Introduction
Molecular-based environmental surveys of bacteria
have revealed a large degree of previously uncharacterized
diversity (1,2). More recently, the use of
culture-independent methods has played a key role in the discovery
of previously unrecognized species in the oral cavity as well as in
redefining the pathogenesis of major oral infections (2–4). The
outcome of these studies indicates that major oral infections are
polymicrobial (5,6). Using culture-independent molecular
methods, nearly 500 bacterial strains have been recovered from the
subgingival crevice, a particularly well-studied microbial niche
(6). Other investigators have used
similar techniques to determine the bacterial diversity of saliva,
subgingival plaque and carious roots (7,8).
Over half of the species detected have not yet been cultivated
(6). Data from these studies have
implicated specific species or phylotypes in a variety of diseases
and oral infections (4,9–13),
but still only limited information is available on species in
subgingival plaque of healthy subjects. Moreover, to date,
culture-independent studies describing the bacterial community of
subgingival plaque in Chinese individuals are limited.
It is known that extensive differences in eating
habits exist between Chinese and American individuals. These
differences may play an important role in alterations in the
colonization of oral bacterial species. Hence, understanding of the
entire bacterial community of subgingival plaque may aid in the
diagnosis and treatment of caries in Chinese people. Aas et
al (6) analyzed the oral flora
of 5 healthy subjects in the US using Sanger sequencing and
concluded that over 50% of the oral bacteria could not be cultured.
Kroes et al (14) studied
the subgingival plaque of a 39-year-old Caucasian male with
cultivation and phenotypic analysis, and found that a significant
proportion of the resident subgingival bacterial flora remained
poorly characterized and uncultured. Paster et al (15) analyzed 5 American healthy
subgingival plaque samples and acquired a preliminary understanding
of the composition of subgingival plaque. Recent investigations of
the human subgingival microbiota based on 16S rRNA gene sequences
indeed have shown that many of the present bacterial species are
novel species or phylotypes. However, we currently do no have a
comprehensive understanding of the subgingival microbiota of
healthy Chinese adults, nor do we know whether pathogenic bacteria
related to oral disease are present or, if there are, what those
bacteria are. Hence, the aim of our study was to determine the
detailed species diversity of the subgingival microbiota of healthy
Chinese adults.
Materials and methods
Subjects and sample collection
Six subjects (4 female and 2 male, ranging in age
from 25 to 35 years) were included in the study. Subjects were
excluded from this study if they had received systemic periodontal
treatment in the preceding year or had taken antibiotics within the
previous 3 months or were pregnant or nursing. They had no clinical
signs of oral mucosal disease, periodontal disease or caries and
did not suffer from halitosis. Their periodontia was healthy, and
all periodontal pockets were <3-mm deep with no redness or
inflammation of the gums. Subjects who were free of systemic
diseases and had 28 teeth in occlusion were subjected to clinical
examinations. The study protocol was approved by the Ethics
Committee of the Ninth People’s Hospital, Shanghai Jiao Tong
University. All participants provided written informed consent.
Saliva was isolated with sterile cotton rolls, after
which supragingival plaque was obtained using a fresh sterile
curette from four sites, usually distolingual, on the 1st or 2nd
molars, and the samples were pooled. All samples were placed in a
sterile plastic tube and stored at −80°C until use.
DNA extraction and amplification of 16S
rRNA genes
DNA in the samples was extracted using the QIAamp
DNA Mini kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions. The 16S rRNA genes were amplified
under standard conditions using a universal primer set. The primer
pair spanned positions 7–1541 of the E. coli 16S rRNA gene
(forward primer, 5′-GAG AGT TTG ATY MTG GCTCAG-3′; reverse primer,
5′-GAA GGA GGT GWT CCA RCC GCA-3′) (6). Each PCR mixture (50 μl) contained 100
ng of the extracted DNA as a template, 20 pmol of each primer, 0.25
μl Takara Ex Taq (5 U/μl), 5 μl 10X Ex Taq buffer and
4 μl dNTP mixture (2.5 mM of each deoxynucleoside triphosphate).
For PCR, the process consisted of an initial denaturation of the
template DNA at 94°C for 5 min, followed by 20 cycles of denaturing
at 94°C for 1 min, the annealing temperature (TA) for 1 min and
72°C for 1 min. The TA was decreased stepwise by 1°C every 2 cycles
from 65°C in the first cycle to 56°C in the 20th cycle, and a final
extension was carried out at 72°C for 5 min. The results of the PCR
amplification were examined by electrophoresis in a 1% agarose gel.
DNA was stained with ethidium bromide and visualized under
short-wavelength UV light. All PCR products were purified by
E.Z.N.A.® Gel Extraction kit (Omega Bio-Tek, USA).
Phylogenetic analysis
All sequences with the same size were aligned using
Clustal X 1.83 and modified by Jalview2.07. Sequences with >97%
identity are typically assigned to the same species, those with
>95% identity are typically assigned to the same genus and those
with >80% identity are typically assigned to the same phylum,
although these distinctions are controversial (16). Therefore, the redundancy cutoff was
set at 97%.
After redundancies were removed, the remaining
sequences were assigned to the most closely related sequence using
the Megablast function of GenBank (http://www.ncbi.nlm.nih.gov/blast/blast.cgi) and
Ribosomal Database Project II (http://rdp.cme.msu.edu). They were clustered by
phylogenetic groups in each library. Chimeric sequences were
detected using the RDP Check-Chimera program (release 8.1) and
Bellerophon (17). The
phylogenetic trees were generated by using MEGA 4.0. The
statistical strength of the Neighbor-Joining method was assessed by
bootstrap resampling (1,000 replicates). The richness of total
bacteria communities of the 6 healthy Chinese human subgingival
plaque was estimated by rarefaction analysis. The total number of
phylotypes that may be present in the sampled subgingival plaque
and its associated confidence interval (CI) were calculated by
using a non-parametric richness estimator, Chao1, as previously
described (18). Double principle
coordinate analysis (DPCoA) (19)
was implemented to investigate the relationships between phylotype
dissimilarities using DPCoA functions within the R statistical
package (www.rproject.org) (19).
Nucleotide sequence accession number
The clone sequences reported in this study were
deposited in the GenBank database (assigned GenBank accession
numbers: FJ470405-FJ470520; FJ470595-FJ470618; FJ470658-FJ470725;
FJ470788-FJ470842; FJ470910-FJ470947; FJ470976-FJ471006).
Results
Classification of the clones
Partial sequences of ∼500 bp were obtained for 2,439
16S rRNA clones in order to identify the predominant bacterial
species present in the subgingival plaque of 6 healthy Chinese
subjects. A markedly high diversity, 383 bacterial species
representing seven bacterial phyla, was observed for the 2,439
clones analyzed. The overall bacterial profile had 160 (85%)
cultivable species within known genera and 373 (15%) sequences from
not-yet-cultivated phylotypes or species that are currently
unrecognized. The term ‘phylotype’ is used for clusters of clone
sequences that differed from known species by ∼45 bases (or 3%) and
were at least 97% similar to members of their cluster.
Approximately 9.8% of clones analyzed in each category of healthy
subjects were novel phylotypes.
Distribution of the clones at the genus
and species level
The subgingival plaque samples from the 6 subjects
yielded sequences representing 49 genera. Ten genera were observed
in all 6 subjects, comprising 2,000 (82%) of the 2,439 clones
analyzed, including Streptococcus (23.8% of all clones), Neisseria
(12.9%), Fusobacterium (9.9%), Haemophilus (7.5%), Veillonella
(5.9%), Granulicatella (5.3%), Aggregatibacter (4.9%), Selenomonas
(3.9%), Capnocytophaga (1.3%) and Derxia (0.9%). The ten most
common species, accounted for 58.8% of all clones (2,439) (Table I). Five species (Haemophilus
pittmaniae, Campylobacter gracilis, Streptococcus
infantis, Streptococcus gordonii and Veillonella
parvula), detected in all 6 subjects, accounted for 37% of the
clones analyzed. By contrast, 25% of the individual species-level
operational taxonomic units (SLOTUs) were found in single subjects
only.
 | Table I.Species represented by 16S rRNA gene
sequences from supergingival plaque ≥4 in 6 healthy Chinese
subjects. |
Table I.
Species represented by 16S rRNA gene
sequences from supergingival plaque ≥4 in 6 healthy Chinese
subjects.
| Phylum | Genus | Species name | Clones (n)
|
|---|
| A | B | C | D | E | F | Total (%) |
|---|
| Proteobacteria | Neisseria | Neisseria
mucosa | 4 | | 9 | 20 | 34 | 10 | 77 (3.16) |
| | Neisseria
zoodegmatis | 11 | 9 | | | 34 | 28 | 82 (3.36) |
| Haemophilus | Haemophilus
pittmaniae | 1 | 56 | 35 | 24 | 25 | 37 | 178 (7.30) |
| Campylobacter | Campylobacter
gracilis | 15 | 10 | 12 | 9 | 16 | 8 | 70 (2.87) |
| Fusobacteria | Fusobacterium | Fusobacterium
canifelinum | 39 | 4 | 31 | 39 | 52 | | 165 (6.77) |
| Firmicutes | Streptococcus | Streptococcus
infantis | 6 | 28 | 168 | 17 | 42 | 112 | 373 (15.29) |
| | Streptococcus
mitis | 4 | 18 | 31 | 19 | | 61 | 133 (5.45) |
| | Streptococcus
gordonii | 10 | 43 | 33 | 15 | 61 | 23 | 185 (7.59) |
| Granulicatella | Granulicatella
elegans | 5 | 10 | 10 | 6 | | 44 | 75 (3.08) |
| Veillonella | Veillonella
parvula | 4 | 12 | 5 | 17 | 16 | 43 | 97 (3.98) |
| Subtotal (n) | | | 99 | 190 | 334 | 166 | 280 | 366 | 1,435 (58.84) |
Detection of previously uncharacterized
phylotypes
Approximately 6% (146 sequences) of the 16S rRNA
gene sequences generated from the subgingival plaque did not match
any known bacterial sequences present in public databases. In
total, 104 previously uncharacterized phylotypes (146 clones) were
detected, corresponding to seven bacterial phyla. The distribution
of the known species and novel phylotypes within each of these
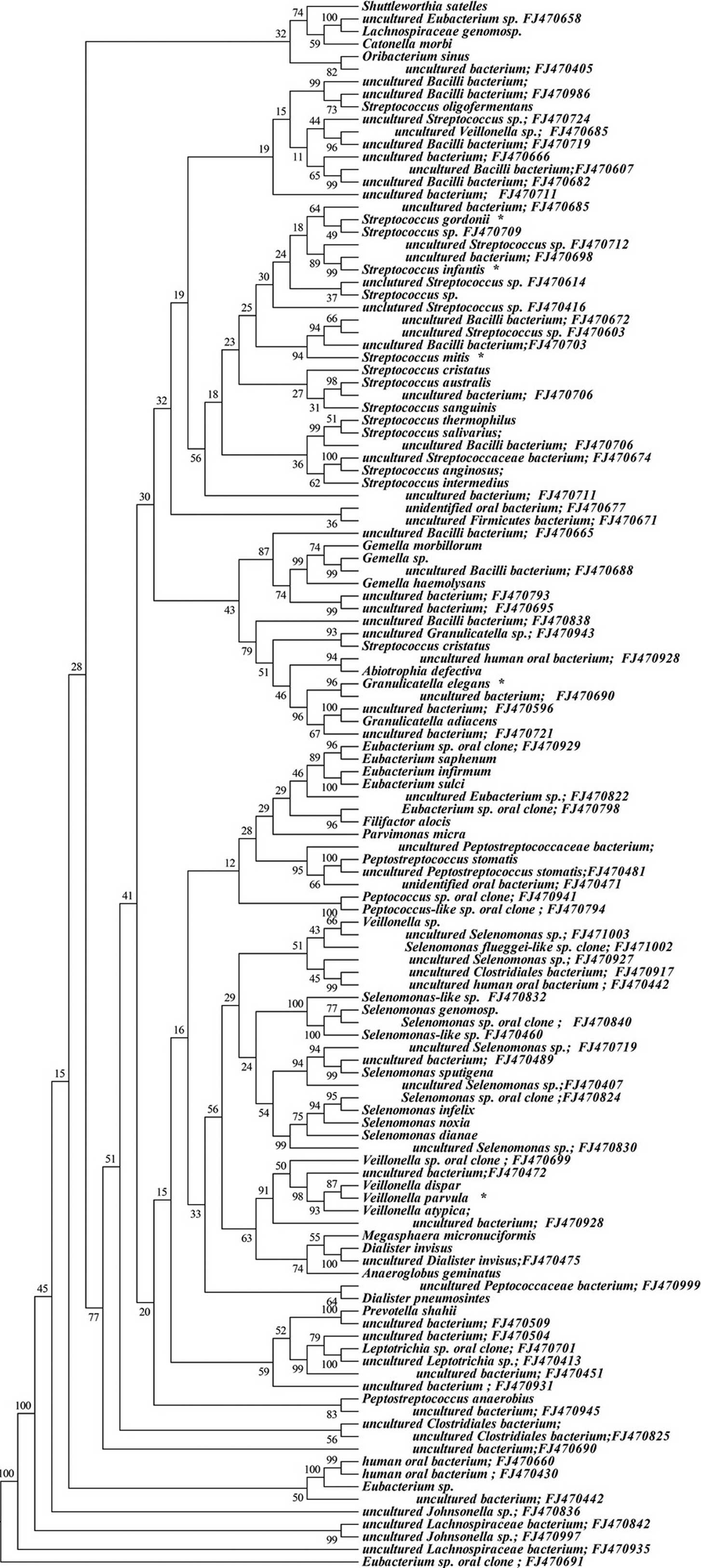
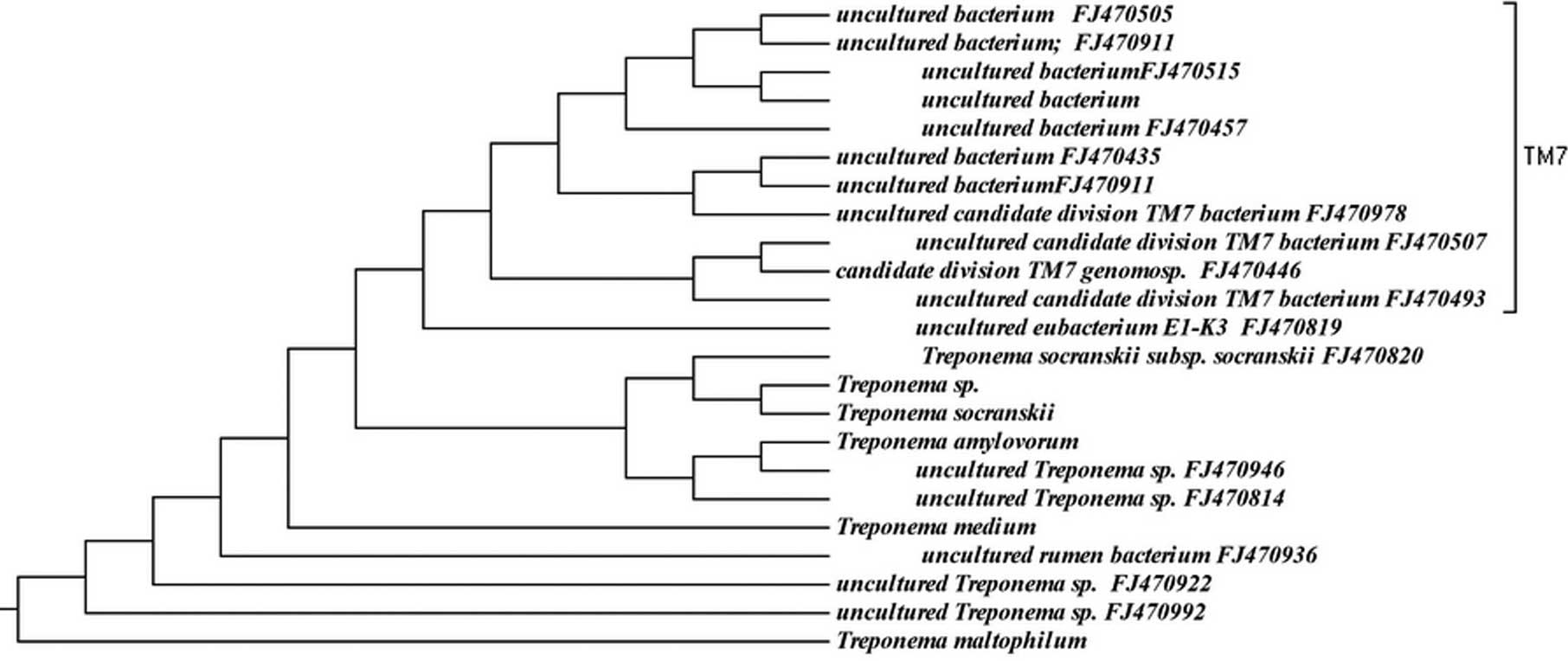
phyla is shown in Figs. 1–6, where the phylogenetic diversity within
each phylum is shown and discussed in detail below. The information
presented includes bacterial species or phylotype, strain or clone
identification and sequence accession number. The first letter of
previously uncharacterized phylotypes is indented in each
dendrogram.
Species richness and diversity with
Sanger sequencing
Estimations of species coverage, richness, evenness
and diversity were calculated for the combined data set from the
oral samples of the 6 subjects using conventional techniques and a
corresponding sequence library.
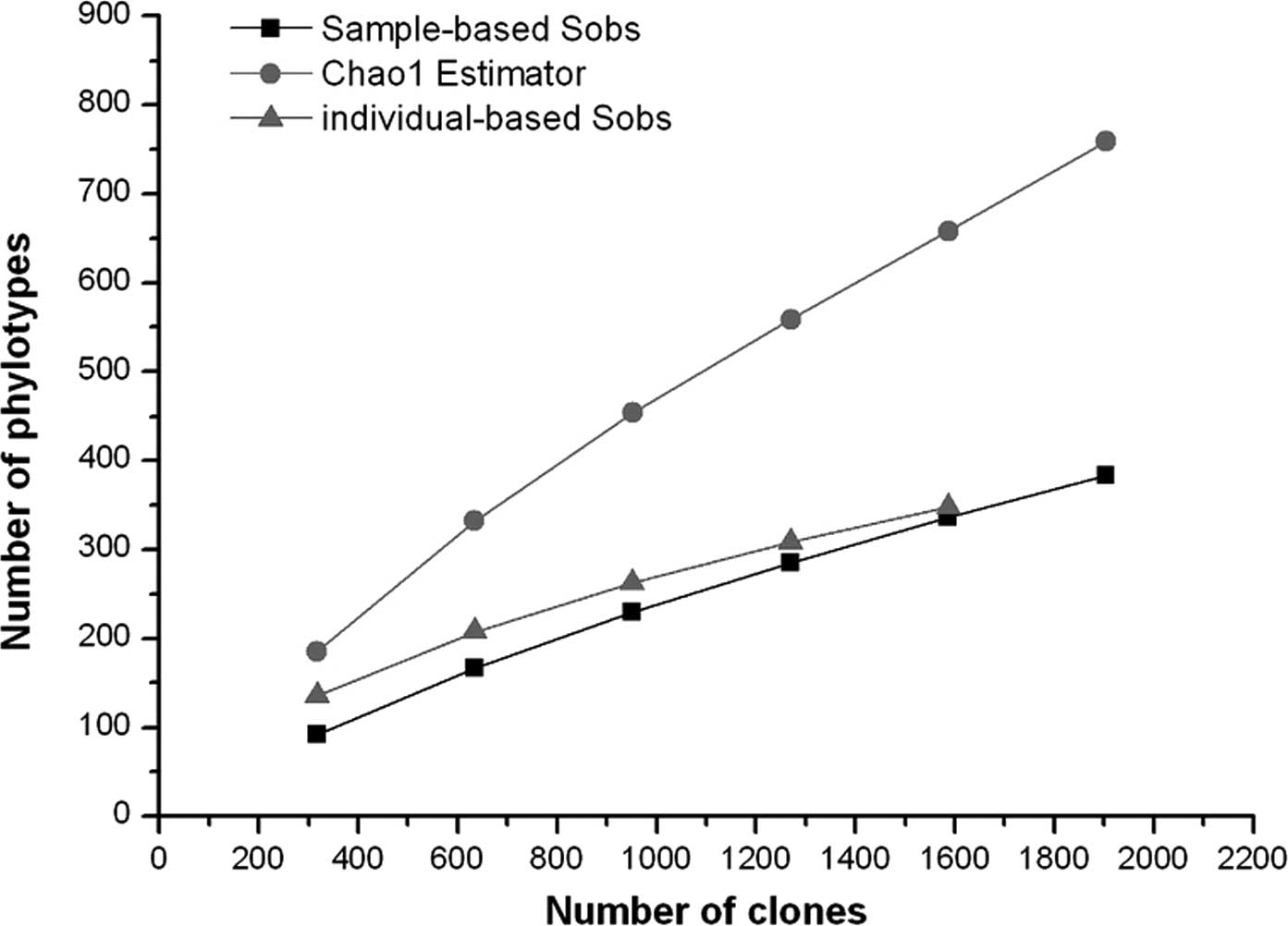
Good’s coverage was 90% for the overall sequence
set, indicating that ten additional phylotypes would be expected
for every 100 additional sequenced clones. This level of coverage
indicated that the 16S rDNA sequences identified in these samples
represent the majority of the bacterial species present in
subgingival samples under study. Rarefaction curves were calculated
for the overall combined data set using the individual-based
Coleman method and the sample-based Mao Tau method (Fig. 7). The total number of SLOTUs
present in the normal bacterial flora of subgingival samples in the
6 subjects was calculated using the Chao1 estimator, based on the
distribution of singletons (18).
We estimated that the bacterial biota from those specimens contains
∼759 SLOTUs (95% CI 637–939); the 383 SLOTUs observed represent 51%
(95% CI 43.9–55.4) of the estimated species or phylotypes (Fig. 7).
Discussion
Gross patterns of bacterial diversity have begun to
emerge in recent years from comparative analyses of 16S rRNA gene
sequences (6,20). In these surveys, 141 different
bacterial taxa representing the bacterial flora of healthy American
oral cavities have been reported (6). In our study, based on the analysis of
2,439 16S rRNA gene clones, the bacterial diversity of the
microbiota from the subgingival plaque of 6 Chinese subjects was
striking; a total of 383 different bacterial SLOTUs representing
seven phyla, with approximately 90% species coverage was estimated.
Hence, sequence-based environmental microbial surveys have
demonstrated that cultivation methods under-represent the true
extent of bacterial diversity.
In the present study, 32.2% of our sequences
belonged to the genus Streptococcus, confirming the prevalence of
Streptococcus species found in the healthy mouth by
molecular methods (1). The most
predominant bacterial genera in our study were Streptococcus,
Neisseria, Granulicatella, Haemophilus, Fusobacterium,
Aggregatibacter, Veillonella and Campylobacter, accounting for
70.7% of the total sequences. Furthermore, ten types of predominant
bacteria were noted in the subgingival samples from the 6 healthy
Chinese subjects (Table I). These
ten species of bacteria were key species found in human subgingival
plaque and, to some extent, affect periodontal health. The
individual role of each species in a biological community is
different; some species are critical and their existence affects
the structure and function of the entire biological community;
these species are called key species or key species groups
(21). The role of dominant
bacteria in subgingival microbiota can also be explained by the
hypothesis regarding keystone species. Classification of key
species in the medical field, mainly key types of biological
pathogens, biological competition and biological mutualism is
important. The key species are responsible for the maintenance of
the ecosystem functions (21).
Destruction of key bacterial species in subgingival plaque may
disrupt the microbiota balance and can easily lead to over-breeding
conditions resulting in pathogenic oral disease.
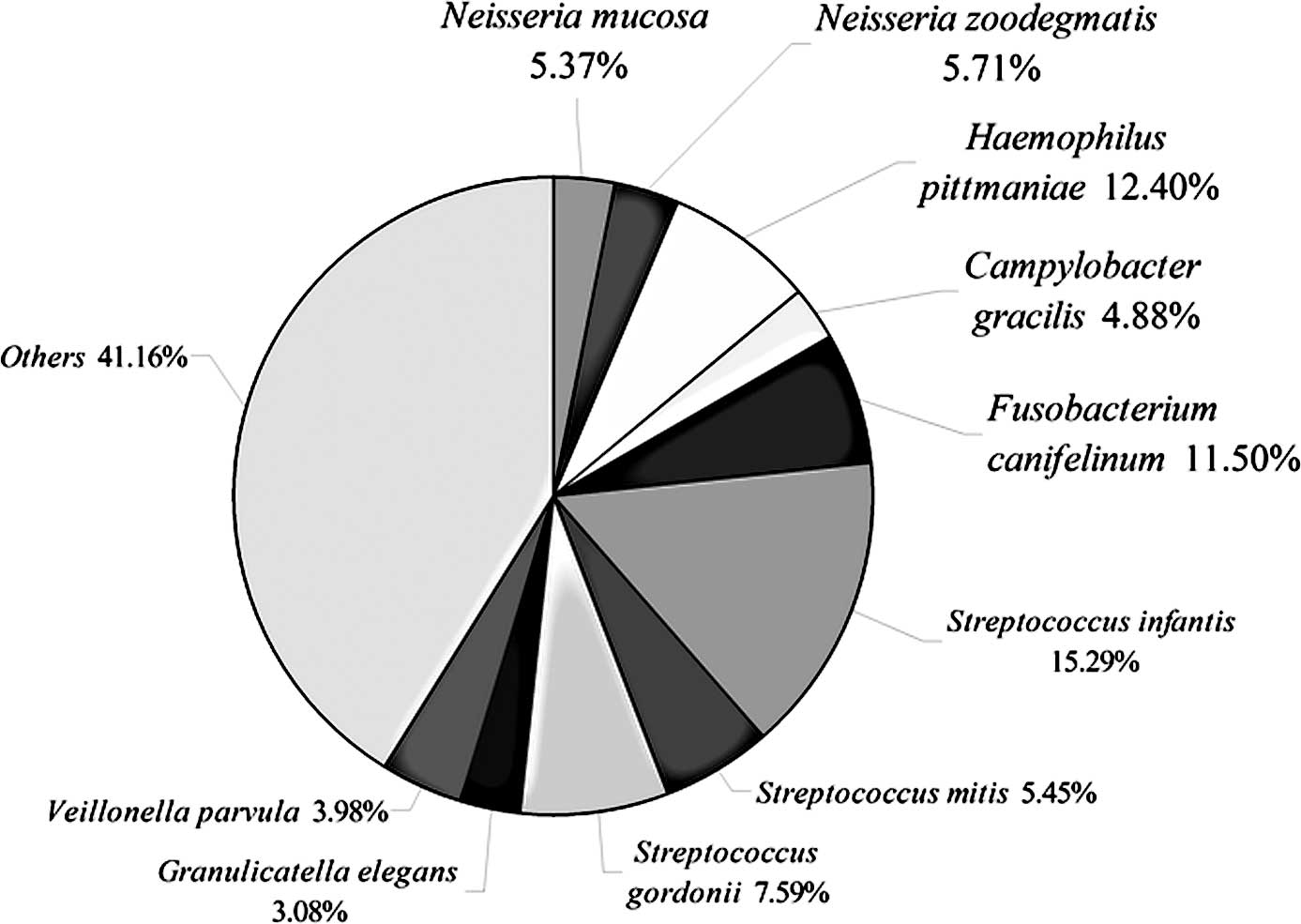
In a recent molecular study, the most predominant
bacterial species found in the subgingival plaque were
Streptococcus spp., Fusobacterium, Leptotrichia
buccalis, Corynebacterium matruchotii, S. oralis
and S. mitis (6). We also
identified the ten most common species that accounted for 58.8% of
all clones. These ten species included Neisseria mucosa,
Neisseria zoodegmatis, Haemophilus pittmaniae,
Campylobacter gracilis, Fusobacterium canifelinum,
Streptococcus infantis, Streptococcus mitis,
Streptococcus gordonii, Granulicatella elegans and
Veillonella parvula (Fig.
8). This discrepancy in results may be due to the three
following reasons. The number of subjects in this study was
relatively small, and inter-individual differences associated with
gender, age or eating habits may become apparent when larger
numbers of subjects are studied. Moreover, there was a deeper
sequencing effort per individual in the present study (average 57.5
clones per subject in the Aas et al study for a total of
2,589 clones, in contrast to an average of 500 clones per
individual in this study). In addition, different DNA extraction
methods and different broad-range PCR primers may also explain the
divergent results.
Among the predominant species, Streptococcus
infantis was most frequently cloned. This species is commonly
found in the mouth of infants (22), which does not match the results of
our experiment. Streptococcus infantis may be characteristic
of the subgingival microflora of the 6 Chinese subjects. In
addition, the predominant species Campylobacter gracilis,
Campylobacter showae, Kingella oralis and
Neisseria mucosa noted in the previous experiment were also
noted in healthy American subjects (15). The pathogens that are known to have
a close link to periodontitis, such as Porphyromonas
endodontalis, Capnocytophaga granulosa and Prevotella
melaninogenica (15,23), were respectively detected in our
experiment in one clone in 1 healthy subject. This suggests that
the oral cavity of a healthy individual is not necessarily
pathogen-free; the absence of symptoms of oral disease does not
indicate the absence of pathogens in the oral cavity. In
conclusion, these reports suggest that, in the subgingival
microbiota of Chinese individuals, there are pathogen and symbiotic
bacteria competing against each other eventually achieving a
balance for a healthy oral environment.
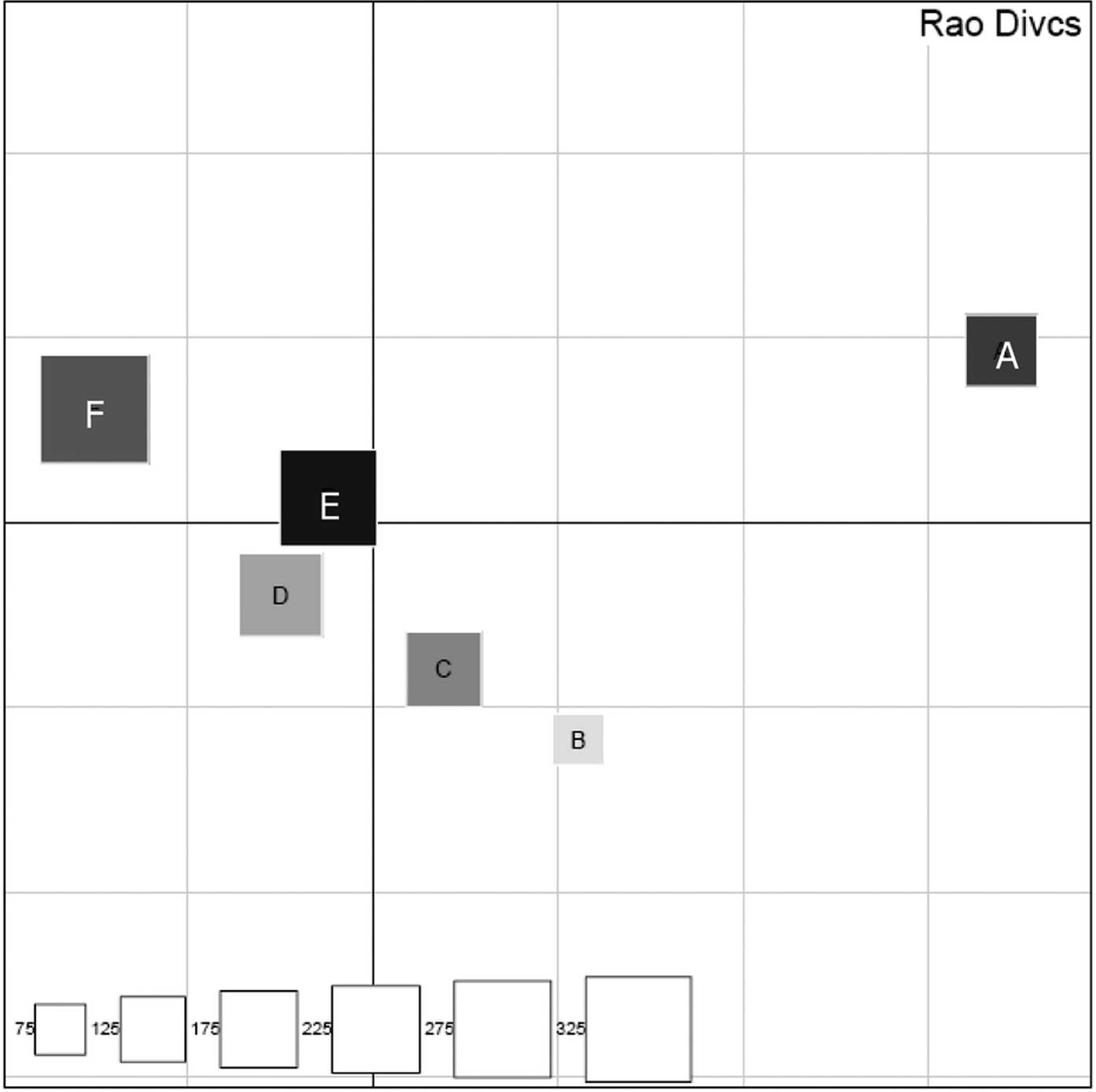
Our results revealed that each individual had a
certain difference in the subgingival microbiota (Fig. 9). Differences in subgingival
microbiota among individuals are caused by various physical and
chemical characteristics of the oral environment, such as
temperature, oxygen tension and availability of nutrients (24). Most oral bacteria are grown in a
neutral environment (pH equal to 7). The oral cavity provides a
relatively constant pH environment,which provides a suitable
foundation for the colonization of bacteria. A variety of exogenous
food, the saliva buffer system and bacterial fermentation may alter
the pH of the oral environment, resulting in different bacterial
colonization in individuals (25).
However, the factors affecting bacterial diversity may include
ethnicity, geographic region, different eating habits, oral hygiene
habits and gender. In addition, this study only preliminarily
analyzed 6 samples of healthy subgingival plaque. To better
represent the characteristics of the healthy subgingival
microbiota, study of a large number of samples is required. In
addition, how the differences in subgingival microbiota fluctuates
over time warrants further analysis.
As determined in previous studies using
culture-independent molecular techniques, over 60% of the bacterial
flora identified was represented by not-yet-cultivated phylotypes
(6,15). In the present study, we detected
104 of 383 SLOTUs (27.2%) as not-yet-cultivable phylotypes with
Sanger sequencing. One hundred and four unknown phylotypes may be
new species of a known genus or unknown species of a yet unkown
genus. There were 104 phylotypes noted in the subgingival
microbiota of the 6 healthy subjects; 92 had only one clone for
each. A portion of the ‘new species’ was crossing bacteria (also
known as foreign bacteria) rather than normal microbiota (also
known as permanent microbiota), relative to the permanent
microbiota found in the oral cavity. The crossing bacteria may be
non-pathogenic or opportunistic pathogens for which the residence
time is short and colonization is caused by adverse effects, while
normal microbiota are relatively fixed and regularly settled in a
specific location and are an integral part of the host. Although a
‘new phylotype’ in a sense can be understood as a new species, the
physiological characteristics of new species and their relationship
with diseases is not yet clear at present, and this has brought
certain difficulties in the study of their role in the ecological
environment. Understanding the nature of the study of new species
is very difficult. The main reason for this is that current
technology cannot meet the conditions of culture.
In conclusion, in comparing the results reported in
the literature (3,14,15),
we found that there was an overlap in the species detected in the
subjects without symptomatic periodontal disease and periodontitis.
Thus, certain key species may be critical for the maintenance of
ecosystem stability and oral health, or may be occasional
pathogens. Hence, pathogens and symbionts are in dynamic
equilibrium, and maintainence or disruption of this equilibrium may
affect oral health or result in disease.
Acknowledgements
This study was supported by the
Shanghai Education Commission Project [project no. (2010)83] and
the Shanghai Leading Academic Discipline Project (project no.
T0202).
References
|
1.
|
Bik EM, Long CD, Armitage GC, et al:
Bacterial diversity in the oral cavity of 10 healthy individuals.
ISME J. 4:962–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Preza D, Olsen I, Willumsen T, Grinde B
and Paster BJ: Diversity and site-specificity of the oral
microflora in the elderly. Eur J Clin Microbiol Infect Dis.
28:1033–1040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Colombo AP, Boches SK, Cotton SL, et al:
Comparisons of subgingival microbial profiles of refractory
periodontitis, severe periodontitis, and periodontal health using
the human oral microbe identification microarray. J Periodontol.
80:1421–1432. 2009. View Article : Google Scholar
|
|
4.
|
Corby PM, Lyons-Weiler J, Bretz WA, et al:
Microbial risk indicators of early childhood caries. J Clin
Microbiol. 43:5753–5759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Riggio MP, Lennon A, Rolph HJ, et al:
Molecular identification of bacteria on the tongue dorsum of
subjects with and without halitosis. Oral Dis. 14:251–258. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Aas JA, Paster BJ, Stokes LN, Olsen I and
Dewhirst FE: Defining the normal bacterial flora of the oral
cavity. J Clin Microbiol. 43:5721–5732. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Aas JA, Griffen AL, Dardis SR, et al:
Bacteria of dental caries in primary and permanent teeth in
children and young adults. J Clin Microbiol. 46:1407–1417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Tanner AC, Kent R Jr, Kanasi E, et al:
Clinical characteristics and microbiota of progressing slight
chronic periodontitis in adults. J Clin Periodontol. 34:917–930.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Sakamoto M, Rocas IN, Siqueira JF Jr and
Benno Y: Molecular analysis of bacteria in asymptomatic and
symptomatic endodontic infections. Oral Microbiol Immunol.
21:112–122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Tanner AC, Paster BJ, Lu SC, et al:
Subgingival and tongue microbiota during early periodontitis. J
Dent Res. 85:318–323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Munson MA, Banerjee A, Watson TF and Wade
WG: Molecular analysis of the microflora associated with dental
caries. J Clin Microbiol. 42:3023–3029. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Sakamoto M, Huang Y, Umeda M, Ishikawa I
and Benno Y: Detection of novel oral phylotypes associated with
periodontitis. FEMS Microbiol Lett. 217:65–69. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Becker MR, Paster BJ, Leys EJ, et al:
Molecular analysis of bacterial species associated with childhood
caries. J Clin Microbiol. 40:1001–1009. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Kroes I, Lepp PW and Relman DA: Bacterial
diversity within the human subgingival crevice. Proc Natl Acad Sci
USA. 96:14547–14552. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Paster BJ, Boches SK, Galvin JL, et al:
Bacterial diversity in human subgingival plaque. J Bacteriol.
183:3770–3783. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Schloss PD and Handelsman J: Introducing
DOTUR, a computer program for defining operational taxonomic units
and estimating species richness. Appl Environ Microbiol.
71:1501–1506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Huber T, Faulkner G and Hugenholtz P:
Bellerophon: a program to detect chimeric sequences in multiple
sequence alignments. Bioinformatics. 20:2317–2319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hughes JB, Hellmann JJ, Ricketts TH and
Bohannan BJ: Counting the uncountable: statistical approaches to
estimating microbial diversity. Appl Environ Microbiol.
67:4399–4406. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Pavoine S, Dufour AB and Chessel D: From
dissimilarities among species to dissimilarities among communities:
a double principal coordinate analysis. J Theor Biol. 228:523–537.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gao Z, Tseng CH, Pei Z and Blaser MJ:
Molecular analysis of human forearm superficial skin bacterial
biota. Proc Natl Acad Sci USA. 104:2927–2932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Pearce DA, van der Gast CJ, Woodward K and
Newsham KK: Significant changes in the bacterioplankton community
structure of a maritime Antarctic freshwater lake following
nutrient enrichment. Microbiology. 151:3237–3248. 2005. View Article : Google Scholar
|
|
22.
|
Bin-Nun A, Bromiker R, Wilschanski M, et
al: Oral probiotics prevent necrotizing enterocolitis in very low
birth weight neonates. J Pediatr. 147:192–196. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Mayanagi G, Sato T, Shimauchi H and
Takahashi N: Detection frequency of periodontitis-associated
bacteria by polymerase chain reaction in subgingival and
supragingival plaque of periodontitis and healthy subjects. Oral
Microbiol Immunol. 19:379–385. 2004. View Article : Google Scholar
|
|
24.
|
Lodhia P, Yaegaki K, Khakbaznejad A, et
al: Effect of green tea on volatile sulfur compounds in mouth air.
J Nutr Sci Vitaminol. 54:89–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Duckworth RM: The science behind caries
prevention. Int Dent J. 43:529–539. 1993.PubMed/NCBI
|