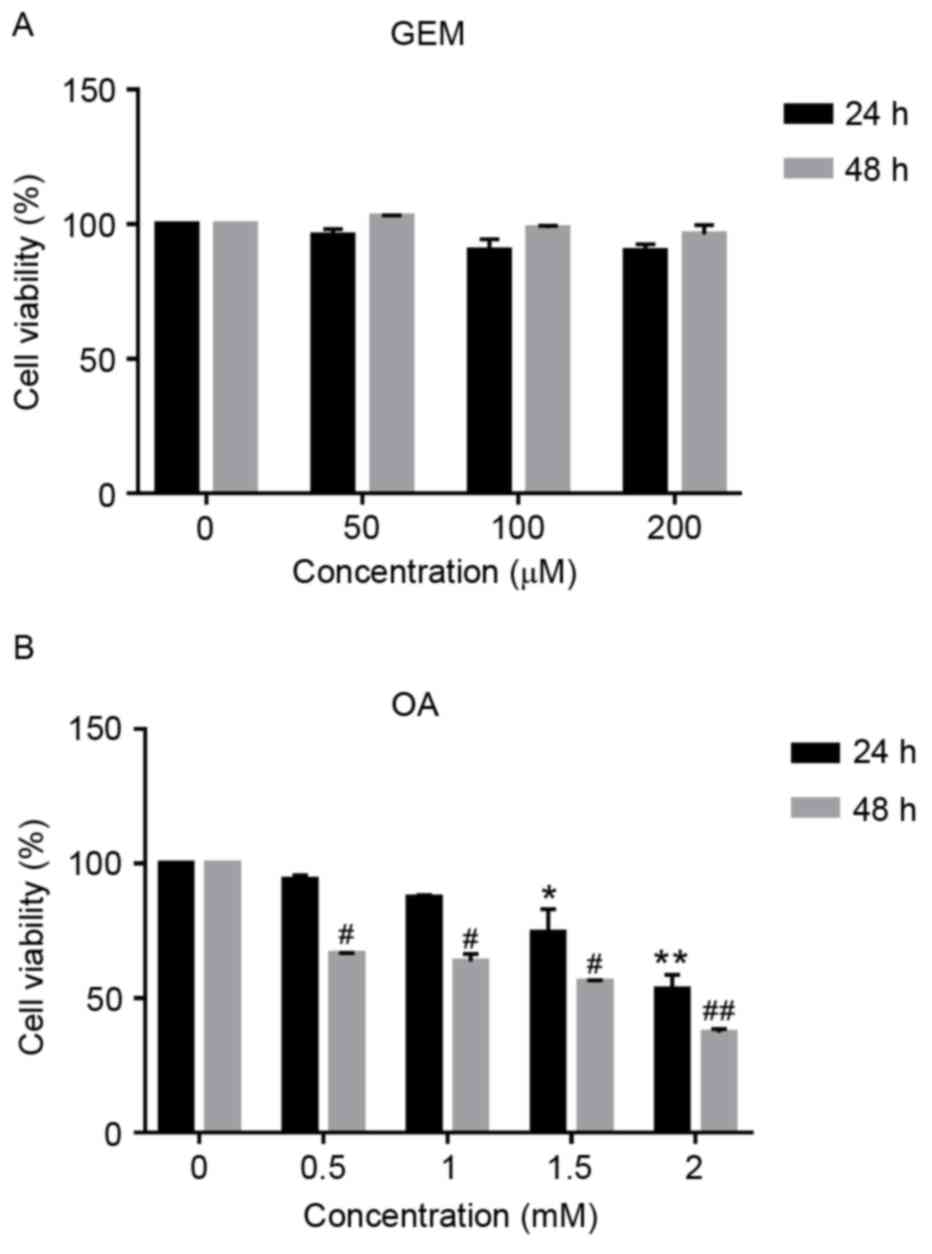
Cytotoxicity of GEM in an in vitro
hepatocellular model
Using the WST-8 based Colorimetric Assay Cell
Counting kit-8, the effect of GEM on cell viability was measured.
As demonstrated in Fig. 1A, no
significant cytotoxic effect was observed in cells following
treatment with GEM. Cell viability of SMMC-7721 cells was inhibited
by 4.22, 9.72 and 9.55% at GEM concentrations of 50, 100 and 200 µM
after 24 h, respectively. Cells were also exposed to different
concentration of OA. The concentrations of OA were chosen according
to our previous work (15). As
demonstrated in Fig. 1B, OA
inhibited cell viability in a dose- and time-dependent manner. When
cells were cultured in the presence of OA at 0.5 and 1 mM for 24 h,
they accumulated intracellular lipids without acute cytotoxic
effect, while cell viability was significantly inhibited when
treated with 1.5 and 2 mM OA for 24 and 48 h compared with 0 mM OA
(P<0.05). Cell viability was decreased by 25.7 and 46.9% at 1.5
mM OA for 24 and 48 h, respectively and 44 and 62.9% at 2 mM OA for
24 and 48 h, respectively. Therefore, cells treated with 1 mM OA
were used as the cellular model of NAFLD.
GEM ameliorates lipid
accumulation
To determine whether GEM affects lipid accumulation
in SMMC-7721 cells, cells were incubated with different
concentrations of GEM and 1 mM OA. As demonstrated in Fig. 2A, a clear dose-dependent decrease in
lipid accumulation was observed in the cells under the microscope.
Furthermore, the total lipid levels were decreased by 12.9, 21 and
16.5% in cells incubated with GEM concentrations of 25, 50 and 100
µM, respectively (Fig. 2B). The
results indicated that 50 µM of GEM significantly reduced levels of
intracellular total lipids and TG compared with cells treated with
1 mM OA only (P<0.05; Fig. 2B and
C). Therefore 50 µM was used for subsequent experiments and
assessments. The level of cellular TG was detected and was observed
to be decreased by 43.8% at 50 µM GEM compared with cells treated
with 1 mM OA only (Fig. 2C). To
further confirm the lipid changes in the cellular model, TLC
analysis was performed. TLC results suggested that there was a
decrease in TG levels in cells treated with 50 µM GEM compared with
those treated with OA only (Fig.
2D). Taken together, GEM may lower the lipid accumulation in an
in vitro model of NAFLD. The optimal concentration is 50
µM.
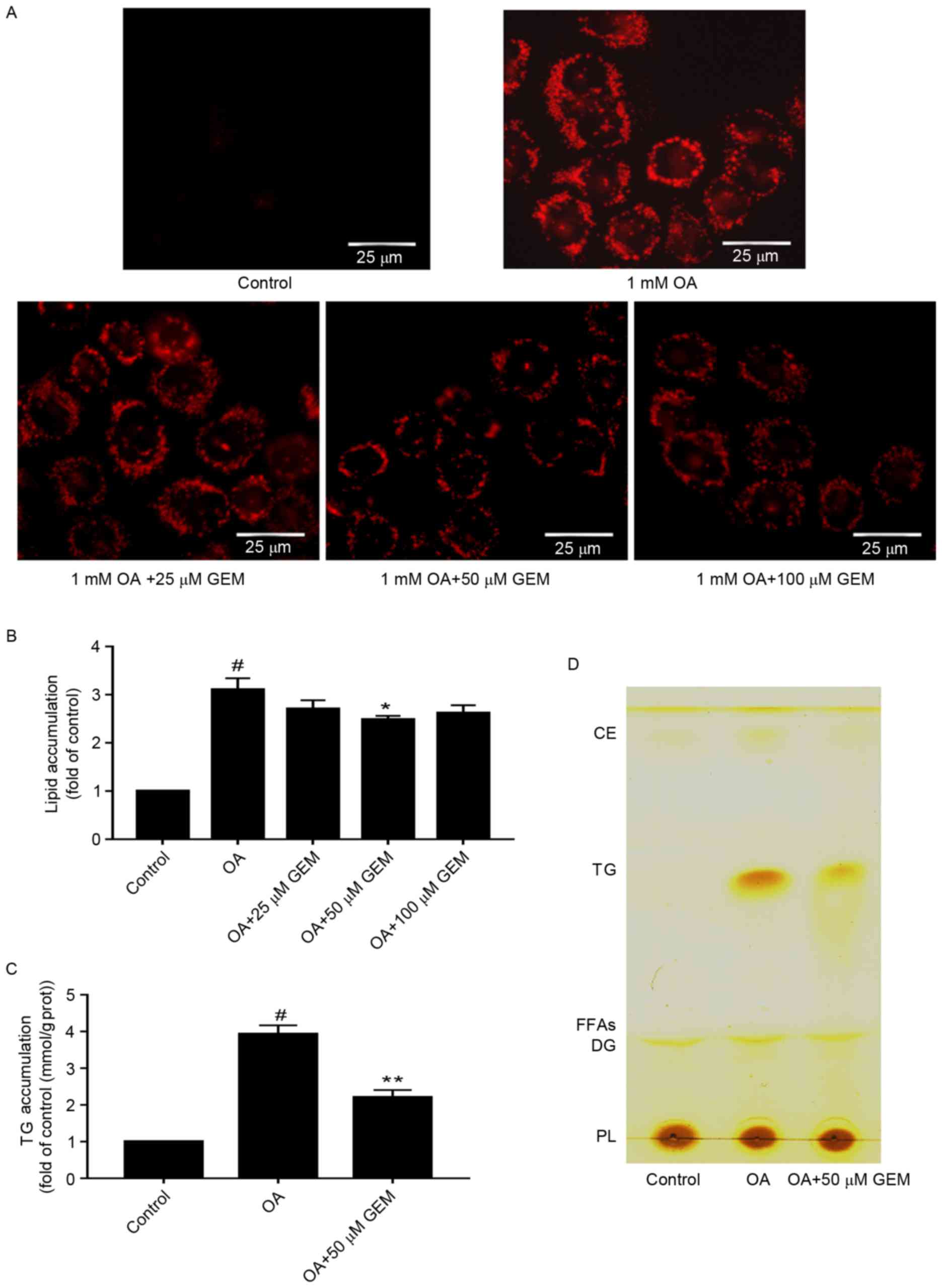 | Figure 2.Effect of GEM on lipid accumulation.
(A) After cells were treated with GEM at various concentrations
(25, 50 and 100 µM) and 1 mM OA for 24 h, intracellular lipid
droplets were stained with Oil red O and photographed by microscopy
(magnification, ×400; scale bar, 25 µm). (B) Quantification of
lipid content in OA-overloaded cells following treatment with
various concentrations (25, 50 and 100 µM) of GEM. Lipid content
was expressed as the fold of control. (C) Quantification of TG
content in OA-overloaded cells following treatment with 50 µM GEM.
TG content was expressed as the fold of control. (D) Total lipid
extraction and separation by thin layer chromatography. Data are
expressed at the mean + standard deviation. #P<0.05
vs. control cells; *P<0.05 and **P<0.01 vs. OA only. GEM,
gemfibrozil; OA, oleic acid; TG, triglyceride; CE, cholesteryl
ester; FFAs, free fat acid; DG, diacylglycerol; PL,
phospholipid. |
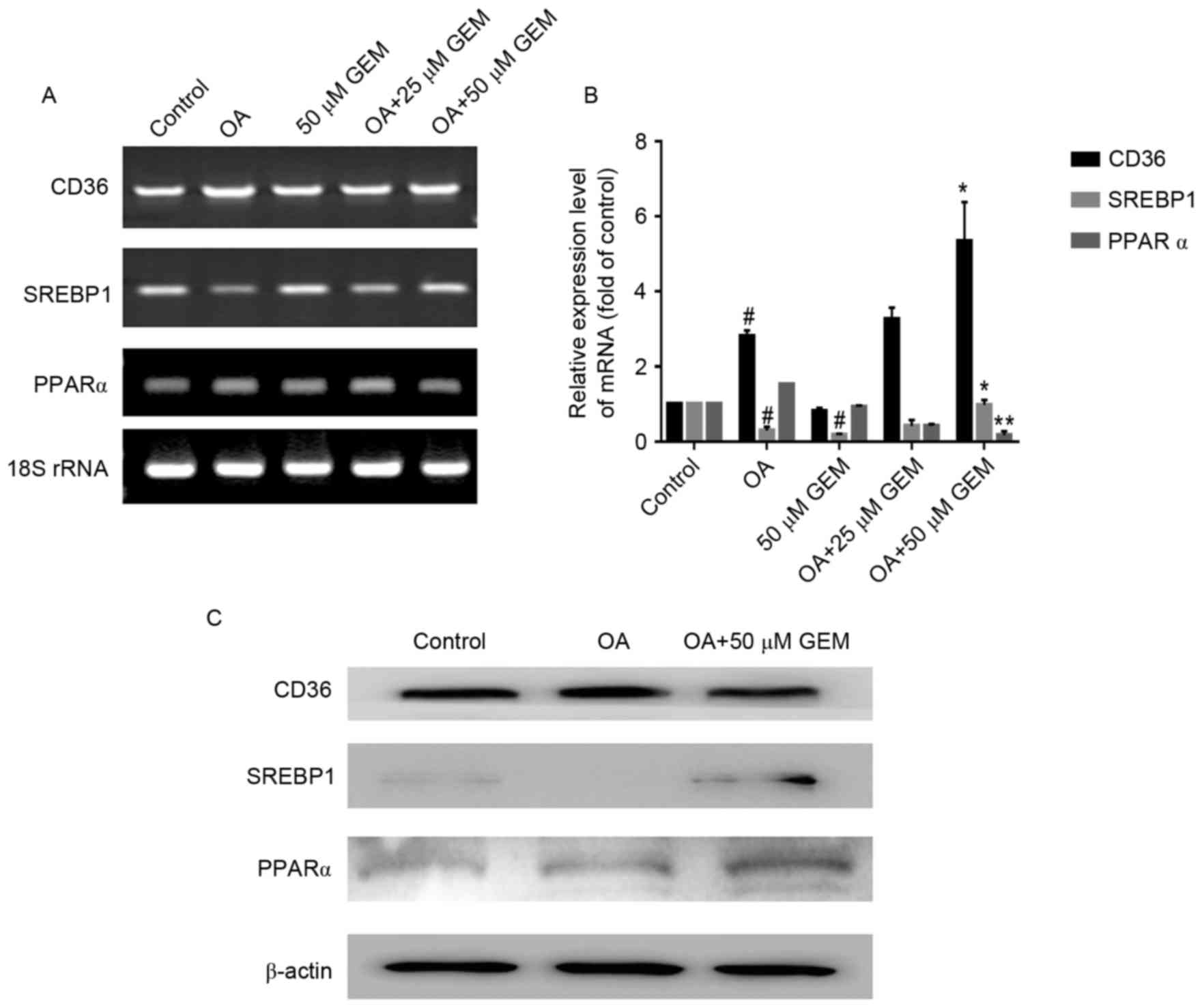
Changes to lipid metabolism-related
mRNA and protein expression
RT-PCR and RT-qPCR analyses were employed to
determine whether GEM was able to regulate lipid metabolism-related
gene expression in SMMC-7721 cells. The results demonstrated that
GEM (50 µM) significantly increased the levels of CD36, SREBP1
(P<0.05) and significantly reduced the levels of PPARα
(P<0.01) in cells treated with 1 mM OA compared with cells
treated with OA only (Fig. 3A and
B). Subsequently, western blotting with specific antibodies was
performed to detect the changes of related protein expression
levels. The expression levels of SREBP1 and PPARα were markedly
upregulated following treatment with GEM in the OA-overloaded
cells. However, the level of CD36 in OA-overloaded cells treated
with GEM showed no marked change compared with cells treated with
OA only (Fig. 3C).
Confirmation of modulation of lipid
synthesis and lipid oxidation
It is well recognized that lipogenic genes are
commonly trans-activated by SREBP1, and this has critical central
roles in the regulation of lipid synthesis (20). To validate the upregulation of
SREBP1, the expression levels of its downstream target genes, such
as LIPIN1, LIPIN2, diacylglycerol O-acyltransferase (DGAT)1
and DGAT2, were examined in the cells. The results
demonstrated that mRNA levels of LIPIN1 and DGAT1
were markedly increased following treatment with GEM in cells
treated with OA. However, mRNA expression levels of LIPIN2
and DGAT2 remained unchanged (Fig. 4A). As a transcription factor, PPARα
has central roles in hepatic lipid oxidation, predominantly through
regulating lipid target genes, such as carnitine
palmitoyltransferase (CPT)1, CPT2, acyl-coA oxidase 1 (ACOX1)
and hydroxyacyl-CoA dehydrogenase (HADHA) (21,22).
Therefore, mRNA expression level changes of these genes were
measured. The data demonstrated that mRNA levels of CPT2,
ACOX1 and HADHA were significantly increased in the
cellular model of NAFLD treated with GEM compared with the OA
control, while the CPT1 mRNA expression level was not
altered (Fig. 4B). Therefore, GEM
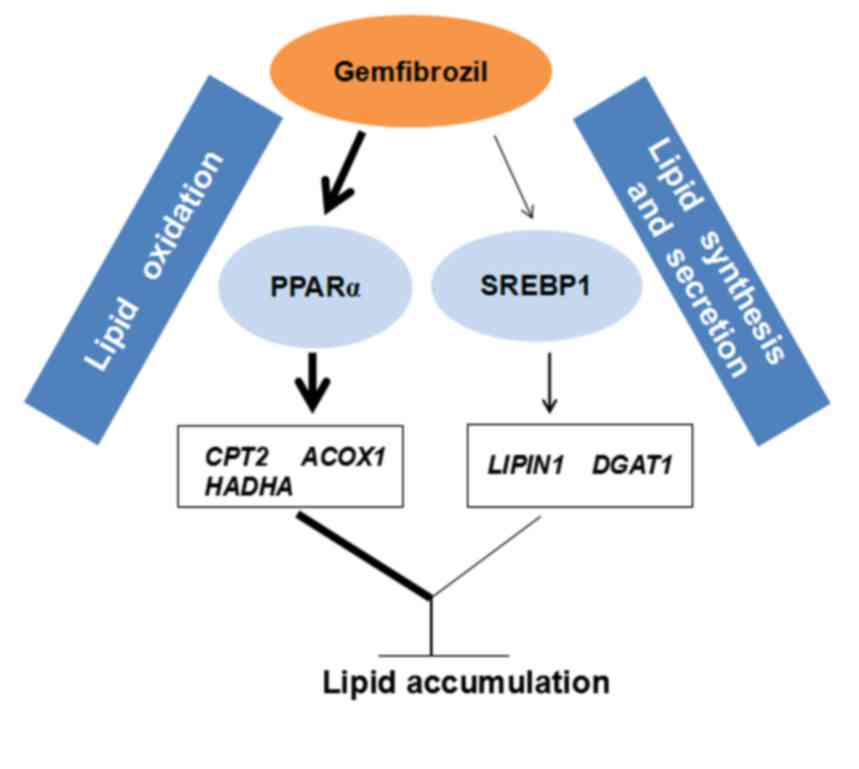
may lower TG accumulation in OA-overloaded SMMC-7721 cells via the
involvement of the PPARα and SREBP1 signaling pathways (Fig. 5).
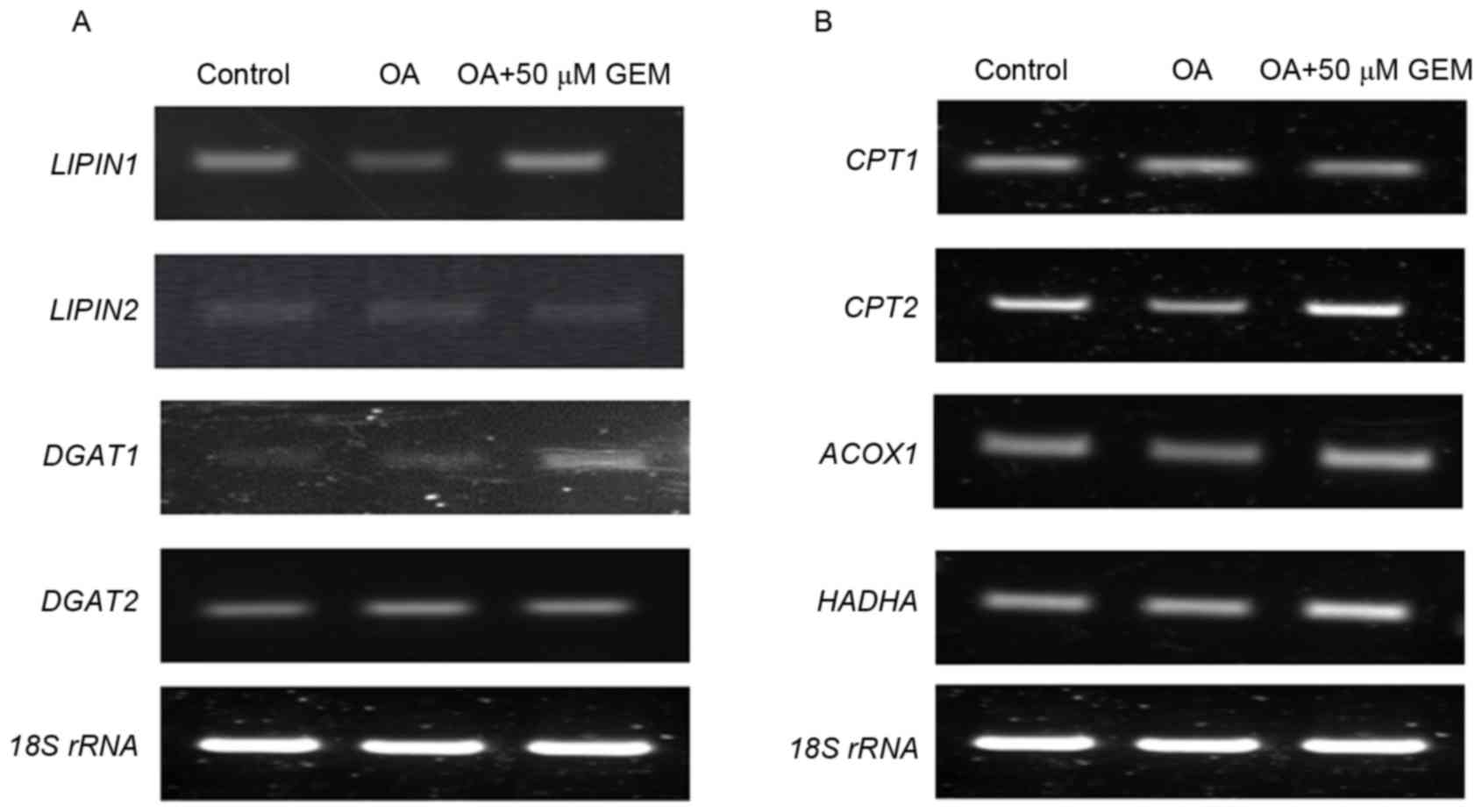 | Figure 4.Effect of GEM on sterol regulatory
element-binding protein 1 and peroxisome proliferator-activated
receptor α targets under the same experimental conditions. (A)
RT-PCR results of the effect of GEM on mRNA expression levels of
LIPIN1, LIPIN2, DGAT1 and DGAT2. (B) RT-PCR results
of effect of GEM on mRNA expression levels of CPT1, CPT2,
ACOX1 and HADHA. RT-PCR, reverse
transcription-polymerase chain reaction; GEM, gemfibrozil; OA,
oleic acid; rRNA, ribosomal RNA; DGAT, diacylglycerol
O-acyltransferase; CPT, carnitine palmitoyltransferase; ACOX1,
acyl-coA oxidase 1; HADHA, hydroxyacyl-CoA dehydrogenase. |
Discussion
Previous studies have demonstrated that GEM
functions as a PPARα agonist and is employed to treat
hyperlipidemia and hypercholesterolemia (1,2,4). A study by Smith et al (23) reported that large significant
declines in TG and smaller but significant declines in total lipids
were the classical response to GEM treatment in patients with
hypertriglyceridemia. In our laboratory, an in vitro
hepatocellular steatosis model has been successfully established in
human SMMC-7721 cells by using OA (15). In the present paper, the effect of
GEM on this cellular model was examined. The present results
demonstrated that GEM affected the expression levels of genes and
proteins related to lipid metabolism, leading to a decrease in the
lipid content and TG level in the fat over-accumulating
hepatocytes.
PPARα belongs to the nuclear receptor family and is
responsible for the regulation of lipid metabolism (24). The roles of PPARα in hepatic lipid
homeostasis are well established, it governs β-oxidation to
decrease lipid storage (25). PPARα
agonist treatment protects wild type mice fed a methionine choline
deficient (MCD) diet from both steatosis and steatohepatitis by
preventing hepatic lipid accumulation (26). The results of the present study
demonstrated that mRNA and protein levels of PPARα were altered in
OA-overloaded SMMC-7721 cells treated with GEM, which was
consistent with an increase in the expression levels of CPT2, ACOX1
and HADHA. A study by Ogata et al (27) observed the increase of hepatic lipid
oxidation in rats on a high fat diet supplemented with GEM. GEM
increased mRNA abundance of PPARα, as well as several of its
downstream targets in the male goldfish and zebrafish (28,29). In
the current study, the mRNA expression levels of PPARα
decreased while the protein expression of PPARα was increased
following treatment with GEM. Therefore, the present results
demonstrated further that GEM functions as a PPARα agonist to
activate lipid oxidation, leading to the reduction of excessive
intracellular TG content in the hepatic steatosis model.
In addition, the present study indicated that GEM
induced an increase in the mRNA and protein levels of SREBP1,
accompanied by an increase in LIPIN1 and DGAT1 mRNA.
SREBP1 is an important regulator of various genes involved in
hepatic lipid metabolism and homeostasis (30). SREBP1 is a transcription factor that
controls the anabolic pathways of cholesterol, free fat acids
(FFAs) and TG (31). In FFA
metabolism, SREBP-1 upregulates the expression of de novo
lipogenesis via fatty acid synthase (32). Elevated SREBP-1c increases lipogenic
gene expression, enhances fatty acid synthesis and accelerates TG
accumulation in mice (33). As one
of the downstream target genes of SREBP-1, LIPIN1 has been reported
to encode significant hepatic phosphatidic acid phosphatase (PAP)
activity (34). LIPIN1 deficiency is
associated with lipodystrophy and hepatic steatosis in mice
(35). DGAT activities catalyze the
synthesis of TG in lipid droplets for storage or in nascent
lipoproteins for secretion (36).
Additionally, inhibition of DGAT1 increased triacylglycerol
secretion, while inactivation of DGAT1 promoted large lipid droplet
formation (37). From the present
results, it is evident that GEM is involved in TG synthesis and
secretion.
CD36 is a membrane-associated protein that
facilitates the uptake of chylomicron and very low density
lipoprotein remnants, as well as long-chain FFAs (38,39).
Elevated CD36 expression is involved in steatosis of animal models
(40,41). In patients with NAFLD and chronic
hepatitis C virus, upregulation of CD36 expression is also detected
(42). However, it appeared that the
expression of CD36 was not affected by GEM in the present in
vitro model.
In conclusion, GEM lowers TG accumulation in
OA-overloaded SMMC-7721 cells via the involvement of the PPARα and
SREBP1 signaling pathways, which enhances lipid oxidation and
interferes with lipid synthesis and secretion. The present results
strongly suggest that GEM may potentially be utilized for the
treatment of NAFLD.
Acknowledgements
The present work was sponsored by grants from
Shanghai Scientific and Technological Innovation Project (grant no.
14520720700) and State Education Ministry and Fundamental Research
Funds for the Central Universities (grant no. 222201313010).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fruchart JC and Duriez P: Mode of action
of fibrates in the regulation of triglyceride and HDL-cholesterol
metabolism. Drugs Today (Barc). 42:39–64. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mandard S, Müller M and Kersten S:
Peroxisome proliferator-activated receptor alpha target genes. Cell
Mol Life Sci. 61:393–416. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chinetti-Gbaguidi G, Fruchart JC and
Staels B: Pleiotropic effects of fibrates. Curr Atheroscler Rep.
7:396–401. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jun M, Foote C, Lv J, Neal B, Patel A,
Nicholls SJ, Grobbee DE, Cass A, Chalmers J and Perkovic V: Effects
of fibrates on cardiovascular outcomes: A systematic review and
meta-analysis. Lancet. 375:1875–1884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Keech A, Simes RJ, Barter P, Best J, Scott
R, Taskinen MR, Forder P, Pillai A, Davis T, Glasziou P, et al:
Effects of long-term fenofibrate therapy on cardiovascular events
in 9,795 people with type 2 diabetes mellitus (the FIELD study):
Randomised controlled trial. Lancet. 366:1849–1861. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al: Design and validation of a histological
scoring system for nonalcoholic fatty liver disease. Hepatology.
41:1313–1321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brunt EM: Pathology of nonalcoholic fatty
liver disease. Nat Rev Gastroenterol Hepatol. 7:195–203. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ajmal MR, Yaccha M, Malik MA, Rabbani MU,
Ahmad I, Isalm N and Abdali N: Prevalence of nonalcoholic fatty
liver disease (NAFLD) in patients of cardiovascular diseases and
its association with hs-CRP and TNF-α. Indian Heart J. 66:574–579.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Narasimhan S, Gokulakrishnan K,
Sampathkumar R, Farooq S, Ravikumar R, Mohan V and Balasubramanyam
M: Oxidative stress is independently associated with non-alcoholic
fatty liver disease (NAFLD) in subjects with and without type 2
diabetes. Clin Biochem. 43:815–821. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duan XY, Zhang L, Fan JG and Qiao L: NAFLD
leads to liver cancer: Do we have sufficient evidence? Cancer Lett.
345:230–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shifflet A and Wu YG: Non-alcoholic
steatohepatitis: An overview. J Formos Med Assoc. 108:4–12. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Day CP and James OF: Steatohepatitis: A
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Musso G, Gambino R and Cassader M: Recent
insights into hepatic lipid metabolism in non-alcoholic fatty liver
disease (NAFLD). Prog Lipid Res. 48:1–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui W, Chen SL and Hu KQ: Quantification
and mechanisms of oleic acidinduced steatosis in HepG2 cells. Am J
Transl Res. 2:95–104. 2010.PubMed/NCBI
|
|
15
|
Wang S, Kuang X, Fang ZJ, Huang Z and Shi
P: Effect of oleic acid on the levels of eight metal ions in human
hepatoma SMMC-7721 cells. Biol Trace Elem Res. 159:445–450. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ramírez-Zacarías JL, Castro-Muñozledo F
and Kuri-Harcuch W: Quantitation of adipose conversion and
triglycerides by staining intracytoplasmic lipids with Oil red O.
Histochemistry. 6:493–497. 1992. View Article : Google Scholar
|
|
17
|
Bergman AC, Benjamin T, Alaiya A, Waltham
M, Sakaguchi K, Franzén B, Linder S, Bergman T, Auer G, Appella E,
et al: Identification of gel-separated tumor marker proteins by
mass spectrometry. Electrophoresis. 21:679–686. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bradford MM: A rapid and sensitive method
for quantitation of microgram quantities of protein utilizing the
principle of protein-dye binding. Anal Biochem. 72:248–254. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown MS and Goldstein JL: The SREBP
pathway: Regulation of cholesterol metabolism by proteolysis of a
membrane-bound transcription factor. Cell. 89:331–340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang H, McIntosh AL, Martin AG, Petrescu
AD, Landrock KK, Landrock D, Kier AB and Schroeder F: Inhibitors of
fatty acid synthesis induce PPAR α-regulated fatty acid β-oxidative
genes: Synergistic roles of L-FABP and glucose. PPAR Res.
2013:804–865. 2013. View Article : Google Scholar
|
|
22
|
Bishop-Bailey D: Peroxisome
proliferator-activated receptors in the cardiovascular system. Br J
Pharmacol. 129:823–834. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smith WJ, Wang J, Dang AQ, Reeves C, Bibbs
D and Faas FH: Gemfibrozil lowers plasma lipids and increases
polyunsaturated fatty acid content and oxidative susceptibility of
lipoproteins in hypertriglyceridemia. Clin Chim Acta. 322:77–84.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Uchida A, Slipchenko MN, Cheng JX and
Buhman KK: Fenofibrate, a peroxisome proliferator-activated
receptor α agonist, alters triglyceride metabolism in enterocytes
of mice. Biochim Biophys Acta. 1811:170–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peters JM, Rusyn I, Rose ML, Gonzalez FJ
and Thurman RG: Peroxisome proliferator-activated receptor alpha is
restricted to hepatic parenchymal cells, not Kupffer cells:
Implications for the mechanism of action of peroxisome
proliferators in hepatocarcinogenesis. Carcinogenesis. 21:823–826.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fernández-Rojo MA, Restall C, Ferguson C,
Martel N, Martin S, Bosch M, Kassan A, Leong GM, Martin SD, McGee
SL, et al: Caveolin-1 orchestrates the balance between glucose and
lipid-dependent energy metabolism: Implications for liver
regeneration. Hepatology. 55:1574–1584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ogata M, Tsujita M, Hossaina MA, Akita N,
Gonzalez FJ, Staels B, Suzuki S, Fukutomi T, Kimura G and Yokoyama
S: On the mechanism for PPAR agonists to enhance ABCA1 gene
expression. Atherosclerosis. 205:413–419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mimeault C, Trudeau VL and Moon TW:
Waterborne gemfibrozil challenges the hepatic antioxidant defense
system and down-regulates peroxisome proliferator activated
receptor beta (PPARbeta) mRNA levels in male goldfish (Carassius
auratus). Toxicology. 228:140–150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Passeri MJ, Cinaroglu A, Gao C and Sadler
KC: Hepatic steatosis in response to acute alcohol exposure in
zebrafish requires sterol regulatory element binding protein
activation. Hepatology. 49:443–452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T,
Zhong Z, McCue PA, Sang N, Ji JY, et al: Repression of endometrial
tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle.
11:2348–2358. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stone SJ, Myers HM, Watkins SM, Brown BE,
Feingold KR, Elias PM and Farese RV Jr: Lipopenia and skin barrier
abnormalities in DGAT2-deficient mice. J Biol Chem.
279:11767–11776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sumida Y, Niki E, Naito Y and Yoshikawa:
Involvement of free radicals and oxidative stress in NAFLD/NASH.
Free Radical Res. 47:869–880. 2004. View Article : Google Scholar
|
|
33
|
Horton JD, Bashmakov Y, Shimomura I and
Shimano H: Regulation of sterol regulatory element binding proteins
in livers of fasted and refed mice. Proc Natl Acad Sci USA.
95:5987–5992. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kok BP, Dyck JR, Harris TE and Brindley
DN: Differential regulation of the expressions of the PGC-1α splice
variants, lipins, and PPARα in heart compared to liver. J Lipid
Res. 54:1662–1677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Péterfy M, Phan J, Xu P and Reue K:
Lipodystrophy in the fld mouse results from mutation of a new gene
encoding a nuclear protein. Lipin Nat Genet. 27:121–124. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yen CLE, Stone SJ, Koliwad S, Harris C and
Farese RV Jr: Thematic review series: Glycerolipids. DGAT enzymes
and triacylglycerol biosynthesis. J Lipid Res. 49:2283–2301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li C, Li L, Lian J, Watts R, Nelson R,
Goodwin B and Lehner R: Roles of Acyl-CoA: Diacylglycerol
acyltransferases 1 and 2 in triacylglycerol synthesis and secretion
in primary hepatocytes. Arterioscl Throm Vas. 35:1080–1091. 2015.
View Article : Google Scholar
|
|
38
|
Ibrahimi A and Abumrad NA: Role of CD36 in
membrane transport of long-chain fatty acids. Curr Opin Clin Nutr.
5:139–145. 2002. View Article : Google Scholar
|
|
39
|
Kennedy DJ, Kuchibhotla S, Westfall KM,
Silverstein RL, Morton RE and Febbraio M: A CD36-dependent pathway
enhances macrophage and adipose tissue inflammation and impairs
insulin signalling. Cardiovasc Res. 89:604–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ouwens DM, Diamant M, Fodor M, Habets DDJ,
Pelsers MMAL, El Hasnaoui M, Dang ZC, van den Brom CE, Vlasblom R,
Rietdijk A, et al: Cardiac contractile dysfunction in
insulin-resistant rats fed a high-fat diet is associated with
elevated CD36-mediated fatty acid uptake and esterification.
Diabetologia. 50:1938–1948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koonen DP, Jacobs RL, Febbraio M, Young
ME, Soltys CL, Ong H, Vance DE and Dyck JR: Increased hepatic CD36
expression contributes to dyslipidemia associated with diet-induced
obesity. Diabetes. 56:2863–2871. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miquilena-Colina ME, Lima-Cabello E,
Sánchez-Campos S, García-Mediavilla MV, Fernández-Bermejo M,
Lozano-Rodríguez T, Vargas-Castrillón J, Buqué X, Ochoa B,
Aspichueta P, et al: Hepatic fatty acid translocase CD36
upregulation is associated with insulin resistance,
hyperinsulinaemia and increased steatosis in non-alcoholic
steatohepatitis and chronic hepatitis C. Gut. 60:1394–1402. 2011.
View Article : Google Scholar : PubMed/NCBI
|