Introduction
Acute lung injury (ALI) is a life-threatening
clinical disease characterized by acute respiratory distress
syndrome, which may be caused by several different factors,
including trauma, sepsis and pneumonia (1,2).
Despite significant advances in antimicrobial supportive care and
therapy to improve the survival rate of patients with ALI, the
mortality rate remains high (~40%) (3). Therefore, the development of novel
and effective measures against ALI is required.
Several studies have demonstrated that numerous
proinflammatory cytokines, including interleukin-6 (IL-6) and tumor
necrosis factor-α (TNF-α), are important in the initiation and
amplification of lung injury (4).
Clinical studies have also demonstrated that elevated levels of
proinflammatory cytokines in the serum of patients with ALI are
associated with an increased mortality rate (5). Lung-protective ventilation and
corticosteroids have been shown to downregulate the level of
inflammatory cytokines as well as decrease the mortality rate of
patients with ALI (6).
Furthermore, previous studies demonstrated that signaling pathways,
including the nuclear factor-κB (NF-κB), mitogen-activated protein
kinase (MAPK) and phosphatidylinositol 3-kinase pathways, were
upregulated in animal models of ALI (7–9). The
c-Jun NH2-terminal kinase (JNK) is a member of the MAPK
family, which has been implicated in the regulation of inflammatory
signals and other extracellular signals to intracellular target
molecules (10,11). JNK has been identified as a
stress-activated protein kinase that phosphorylates c-Jun at two
sites on the NH2-terminal domain. Inhibition of the JNK
signaling pathway leads to the inactivation of transcription
factors and other regulatory cellular proteins (12,13).
SP600125 is a small molecule that acts as a reversible,
ATP-competitive inhibitor of JNK1/2 (14). Due to the effectiveness and
specificity of SP600125 in cells and animals experiments, it has
been widely used as a pharmacological inhibitor for assessing the
role of JNK in the regulation of biological processes (15). In the present study, the
therapeutic effect and associated mechanism of SP600125 was
analyzed in lipopolysaccharide (LPS)-induced ALI in vivo and
in vitro.
Materials and methods
Model establishment
A total of 40 male Wistar rats were randomly divided
into four groups (n=10): the control group, LPS group, normal
saline group (NS) and the SP600125 group. ALI was induced via
intratracheal injection of LPS (Sigma, St. Louis, MO, USA) as
previously described (8). Briefly,
the rats were anesthetized with pentobarbital sodium followed by
intratracheal injection of 5 mg/kg LPS. The rats were then placed
in a vertical position and rotated for 1 min to distribute the LPS
in the lungs. Normal saline or SP600125 was administered via
intraperitoneal injection (15 mg/kg) 10 min after the LPS
injection. All experiments were approved by the institutional
animal care and research committee of Zhejiang Provincial People’s
Hospital (Hangzhou, China).
Histological examination
The rats were anesthetized 24 h after injury and
sacrificed transcardially with saline, followed by treatment with
4% paraformaldehyde. The lungs were immediately removed and
post-fixed in 4% paraformaldehyde for 24 h. Paraffin-embedded
sections (3 mm thick) were stained with hematoxylin and eosin
(H&E) for visualization under a light microscope
(magnification, ×200; Leica Microsystems, Wetzlar, Germany).
Enzyme-linked immunosorbent assay
(ELISA)
The levels of claudin-4, TNF-α and IL-6 in the lung
tissue samples were measured using an ELISA according to the
manufacturer’s instructions (R&D Systems, Minneapolis, MN,
USA). The absorbance was measured at 450 nm using a microplate
assay (FluoDia T70, Photon Technology International, Lawrenceville,
NJ, USA).
Lung wet to dry (W/D) weight ratio
The severity of pulmonary edema was assessed using
the W/D ratio. The left lower lungs were weighed and then
dehydrated at 60°C for 72 h in an oven.
Cell culture and treatments
Human type II-like alveolar epithelial cells (A549)
were cultured in RPMI-1640 medium supplemented with 10% fetal
bovine serum, 2 mmol/l glutamine, 100 U/ml penicillin and 100 mg/ml
streptomycin, and maintained in a humid environment at 37°C and 5%
CO2. The cells were then treated with LPS (10 μg/ml) and
different concentrations of SP600125 (10, 20 and 40 nM). Following
24 h, the cells were collected for further analysis.
Cell viability assay
Cell viability was evaluated using the Cell Counting
kit (CCK)-8 assay (Sigma). In brief, the cells were seeded into
96-well plates at a density of 3×103 cells/well and left
to adhere overnight. The cells were then incubated with or without
0–40 nM SP600125. Then, 100 μl CCK-8 was added and incubated in the
dark at 37°C for 3 h. The absorbance was determined using the MRX
II microplate reader (Dynex, Chantilly, VA, USA) at a wavelength of
450 nm.
5-Ethynyl-2′-deoxyuridine (EdU)
incorporation assay
A549 cells were exposed to EdU (Invitrogen Life
Technologies, Carlsbad, CA, USA) for 2 h at 37°C. The cells were
fixed with 4% formaldehyde for 15 min and then treated with 0.5%
Triton X-100 for 20 min at room temperature. Following washing with
phosphate-buffered saline (PBS) three times, the cells of each well
were treated with 100 μl 1 X Apollo reaction cocktail for 30 min.
Subsequently, the DNA contents of the cells in each well were
stained with 100 μl of Hoechst 33342 (5 μg/ml) for 30 min and
visualized using a fluorescent microscope (Leica Microsystems).
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted using a TRIzol®
kit (Invitrogen Life Technologies) and converted to cDNA using the
cDNA Synthesis kit (Takara, Otsu, Shiga, Japan). qPCR was performed
using the SYBR-Green Supermix (Invitrogen Life Technologies). The
primer sequences used for the PCR reactions were as follows:
claudin-4, forward 5′-ACGAGACCGTCAAGGCCAAG-3′ and reverse
5′-GTCCAGGACACAGGCACCATAA-3′; β-actin, forward
5′-GGAGATTACTGCCCTGGCTCCTA-3′ and reverse
5′-GACTCATCGTACTCCTGCTTGCTG-3′. Amplification was performed at 50°C
for 2 min, at 95°C for 2 min, followed by 40 cycles of denaturing
at 95°C for 15 sec and annealing at 60°C for 30 sec. All the
reactions were performed in triplicate. GAPDH was used as a
reference gene.
Western blot analysis
The total protein (20 μg) was separated in each
sample using electrophoresis on a 4–20% sodium dodecyl
sulfate-polyacrylamide gel and electroblotted onto polyvinylidene
difluoride membranes. The membranes were inhibited in a blocking
solution and incubated overnight with primary antibodies, including
anti-claudin-4, anti-phospho-JNK, anti-JNK and anti-GAPDH (Cell
Signaling Technology, Beverly, MA, USA). The membranes were then
incubated with anti-rabbit or anti-mouse secondary antibody
conjugated with horseradish peroxidase (Pierce Chromatography
Cartridges, Thermo Fisher Scientific, Waltham, MA, USA).
Immunoreactive bands were detected using the enhanced
chemiluminescence kit for western blotting detection by using a
ChemiGenius bioimaging system (Syngene, Frederick, MD, USA). Band
densities for each protein were determined using Image-Pro Plus 6.0
software (Media Cybernetics, Inc., Bethesda, MD, USA) with GAPDH as
a control.
Flow cytometric analysis
Cell apoptosis was determined using flow cytometry.
Briefly, A549 cells were washed with PBS, detached with trypsin and
then harvested. The cells were resuspended in 1 ml Hoechst 33258
for 5 min and then washed three times with PBS. Cell apoptosis was
detected using the Annexin V-fluorescein isothiocyanate cell
Apoptosis Detection kit according to the manufacturer’s
instructions (BD Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
The data are presented as the mean ± standard
deviation and were analyzed using the SPSS statistical software
program (SPSS, Inc., Chicago, IL, USA). Comparison between groups
was performed using analysis of variance and P<0.05 was
considered to indicate a statistically significant difference.
Results
SP600125 attenuates LPS-induced ALI in
rats in vivo
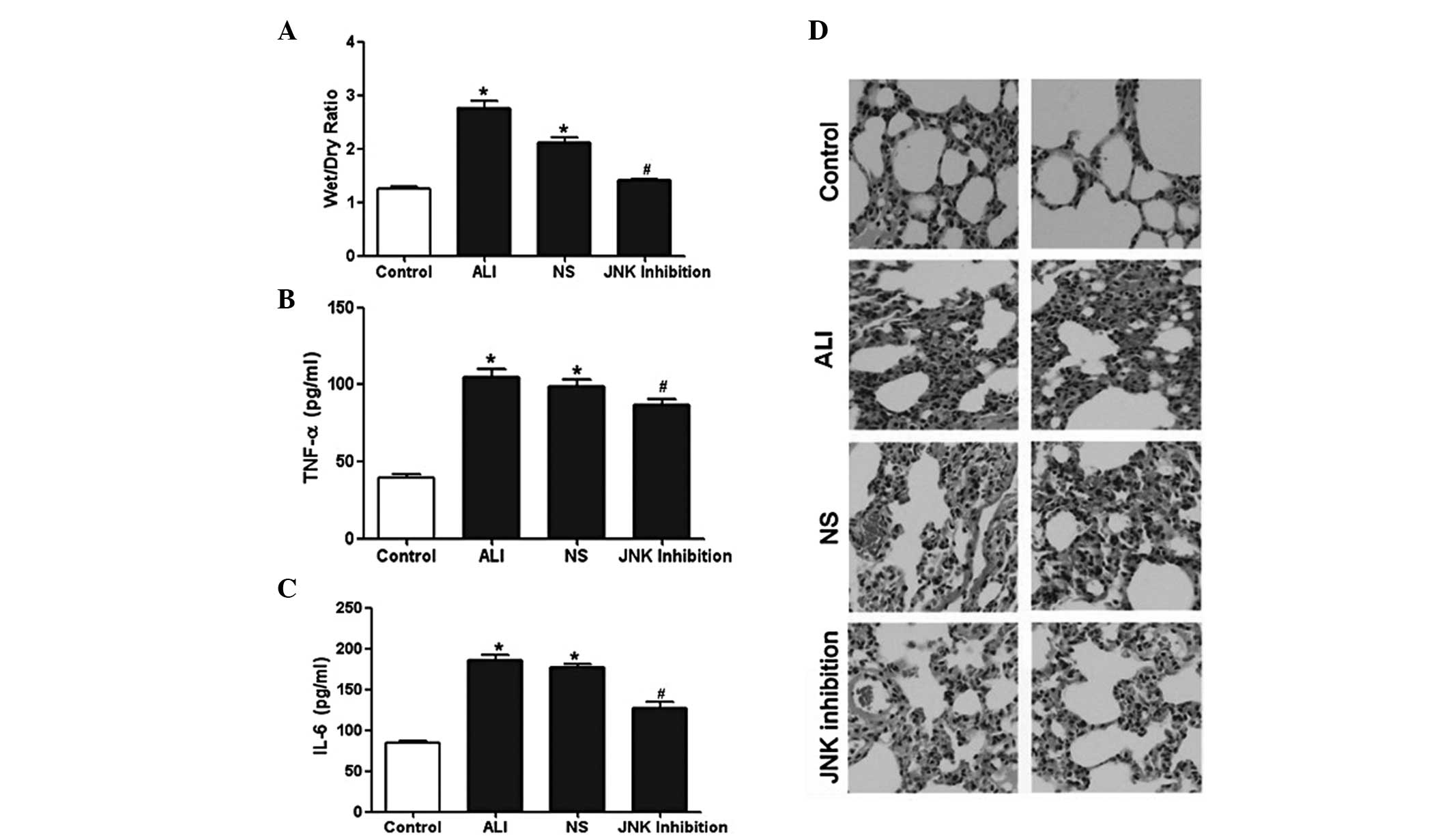
The lung W/D ratio was analyzed to evaluate
pulmonary edema. The LPS-treated rats had higher W/D ratios
compared with the control rats. However, the W/D ratio was
significantly decreased following administration of SP600125
(Fig. 1A). Furthermore, the
results from the ELISA demonstrated that the expression of TNF-α
and IL-6 in the bronchoalveolar lavage fluid (BALF) in the
LPS-treated rats was markedly increased compared with the rats in
the control group. However, the expression levels of TNF-α and IL-6
in the BALF in rats in the SP600125 group were significantly
decreased (Fig. 1B and C). To
assess the pathological alterations, H&E staining was performed
and the results revealed evidence of infiltration of inflammatory
cells, interstitial edema and interalveolar septal thickening, as
well as intra-alveolar and interstitial hemorrhage. However,
following treatment with SP600125, the pathological changes in the
lung tissues of the rats markedly decreased (Fig. 1D). These results demonstrated that
SP600125 treatment alleviated LPS-induced ALI in vivo.
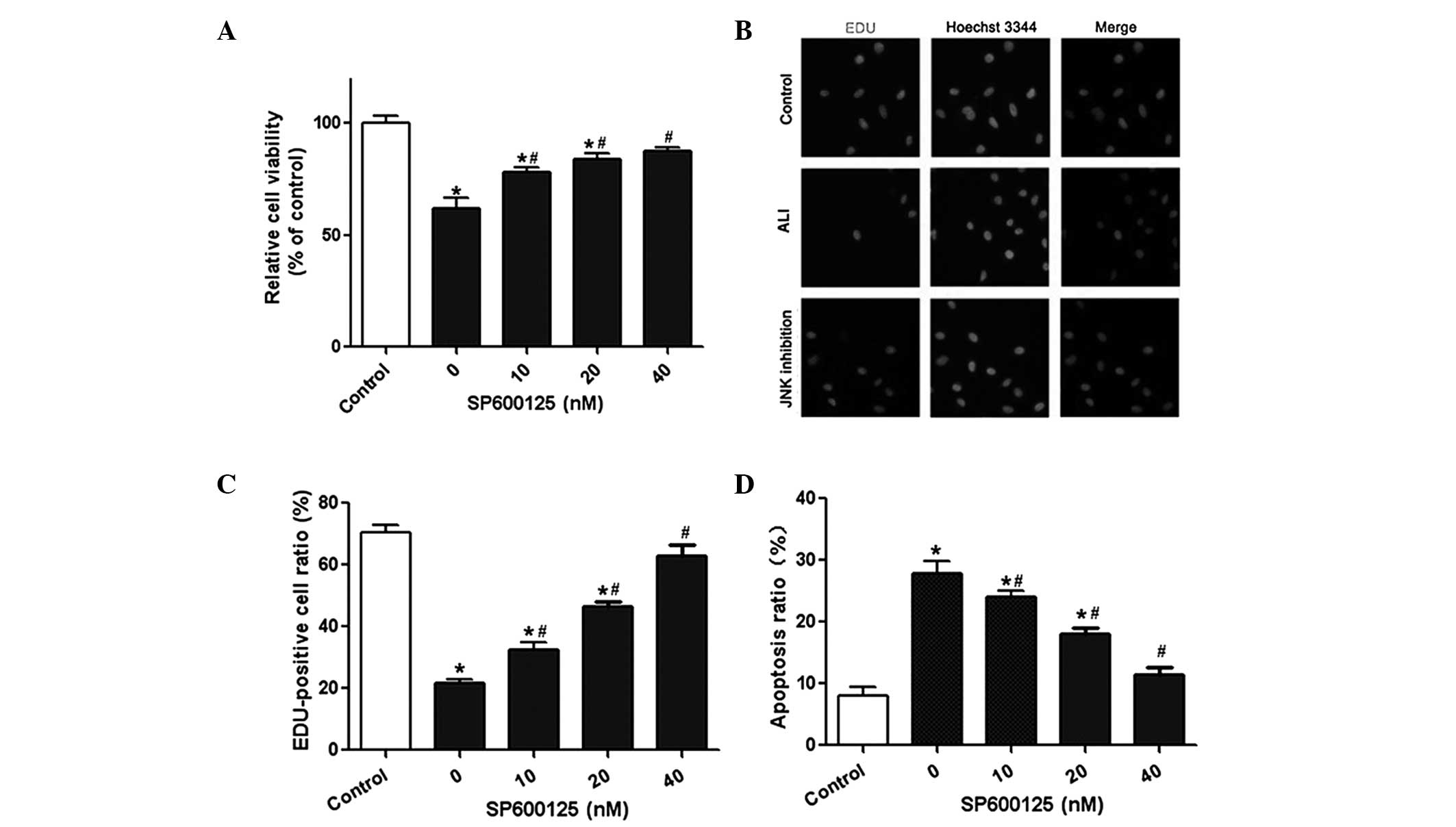
Effect of SP600125 administration on A549
cell viability and apoptosis
The lung epithelial cell line A549 was used in the
present study to investigate the effect of SP600125 on A549 cell
viability and apoptosis. The results from the CCK-8 assay revealed
that cell viability was significantly reduced following LPS
treatment. By contrast, SP600125 administration significantly
improved the viability of A549 cells in a dose-dependent manner
(Fig. 2A). The results from the
EdU assay were consistent with these results (Fig. 2B and C). Flow cytometry was used to
determine the effect of SP600125 on cell apoptosis. The results
demonstrated that SP600125 treatment significantly reduced the
apoptotic rate of A549 cells in a dose-dependent manner (Fig. 2D). In combination, these results
suggested that SP600125 was able to protect against LPS-induced ALI
in rats in vitro.
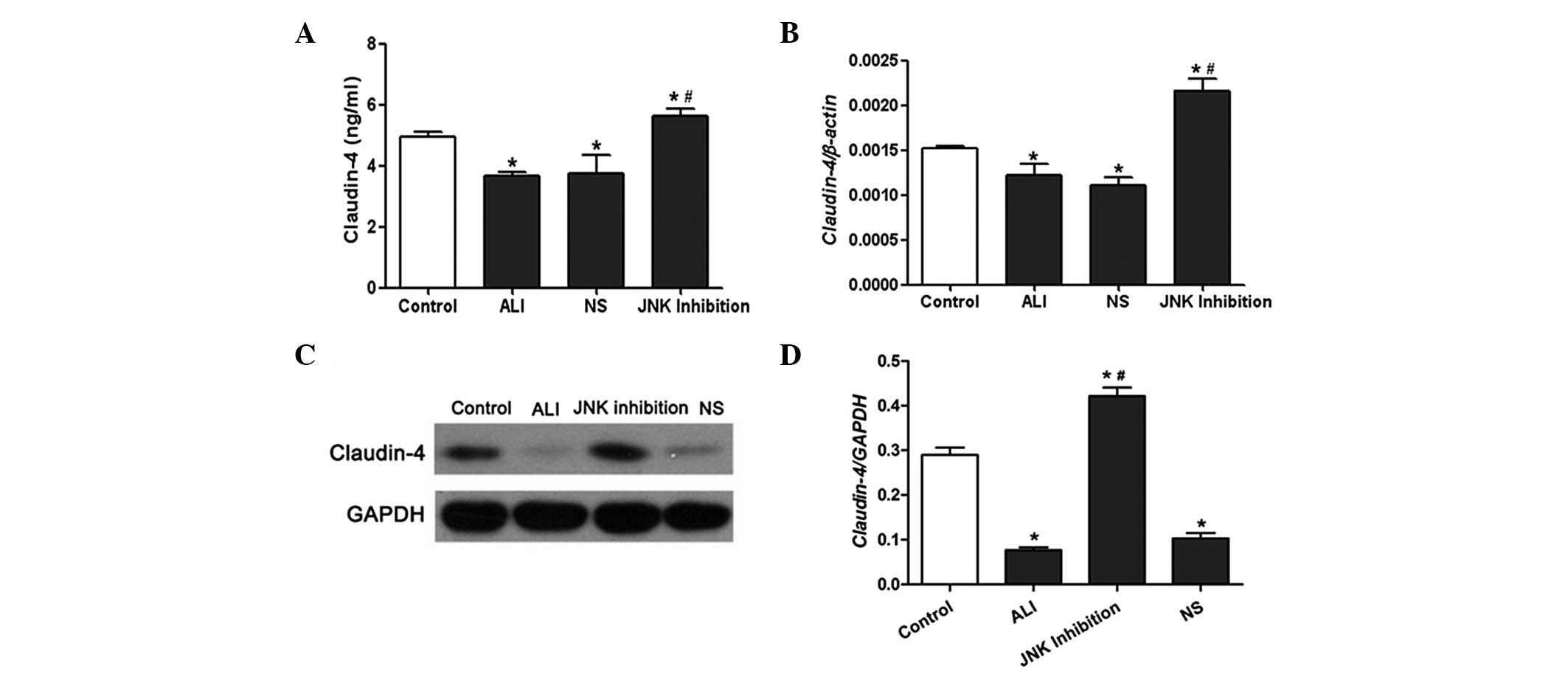
Effect of SP600125 on claudin-4
expression and JNK phosphorylation in vivo and in vitro
The results from the ELISA demonstrated that the
expression of claudin-4 in BALF in the LPS-treated rats was
markedly lower compared with the rats in the control group.
However, the expression levels of claudin-4 in BALF in the rats
from the SP600125 group were significantly increased (Fig. 3A). The mRNA and protein expression
of claudin-4 was significantly reduced in rat lung tissues treated
with LPS; however, this was reversed following administration of
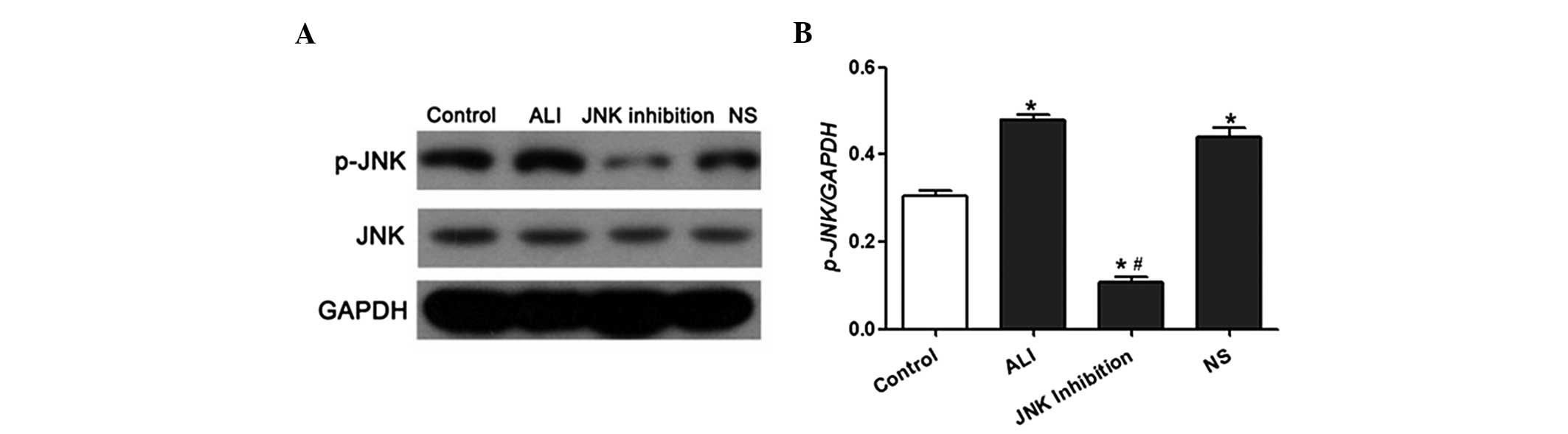
SP600125 (Fig. 3B–D). Western blot
analysis demonstrated that JNK phosphorylation in lung tissues was
significantly increased following LPS treatment. However, SP600125
administration led to a reduction in JNK phosphorylation in the
lung tissues of LPS-induced ALI rats (Fig. 4A and B).
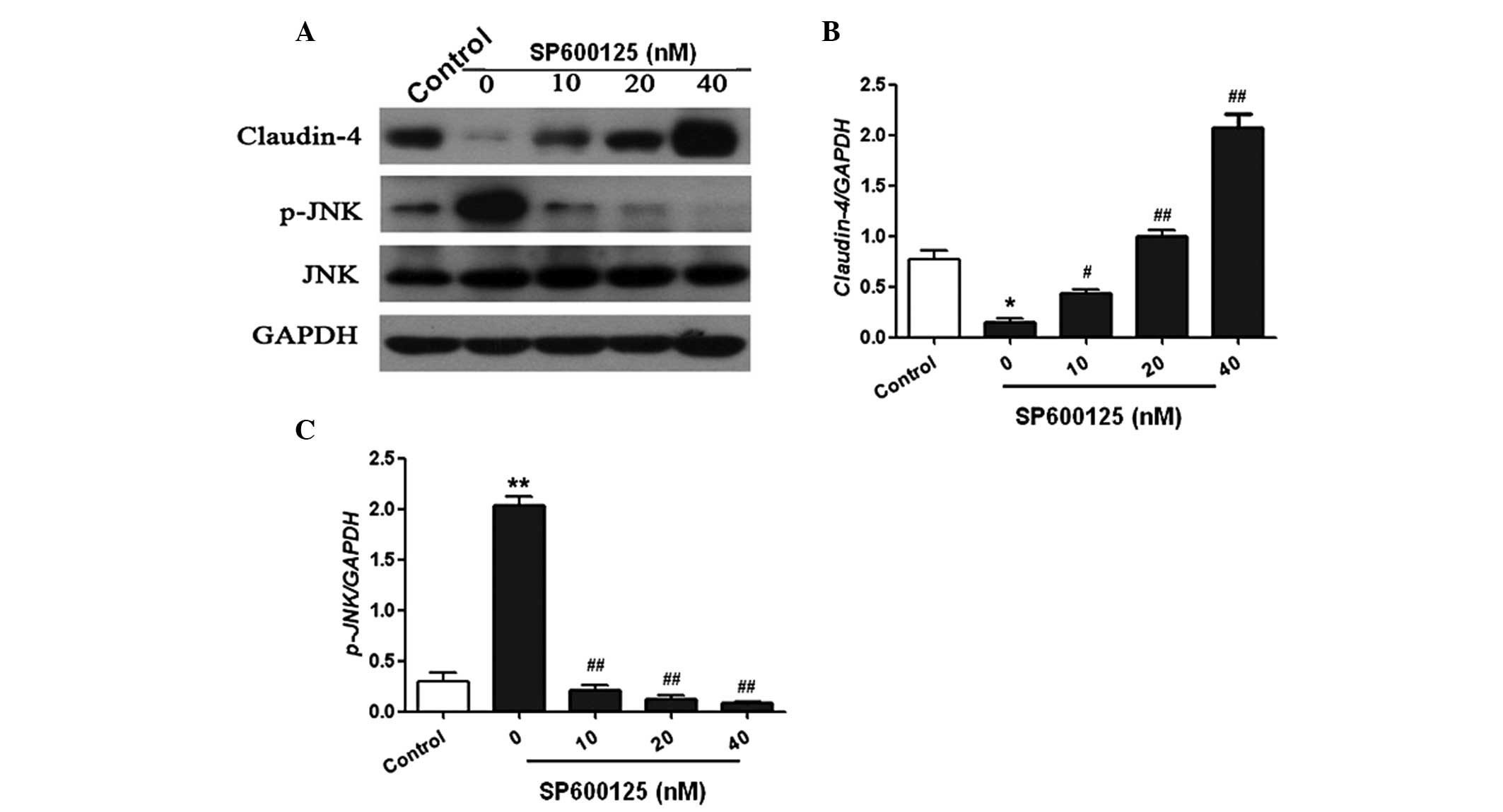
Furthermore, the in vitro study demonstrated
that the expression of claudin-4 was markedly reduced following LPS
injection in A549 cells. However, treatment with SP600125
significantly increased the expression of claudin-4 in a
dose-dependent manner (Fig. 5A and
B). In addition, JNK phosphorylation was significantly
increased following LPS treatment. However, SP600125 treatment
dose-dependently reduced JNK phosphorylation in A549 cells
(Fig. 5C). These results
demonstrated that SP600125 inhibited JNK activation and upregulated
claudin-4 expression levels in vivo and in vitro.
Discussion
ALI is an acute and progressive respiratory disease,
which is characterized by progressive diffuse bilateral pulmonary
edema and inflammation (16). ALI
has a high mortality rate and 40–50% of patients succumb to
multiple organ failure (3). In the
present study, the effect of SP600125, a JNK selective inhibitor,
in LPS-induced ALI and its underlying mechanism was
investigated.
A previous study demonstrated that, LPS, located on
the outer membrane of Gram-negative bacteria, acted as a
pro-inflammatory reaction factor in infectious diseases and
resulted in inflammatory reactions in vivo (17). Pulmonary edema, a typical
pathological change observed in ALI, was found to reduce lung
compliance and decrease pulmonary gas exchange (18). The present study found that
SP600125 significantly attenuated LPS-induced pulmonary edema in
vivo, as shown by the lung W/D ratio. Numerous studies have
demonstrated that various inflammatory mediators are involved in
the pathogenesis of ALI. Among them, TNF-α and IL-6 are considered
to be the most important inflammatory mediators in the innate
immune response (4). According to
the results from the present study, SP600125 treatment markedly
decreased the expression of TNF-α and IL-6 in BALF induced by LPS
stimulation. In addition, the results demonstrated that typical ALI
pathological alterations were reduced by SP600125 administration.
These results indicated that SP600125 effectively inhibited
LPS-induced pulmonary edema and inflammation in vivo.
Next, the effect of SP69600125 in cultured cells was
investigated. The lung epithelial cell line A549 was used in the
present study to investigate the effect of SP600125 on A549 cell
viability and apoptosis. The results demonstrated that SP600125
significantly improved A549 cell viability in a dose-dependent
manner using the CCK-8 and EdU incorporation assays. Apoptosis is
the process of programmed cell death that is important in cell
growth and maintaining cellular homeostasis, and is regulated by
numerous signaling pathways (19,20).
The present study demonstrated, using flow cytometry, that SP600125
treatment dose dependently reduced the apoptotic rate of A549
cells. The results from the present study demonstrated in
vitro that SP600125 administration promoted cell viability and
reduced cell apoptosis, therefore protecting against LPS-induced
lung injury.
In addition, the present study investigated the
molecular mechanism underlying the protective effect of SP600125 on
LPS-induced ALI. JNK is known to be an important upstream regulator
of the induced expression of inflammatory mediators in response to
stress, cytokines and cytoskeletal reorganization (21,22).
Previous studies have demonstrated that JNK inhibition by SP600125
suppressed the release of TNF-α into BALF, as well as inhibited the
expression of matrix metalloproteinases in synoviocytes and in
inflammatory arthritis (23,24).
Another study revealed that the inhibition of JNK suppressed
LPS-induced increases in the DNA binding activity of NF-κB by
downregulation of the phosphorylation of inhibitor κB-α (25). The results from the present study
demonstrated that SP600125 significantly inhibited the JNK
signaling pathway in vivo and in vitro. Claudins are
a family of proteins that are important components of tight
junctions, where they establish the paracellular barrier
controlling the molecular flow between the cells of an epithelium
(26). Claudin-4, one member of
the claudin family, has been previously reported to be required for
maximal epithelial barrier function, including alveolar fluid
clearance in mice (27,28). The levels of claudin-4 affect
paracellular transport by conferring relative chloride selectivity
and decreasing ion conductance, suggesting that claudin-4 is
important in determining alveolar fluid clearance rates in human
lungs (29). In the present study,
claudin-4 expression was found to be significantly increased
following SP600125 administration in vivo and in
vitro, suggesting that the protective effect of SP600125 was
partly mediated by claudin-4.
In conclusion, the present study demonstrated for
the first time, to the best of our knowledge, that the JNK
inhibitor SP600125 protected against LPS-induced ALI in vivo
and in vitro, possibly by upregulating the expression of
claudin-4.
Acknowledgements
This study was supported by the public welfare funds
from the Science and Technology Department of Zhejiang province
(grant no. 2013C33202), the general project funds from the Health
Department of Zhejiang province (grant no. 2012Kyb012) and the
general project funds from the Administration of Traditional
Chinese Medicine of Zhejiang province (grant no. 2012ZA018).
References
|
1
|
Yang B, Huang W, Han J and Liang Z: Study
of the role of epidermal growth factor on lung fluid transport in
rabbits with acute lung injury caused by endotoxin. Exp Ther Med.
4:611–614. 2012.PubMed/NCBI
|
|
2
|
Vincent JL, Sakr Y and Ranieri VM:
Epidemiology and outcome of acute respiratory failure in intensive
care unit patients. Crit Care Med. 31:S296–S299. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou GJ, Jiang SY, Zhang M, et al:
Evaluation of the inflammatory response in a two-hit acute lung
injury model using [18F]FDG microPET. Exp Ther Med. 6:894–898.
2013.PubMed/NCBI
|
|
4
|
Kolliputi N and Waxman AB: IL-6
cytoprotection in hyperoxic acute lung injury occurs via suppressor
of cytokine signaling-1-induced apoptosis signal-regulating
kinase-1 degradation. Am J Respir Cell Mol Biol. 40:314–324. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mutlu GM and Budinger GR: Incidence and
outcomes of acute lung injury. N Engl J Med. 354:416–417. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meade MO, Cook DJ, Guyatt GH, et al:
Ventiliation Study Investigators: Ventilation strategy using low
tidal volumes, recruitment maneuvers, and high positive
end-expiratory pressure for acute lung injury and acute respiratory
distress syndrome: a randomized controlled trial. JAMA.
299:637–645. 2008.
|
|
7
|
Sio SW, Ang SF, Lu J, et al: Substance P
upregulates cyclooxygenase-2 and prostaglandin E metabolite by
activating ERK1/2 and NF-kappaB in a mouse model of burn-induced
remote acute lung injury. J Immunol. 185:6265–6276. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang X, Liu F, Liu H, et al: Urinary
trypsin inhibitor attenuates lipopolysaccharide-induced acute lung
injury by blocking the activation of p38 mitogen-activated protein
kinase. Inflamm Res. 60:569–575. 2011. View Article : Google Scholar
|
|
9
|
Ong E, Gao XP, Predescu D, et al: Role of
phosphatidylinositol 3-kinase-gamma in mediating lung neutrophil
sequestration and vascular injury induced by E. coli sepsis.
Am J Physiol Lung Cell Mol Physiol. 289:L1094–L1103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee N, Maurange C, Ringrose L and Paro R:
Suppression of Polycomb group proteins by JNK signalling induces
transdetermination in Drosophila imaginal discs. Nature.
438:234–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gavrilescu LC, Molnár Á, Murray L, et al:
Retraction for Zhong et al. GCK is essential to systemic
inflammation and pattern recognition receptor signaling to JNK and
p38. Proc Natl Acad Sci USA. 109:51342012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of propyl gallate-induced calf pulmonary arterial
endothelial cell death by MEK and JNK inhibitors. Mol Med Rep.
2:825–830. 2009.PubMed/NCBI
|
|
13
|
Nacken W, Ehrhardt C and Ludwig S: Small
molecule inhibitors of the c-Jun N-terminal kinase (JNK) possess
antiviral activity against highly pathogenic avian and human
pandemic influenza A viruses. Biol Chem. 393:525–534. 2012.
View Article : Google Scholar
|
|
14
|
Yeste-Velasco M, Folch J, Casadesús G, et
al: Neuroprotection by c-Jun NH2-terminal kinase inhibitor SP600125
against potassium deprivation-induced apoptosis involves the Akt
pathway and inhibition of cell cycle reentry. Neuroscience.
159:1135–1147. 2009. View Article : Google Scholar
|
|
15
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu T, Wang DX, Zhang W, et al:
Andrographolide protects against LPS-induced acute lung injury by
inactivation of NF-κB. PLoS One. 8:e564072013.PubMed/NCBI
|
|
17
|
Suresh R, Kupfer Y and Tessler S: Acute
respiratory distress syndrome. N Engl J Med. 343:660–661. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shaham S: Apoptosis: a process with a
(beta)NAC for complexity. Cell. 114:659–661. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inoue S, MacFarlane M, Harper N, et al:
Histone deacetylase inhibitors potentiate TNF-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid
malignancies. Cell Death Differ. 11(Suppl 2): S193–S206. 2004.
View Article : Google Scholar
|
|
20
|
Lee SJ and Lim KT: Inhibitory effect of
ZPDC glycoprotein on the expression of inflammation-related
cytokines through p38 MAP kinase and JNK in
lipopolysaccharide-stimulated RAW 264.7 cells. Inflamm Res.
58:184–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumar A, Byun HS, Bittman R and Saba JD:
The sphingolipid degradation product trans-2-hexadecenal induces
cytoskeletal reorganization and apoptosis in a JNK-dependent
manner. Cell Signal. 23:1144–1152. 2011. View Article : Google Scholar
|
|
22
|
Kim BW, Koppula S, Kim IS, et al:
Anti-neuroinflammatory activity of Kamebakaurin from Isodon
japonicus via inhibition of c-Jun NH2-terminal
kinase and p38 mitogen-activated protein kinase pathway in
activated microglial cells. J Pharmacol Sci. 116:296–308.
2011.PubMed/NCBI
|
|
23
|
Han Z, Boyle DL, Chang L, et al: c-Jun
N-terminal kinase is required for metalloproteinase expression and
joint destruction in inflammatory arthritis. J Clin Invest.
108:73–81. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee HS, Kim HJ, Moon CS, et al: Inhibition
of c-Jun NH2-terminal kinase or extracellular
signal-regulated kinase improves lung injury. Respir Res. 5:232004.
View Article : Google Scholar
|
|
25
|
Cording J, Berg J, Käding N, et al: In
tight junctions, claudins regulate the interactions between
occludin, tricellulin and marvelD3, which, inversely, modulate
claudin oligomerization. J Cell Sci. 126:554–564. 2013. View Article : Google Scholar
|
|
26
|
Wray C, Mao Y, Pan J, et al: Claudin-4
augments alveolar epithelial barrier function and is induced in
acute lung injury. Am J Physiol Lung Cell Mol Physiol.
297:L219–L227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lipschutz JH, Li S, Arisco A and Balkovetz
DF: Extracellular signal-regulated kinases 1/2 control claudin-2
expression in Madin-Darby canine kidney strain I and II cells. J
Biol Chem. 280:3780–3788. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Itallie C, Rahner C and Anderson JM:
Regulated expression of claudin-4 decreases paracellular
conductance through a selective decrease in sodium permeability. J
Clin Invest. 107:1319–1327. 2001.PubMed/NCBI
|
|
29
|
Rokkam D, Lafemina MJ, Lee JW, et al:
Claudin-4 levels are associated with intact alveolar fluid
clearance in human lungs. Am J Pathol. 179:1081–1087. 2011.
View Article : Google Scholar : PubMed/NCBI
|