Introduction
By inhibiting the synthesis of
mycobacterium-specific mycolic acid, isonicotinic acid hydrazide
(INH) causes the loss of acid resistance and hydrophobicity,
followed by proliferation arrest and death of bacteria. It kills
bacteria inside and outside of host cells in the breeding and
resting state (1). INH is an
irreplaceable first-line drug recommended by the World Health
Organization as a standard chemotherapy for tuberculosis (TB)
(2). However, INH causes changes of
liver function and even serious liver injury in certain patients,
resulting in the interruption of chemotherapy and TB drug
resistance, therefore bringing great difficulties to the control of
TB (3). The mechanisms by which
anti-TB drugs induce liver toxicity have remained to be fully
elucidated (4), making it
challenging to develop targeted liver-protective therapies.
Therefore, there is an urgent need to explore the mechanisms of
anti-TB drug-induced liver injury. The involvement of mitochondria
in the pathogenesis of anti-TB drug-induced hepatotoxicity (ADIH)
has gained much attention in recent years. Mitochondria are
important organelles within eukaryotic cells, which generate
adenosine triphosphate (ATP) through an oxidative phosphorylation
reaction with electron transfer for the body (5). Mitochondria have further important
functions, including the generation of reactive oxygen species,
regulation of the cell's redox potential, signal transduction,
apoptosis and regulation of gene expression. Therefore,
mitochondria have significant roles in cell growth, metabolism and
disease development (6).
Mammalian liver cells contain abundant mitochondria.
As the liver has a major role in the metabolization of INH, the
liver mitochondria are exposed to the toxic metabolites of INH and
are easily damaged (7). As
mitochondrial DNA (mtDNA) is bare, lacking the protection of
histones and DNA-binding proteins as well as effective repair
mechanisms, they are vulnerable to damage (8,9).
Numerous studies have identified that mtDNA is closely associated
with oxidative damage and mitochondrial myopathy, neurodegenerative
diseases and aging (10–12). The present study examined the effect
of INH on mitochondrial anti-oxidant enzymes and mitochondrial DNA,
aiming to provide clues regarding the etiology of drug-induced
liver injury.
Materials and methods
Instruments and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum and 0.25% trypsin were obtained from Gibco (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). A Mitochondrial
Extraction kit (CA: SM0020) was purchased from Beijing Solarbio
Co., Ltd. (Beijing, China). MDA (CA: A003-2), SOD (CA: A001-3) and
GSH-PX (CA: A005) assay kit were from Nanjing Jiancheng
Bioengineering Institute (Nanjing, China). 8-OHdG ELISA kit (CA:
ab201734; Abcam, Cambridge, MA, USA). INH injection was obtained
from Tianjin Kingyork Group Co., Ltd (Tianjin, China). HepG2 cells
were supplied by Obio Technology Co., Ltd. (Shanghai, China). The
plate reader was from Molecular Devices (Sunnyvale, California USA;
SpectraMax M5).
Cell culture and treatments
HepG2 cells were routinely cultured in DMEM complete
culture medium containing 10% fetal bovine serum, with culture
conditions of 37°C and 5% CO2. Based on the INZ
concentrations added (13), cells
were divided into control group, low-dose group (1 mg/ml),
medium-dose group (2 mg/ml) and high-dose group (4 mg/ml), with a
treatment time of 24, 48, 72 or 96 h. Each group had six
replicates.
Mitochondrial protein
quantification
After 48 or 96 h of treatment, cells were collected
by centrifugation at 800 × g for 5 min. The protein concentration
of each extracted mitochondrial sample was measured strictly
following the instructions of the measurement kit. The absorbance
was measured at 562 nm on a microplate reader, and the protein
concentration of each sample was calculated according to a standard
curve. The extracted mitochondria were used immediately or stored
at −70°C in storage buffer for later detection.
Determination of malondialdehyde (MDA)
and anti-oxidant enzymes in mitochondria
MDA and the enzymatic activity of superoxide
dismutase (SOD) and glutathione peroxidase (GSH-Px) were measured
using the above mentioned commercial kits following the
manufacturer's instructions.
Determination of mitochondrial
8-hydroxy-2-deoxyguanosine (8-OHdG) content
Mitochondrial 8-OHdG was measured by 8-OHdG ELISA
kit following the instructions of the manufacturer. Absorbance
values were measured on a microplate reader.
Ultrastructural observation
Following treatment, cells were collected by
centrifugation at 800 × g for 5 min at 4°C. Cells were fixed in
2.5% glutaraldehyde (v/v) immediately on ice for 2 h, post-fixed
for 1 h in 1% (v/v) osmium tetroxide, dehydrated in acetone and
embedded in Epon 812 (02635-AB; SPI Supplies; West Chester,
Pennsylvania, USA). From appropriate locations, ultrathin sections
(50 nm) were obtained and the sections were stained with 3% uranyl
acetate and lead citrate, followed by ultrastructural observation
under an electron microscope (HitachiH-7650; Hitachi, Ltd., Tokyo,
Japan) operated at 80 KV.
Statistical analysis
Values are expressed as the mean ± standard error.
Differences among groups were tested by one-way analysis of
variance and least-significant differences post-hoc test using SPSS
13 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Mitochondrial anti-oxidant enzyme and
MDA measurements SOD activity
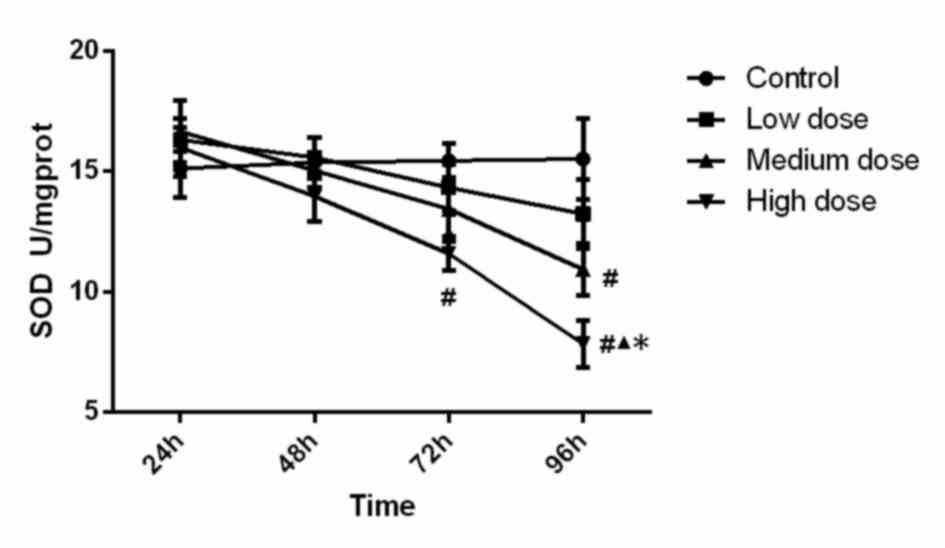
SOD activity in the different groups is displayed in
Fig. 1. Statistical analysis showed
that SOD activity in the high-dose group at 72 h was significantly
lower (P<0.001) than that in the control group, SOD activity in
the high-dose group at 96 h was significantly lower than that in
all other three groups (compared with control, low-dose and
medium-dose groups; P<0.001, P<0.001 and P=0.015). In
addition, the activity of SOD in the medium-dose group at 96 h was
significantly lower (P=0.005) than that in the control group.
Analysis of variance showed that the concentration (F=6.099,
P=0.001), time (F=11.088, P<0.001) and the interaction between
concentration and time (F=2.135, P=0.036) were significant, which
demonstrated that time and concentration both have an effect on SOD
activity. These findings suggest that SOD activity was reduced
along with the increase of the INH concentration and incubation
time.
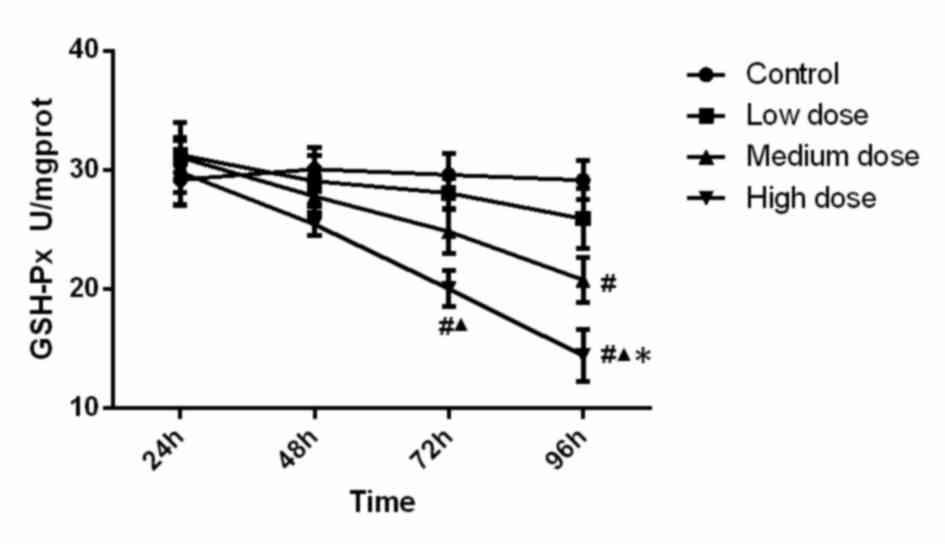
GSH-Px activity
The activity of GSH-Px in the different experimental
groups is presented in Fig. 2.
Statistical analysis indicated that GSH-Px activity in the
high-dose group at 72 h was significantly lower than that in the
control group (P<0.001) and low dose group (P=0.015). GSH-Px
activity in the high-dose group at 96 h was significantly lower
than that in the all other three groups (compared with control,
low-dose and medium-dose groups; P<0.001, P<0.001, P=0.048).
Furthermore, GSH-Px activity in the medium-dose group at 96 h was
significantly lower (P=0.001) than that in the control group.
Analysis of variance revealed that the concentration (F=10.233,
P<0.001), time (F=11.448, P<0.001) and the interaction
between concentration and time (F=2.077, P=0.041) were significant,
which demonstrated that time and concentration both have effect on
GSH-PX activity. These results demonstrated that GSH-PX activity
was reduced along with the increase of INH concentration and
incubation time.
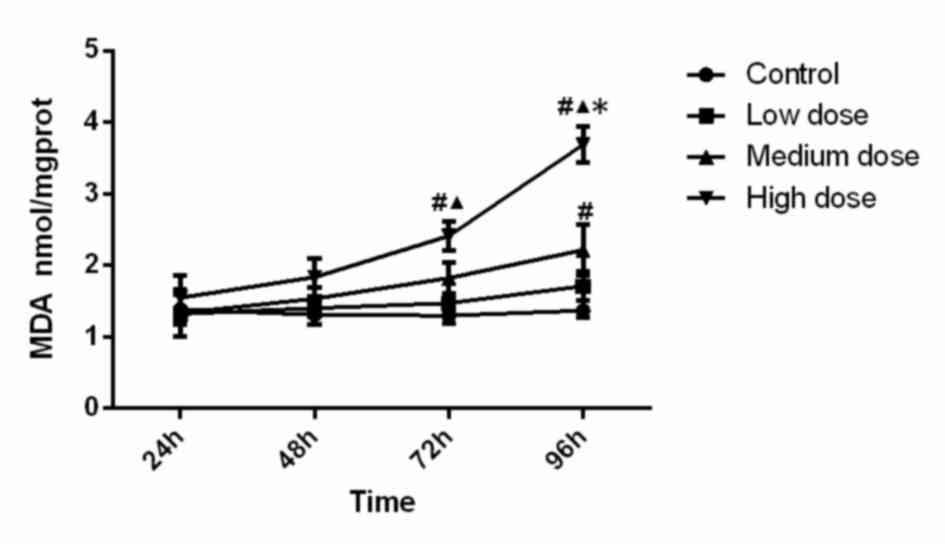
MDA concentration
The MDA concentration in the different experimental
groups is displayed in Fig. 3.
Statistical analysis suggested that MDA level in the high-dose
group at 72 h was significantly higher than that in the control
group (P=0.005) and low-dose group (P=0.048). MDA level in the
high-dose group at 96 h was significantly higher than that in all
other three groups (compared with control, low-dose and medium-dose
groups; P<0.001, P<0.001, P<0.001). In addition, MDA
activity in the medium-dose group at 96 h was significantly higher
(P=0.006) than that in the control group. Analysis of variance
showed that the concentration (F=18.126, P<0.001), time
(F=12.110, P<0.001) and the interaction between concentration
and time (F=3.724, P=0.001) was significant, which demonstrated
that time and concentration both have effects on MDA level. The MDA
content increased along with the increase of INH concentration and
incubation time.
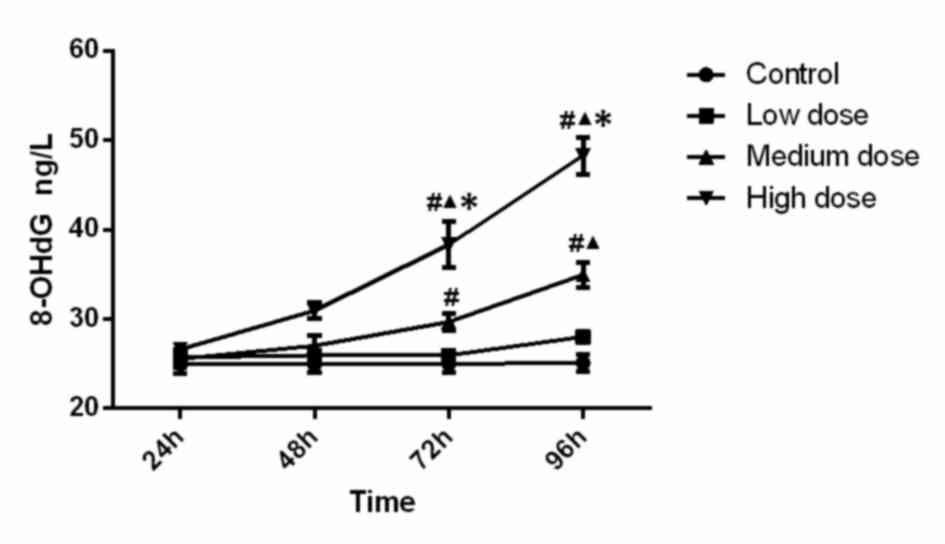
Mitochondrial 8-OHdG levels
The levels of 8-OHdG are presented in Fig. 4. Statistical analysis indicated that
the levels of 8-OHdG in the high-dose group at 72 h (compared with
control, low-dose and medium-dose groups; P<0.001, P<0.001,
P=0.002) and 96 h (compared with control, low-dose and medium-dose
groups; P<0.001, P<0.001 and P<0.001) were significantly
higher. Furthermore, 8-OHdG activity in the medium-dose group at 72
h was significantly higher than that in the control group
(P=0.016), and the medium-dose group at 96 h was significantly
higher than that in the control group (P<0.001) and low-dose
group (P<0.001). Analysis of variance shows that the
concentration (F=68.323, P<0.001), time (F=38.135, P<0.001)
and the interaction between concentration and time (F=12.929,
P<0.001) was significant, which demonstrated that time and
concentration both have effects on 8-OHdG level. The results
indicated that the mitochondrial 8-OHdG content increased along
with the increase of INH dose and incubation time.
Effect of isoniazid on mitochondrial
morphology of HepG2 cells
As displayed in Fig.
5, swelling of mitochondria of the HepG2 cells was observed in
the low-dose 96-h group, medium-dose 72-h group and high-dose 48-h
group. In the medium-dose 96-h group, high-dose 72 h group and
high-dose 96-h group, mitochondrial swelling and vacuoles within
certain mitochondria were visible. The mitochondrial vacuolation
was most severe in the high-dose 96-h group. There were no obvious
changes in mitochondrial morphology in the remaining groups.
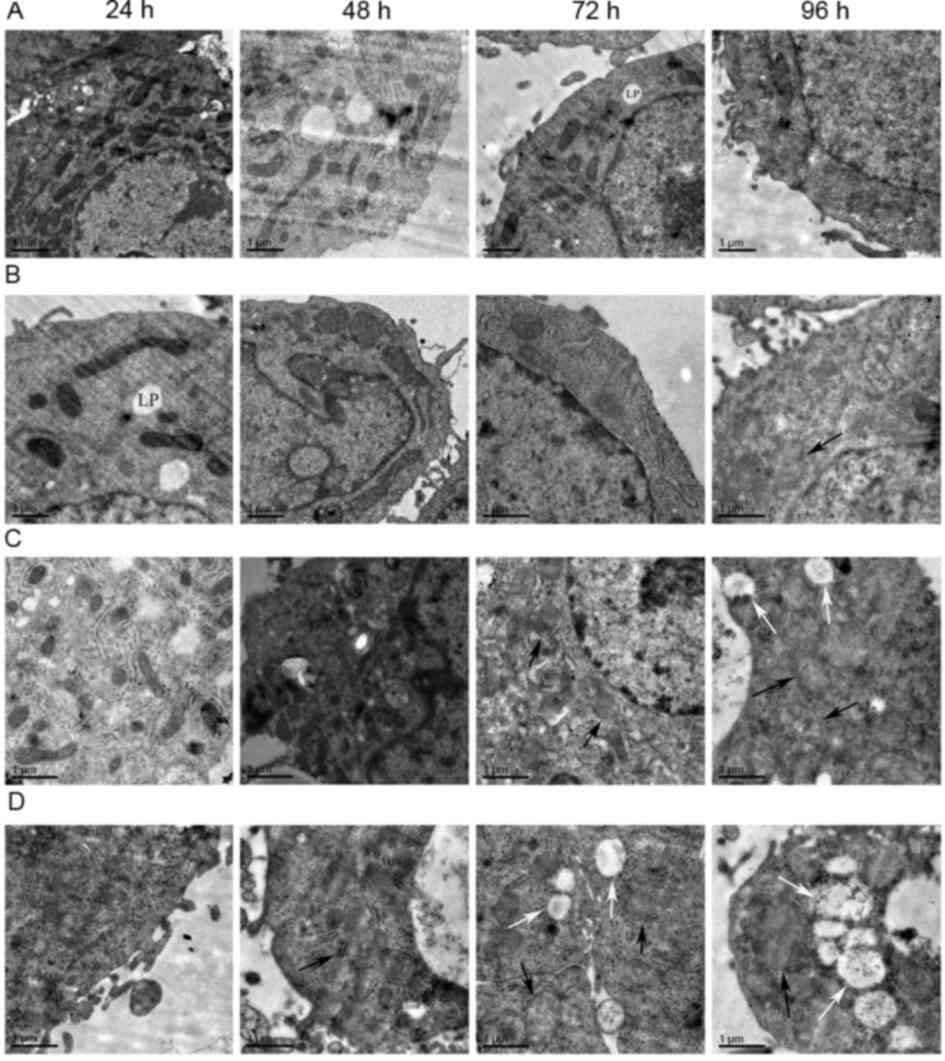 | Figure 5.Electron microscopy images revealing
HepG2 cell mitochondrial morphology. (A) Control group
(magnification, ×8,000). (B) low-dose group (magnification,
×10,000). (C) medium-dose group (magnification, 10,000). (D)
high-dose group (magnification, ×10,000). From left to right the
time point is 24, 48, 72 and 96 h. Subfigures of (A-D) showed the
morphological changes of mitochondria in different treatment groups
at different time points. The morphology of mitochondria notably
changed with the increase of dosage and the prolongation of the
time. Some mitochondria appeared swollen (black arrows) and some
were vacuolated (white arrows). Lipid droplets (LP) were
occasionally visible. Scale bar=1 µm. |
Discussion
As the major organelles for energy production
(14,15), mitochondria are the main cellular
site of the Krebs cycle, electron transport and oxidative
phosphorylation. Therefore, they are the main source of reactive
oxygen radicals and the primary target of oxidative damage
(16,17). Under normal circumstances, the
generation and clearance of oxygen radicals is in a dynamic
equilibrium state. However, mitochondrial oxidative stress occurs
if this balance is disrupted, resulting in mitochondrial damage
(18).
SOD and GSH-Px are important members of the
anti-oxidant defense system. SOD is the first line of defense
against oxygen free radicals and catalyzes the conversion of
superoxide anion radicals into H2O2, which is
further cleared by GSH-Px. MDA is a product of lipid peroxidation,
and reflects the level of free radical production and oxidative
damage (19). The present study
demonstrated that the MDA content increased along with the increase
of the INH concentration and incubation time, indicating that INH
dose- and time-dependently induced mitochondrial oxidative damage.
In consistency with that, SOD and GSH-PX activity were reduced
along with the increase of INH concentration and incubation time.
These results suggested that INH impairs the ability of these
enzymes to scavenge oxygen radicals, causing lipid peroxidation and
mitochondrial damage.
Compared to nuclear DNA, mtDNA is more sensitive to
oxidative damage. mtDNA is a bare DNA lacking protein protection
(20), and its ability to repair
damage is therefore poor (21).
mtDNA is connected with the mitochondrial inner membrane, which is
full of lipids, rendering mtDNA sensitive to damage. 8-OHdG is the
most commonly used biomarker for mtDNA damage (22). The results of the present study
demonstrated that the mitochondrial 8-OHdG content increased along
with the increase of the INH dose and incubation time, suggesting
that INH causes mtDNA damage. Since mtDNA has no intron sequence
except for a short region of replication and transcription, any
damage to mtDNA may significantly affect mitochondrial function.
Studies have suggested that the vast majority of mtDNA mutations
may affect the respiratory chain function and further aggravate the
decrease of the activity of respiratory chain complexes, resulting
in increased oxygen radical levels and reduction in ATP synthesis.
As the oxidation products continue to accumulate, mtDNA loss,
mutation or large fragmentation occurs, leading to the loss of its
important function to various extents (23,24).
Furthermore, mtDNA fragment deletion was reported to be closely
associated with the loss of direct repeat sequences during the
process of mtDNA replication and damage through oxygen free
radicals (25). Mitochondrial DNA
fragment deletion may cause the lack of genes encoding transfer
RNAs and proteins involved in the mitochondrial respiratory chain,
resulting in the dysfunction of the electronic chain, oxidative
phosphorylation and electron leakage. This may also cause immature
electron breathing and increase the generation of oxygen free
radicals, which further promotes mitochondrial damage.
Projection electron microscopy results of the cell
mitochondrial ultrastructure demonstrated that mitochondria of
INH-treated HepG2 cells were swollen in the low-dose 96-h group,
medium-dose 72-h group and high-dose 48-h group. Mitochondria
swelling and vacuoles were found in certain mitochondria in the
medium-dose 96-h group, high-dose 72 h group and high-dose 96-h
group. These results confirmed at the ultrastructural level that
the mitochondrial damage increased along with the increase of INH
dose and incubation time.
A previous study on the toxic effects of INH mainly
focused on apoptosis of HepG2 cells (13). The underlying mechanisms have
remained to be fully elucidated. The present study investigated the
effects of INH on mitochondrial damage in HepG2 cells and the
underlying molecular mechanisms. It was revealed that INH-induced
hepatic cell apoptosis and liver injury are likely caused by mtDNA
damage.
The limitations of the present study investigation
include the lack of an in vivo study and further studies
using animal models are required to confirm the conclusions.
However, to the best of our knowledge, the present study was the
first to provided direct evidence for INH-induced mitochondrial
damage and may enhance the current understanding on the mechanisms
of ADIH. The present study may provide a theoretical basis for the
development of novel therapeutic strategies for ADIH.
Acknowledgements
The authors would like to acknowledge the support of
the present study by Tangshan Science and Technology Bureau (grant
no. 08150201A-1-8).
References
|
1
|
Zhang Z, Song L, He L, Gao L, Shi Z, Tian
X, Zhang G and Feng F: Experimental study on the expression of
nuclear factor erythroid 2-related factor-2 and superoxide
dismutase during the isoniazid-induced liver injury. Chin J Infect
Dis. 32:80–84. 2014.
|
|
2
|
Boelsterli UA and Lee KK: Mechanisms of
isoniazid-induced idiosyncratic liver injury: Emerging role of
mitochondrial stress. J Gastroenterol Hepatol. 29:678–687. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Corbett EL, Watt CJ, Walker N, Maher D,
Williams BG, Raviglione MC and Dye C: The growing burden of
tuberculosis: Global trends and interactions with the HIV epidemic.
Arch Intern Med. 163:1009–1021. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ramappa V and Aithal GP: Hepatotoxicity
related to anti-tuberculosisdrugs: Mechanisms and management. J
Clin Exp Hepatol. 3:37–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Misu H, Takamura T, Matsuzawa N, Shimizu
A, Ota T, Sakurai M, Ando H, Arai K, Yamashita T, Honda M, et al:
Genes involved in oxidative phosphorylation are coordinately
upregulated with fasting hyperglycaemia in livers of patients with
type 2 diabetes. Diabetologia. 50:268–277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Srinivasula SM, Hegde R, Saleh A, Datta P,
Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y
and Alnemri ES: A conserved XIAP-interaction motif in caspase-9 and
Smac/DIABLO regulates caspase activity and apoptosis. Nature.
410:112–116. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Labbe G, Pessayre D and Fromenty B:
Drug-induced liver injury through mitochondrial dysfunction:
Mechanisms and detection during preclinical safety studies. Fundam
Clin. Pharmacol. 22:335–353. 2008.
|
|
8
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chien KR and Karsenty G: Longevity and
lineagea: Toward the integrative biology of degenerative diseases
in heart, muscle, and bone. Cell. 120:533–544. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanahashi C, Nakayama A, Yoshida M, Ito M,
Mori N and Hashizume Y: MELAS with the mitochondrial DNA 3243 point
mutation: A neuropathological study. Acta Neuropathol. 99:31–38.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barthélémy C, Ogier de Baulny H, Diaz J,
Cheval MA, Frachon P, Romero N, Goutieres F, Fardeau M and Lombès
A: Late-onset mitochondrial DNA depletion: DNA copy number,
multiple deletions and compensation. Ann Neurol. 49:607–617. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leonard JV and Schapira AH: Mitochondrial
respiratory chain disorders I: Mitochondrial DNA defects. Lancet.
355:299–304. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang B, Zhang L, Liu JW and An Y: Study on
the isoniazid induced cellular damage and the expressions of
Fas/Fas ligand of HepG2. Chin J Infect Dis. 30:402–406. 2012.
|
|
14
|
Yang L and Yu T: Prolonged donor heart
preservation with pinacidil: The role of mitochondria and the
mitochondrial adenosinetriphosphate-sensitive potassium channel. J
Thorac Cardiovasc Surg. 139:1057–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Simon DK, Pankratz N, Kissell DK, Pauciulo
MW, Halter CA, Rudolph A, Pfeiffer RF, Nichols WC and Foroud T;
Parkinson Study Group-PROGENI Investigators, : Maternal inheritance
and mitochondrial DNA variants in familial Parkinson's disease. BMC
Med Genet. 11:532010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng TI and Jou MJ: Mitochondrial swelling
and generation of reactive oxygen species induced by
photoirradiation are heterogeneously distributed. Ann N Y Acad Sci.
1011:112–122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choksi KB, Boylston WH, Rabek JP, Widger
WR and Papaconstantinou J: Oxidatively damaged proteins of heart
mitochondrial electron transport complexes. Biochim Biophys Acta.
1688:95–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cortes-Rojo C and Rodríguez-Orozco AR:
Importance of oxidative damage on the electon transport chain for
the rational use of mitochondria-targeted antioxidants. Mini Rev
Med Chem. 11:625–632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fattman CL, Schaefer LM and Oury TD:
Extracellular superoxide dismutase in biology and medicine. Free
Radie Biol Med. 35:236–256. 2003. View Article : Google Scholar
|
|
20
|
Clayton DA: Replication of animal
mitochondrial DNA. Cell. 28:693–705. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bandy B and Davison AJ: Mitochondrial
mutations may increase oxidative stress: Implications for
carcinogenesis and aging? Free Radic Biol Med. 8:523–539. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cooke MS, Evans MD, Burd RM, Patel K,
Barnard A, Lunec J and Hutchinson PE: Induction and excretion of
ultraviolet-induced 8-OXO-2′-deoxyguanosine and thymine dimers in
vivo: Implications for PUVA. J Invest DermatoI. 116:281–285. 2001.
View Article : Google Scholar
|
|
23
|
Ingman M and Gyllensten U: Analysis of the
complete human mtDNA genome: Methodology and inferences for human
evolution. J Hered. 92:454–461. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanaka M, Kovalenko SA, Gong JS, Borgeld
HJ, Katsumata K, Hayakawa M, Yoneda M and Ozawa T: Accumulation of
deletions and point mutations in mitochondrial genome in
degenerative diseases. Ann N Y Acad Sci. 786:102–111. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Berneburg M, Grether-Beck S, Kürten V,
Ruzicka T, Briviba K, Sies H and Krutmann J: Singlet oxygen
mediates the UVA-induced generation of the photoaging-associated
mitochondrial common deletion. J Biol Chem. 274:15345–15395. 1999.
View Article : Google Scholar : PubMed/NCBI
|