Introduction
Lymphomatoid papulosis (LyP) is a chronic
papulonecrotic or papulonodular skin disease with histological
features suggestive of a malignant lymphoma (1). The disease is characterized by
recurrent crops of pruritic papules, predominantly on the trunk and
limbs, at different stages of development. The papules heal
spontaneously over 1–2 months, usually leaving slightly depressed
oval scars (2–4). The clinical manifestation of the
disease is similar to that of pityriasis lichenoides et
varioliformis acuta; as a result, LyP is easily misdiagnosed. The
diagnosis is primarily dependent on the histopathology and
immunohistochemical examination. The present study reports two
cases of LyP confirmed by the Second Affiliated Hospital of Xi’an
Jiaotong University (Xi’an, China), with a literature review. The
study was approved by the Ethics Committee of the Second Affiliated
Hospital of Xi’an Jiaotong University. Written informed consent was
provided by the patients involved.
Case reports
Case 1
A 17-year old male who had multiple papules and
nodules with necrosis on the trunk and limbs that had persisted for
one month was admitted to the Second Affiliated Hospital of Xi’an
Jiaotong University. One month prior to admission, no evident red
papules or nodules were visible on the torso and limbs. However,
papules and nodules measuring 0.8×0.4 cm had started to appear,
which gradually increased in number, developed an ulcerated,
crusted surface and were slightly itchy but painless. Accordingly,
this case was initially diagnosed as mite dermatitis at another
hospital. As prescribed, the patient applied compound indomethacin
tincture and mometasone furoate cream to the skin twice a day for
two weeks. However, the curative effect was poor. Subsequently, in
the Second Affiliated Hospital of Xi’an Jiaotong University, this
case was diagnosed as pityriasis lichenoides et varioliformis acuta
and the patient was treated with an antibiotic (clarithromycin, 0.5
g orally once a day for one week), and corticosteroid cream (twice
a day for two weeks). Histopathological and immunohistochemical
examination was also performed.
From the onset of the disease, the patient remained
normal in spirit, food intake, night rest and urinary function, and
underwent no significant changes in body weight. When he was two
years old, the patient suffered from urticaria with severe itch,
and following treatment with calamine lotion, the rash subsided.
Every two years subsequent to this, similar urticarial lesions
recurred in the summer or autumn. The symptoms were relieved
following the administration of astemizole and calamine lotion. The
patient’s grandfather was affected by psoriasis. Other than this,
the patient was unaware of other genetic disorders in his family
history.
Physical examination revealed that the patient had
stable vital signs, and his superficial lymph nodes were not
palpably enlarged. In addition, a physical examination revealed no
abnormalities, with the exception of the large red papules and
nodules, 0.2–1.0 cm in diameter, that were scattered on the trunk
and limbs. Parts of these exhibited necrosis, ulceration, overlying
black crusts in the center and white scales on the surface. Old
lesions were visible as atrophic scars of various sizes with
different degrees of pigmentation. When palpated, the lesions were
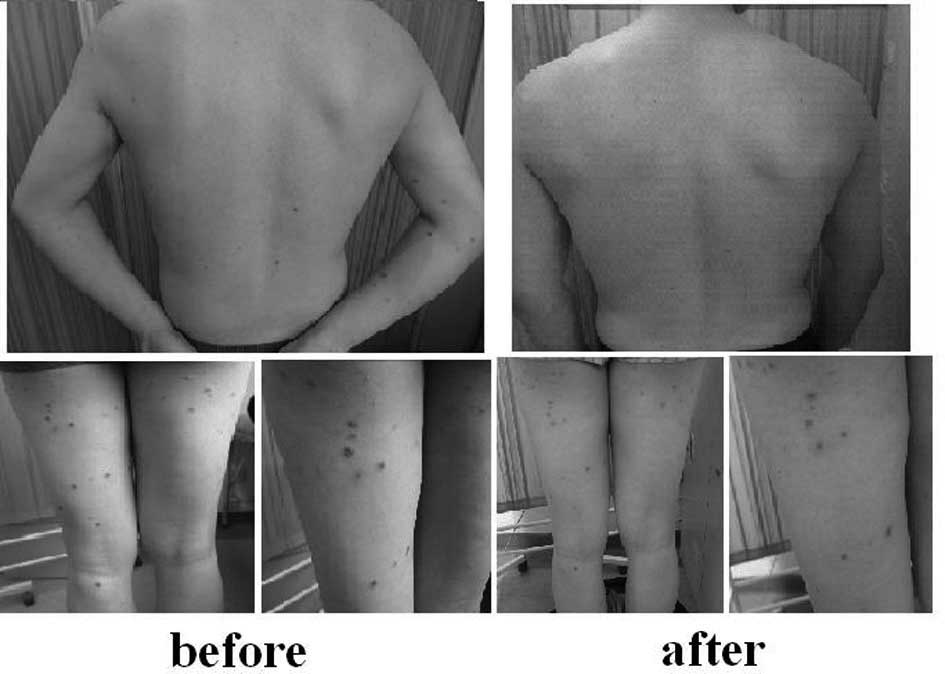
hard and not tender (Fig. 1).
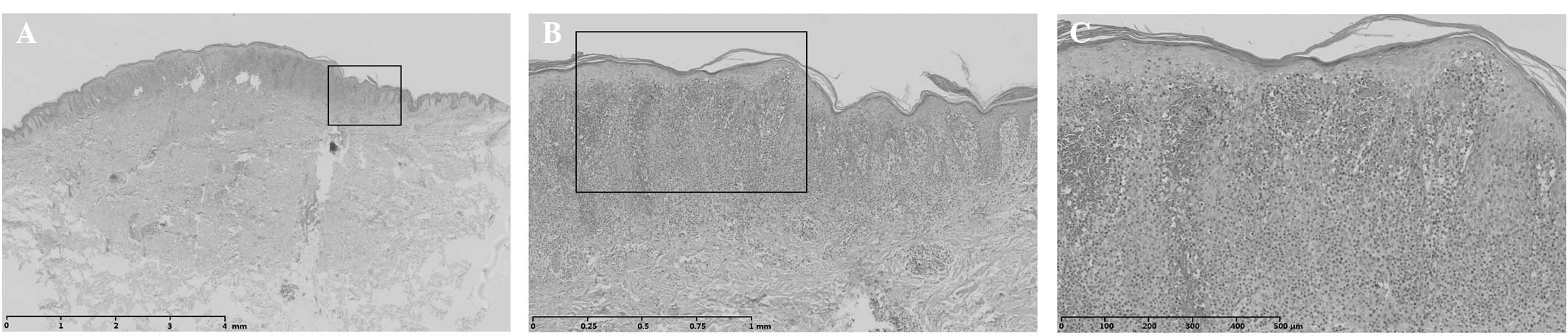
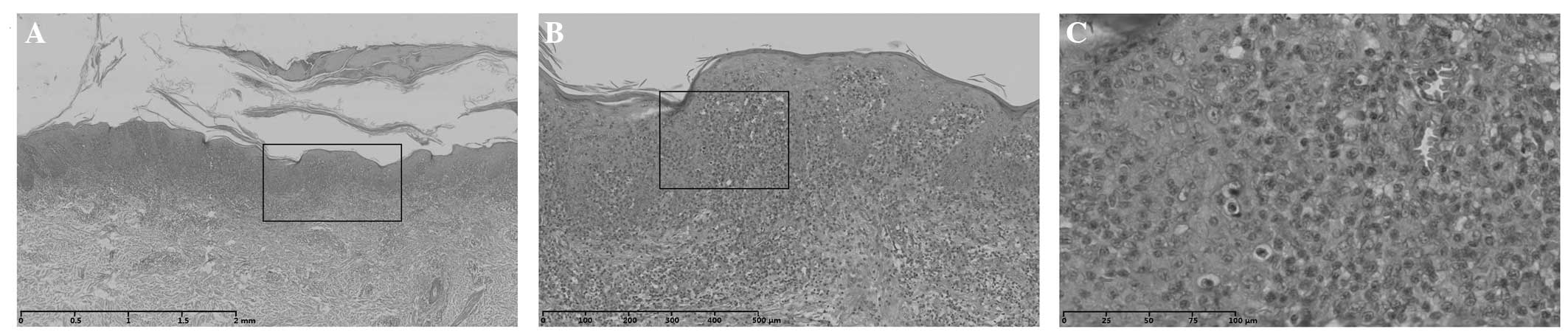
Histopathological examination showed that the
patient had hyperkeratosis of the epidermis, dyskeratosis,
acanthosis and dermal edema. In addition, dense, band-like
lymphocytic infiltration was observed and a large number of
atypical lymphocytes had infiltrated into the epidermis. The
papillary layer of the dermis showed evident edema. Red blood cells
appeared to be undergoing extravasation. Scattered malignant lymph
cells infiltrated into the mid-dermis. Certain lymphocytes showed
nuclear invagination with cerebriform nuclei (Fig. 2).
Immunohistochemical staining demonstrated that this
case was cluster of differentiation (CD)3-, CD4- and T-cell
intracellular antigen-1 (TIA-1)-positive. The Ki67-positive rate
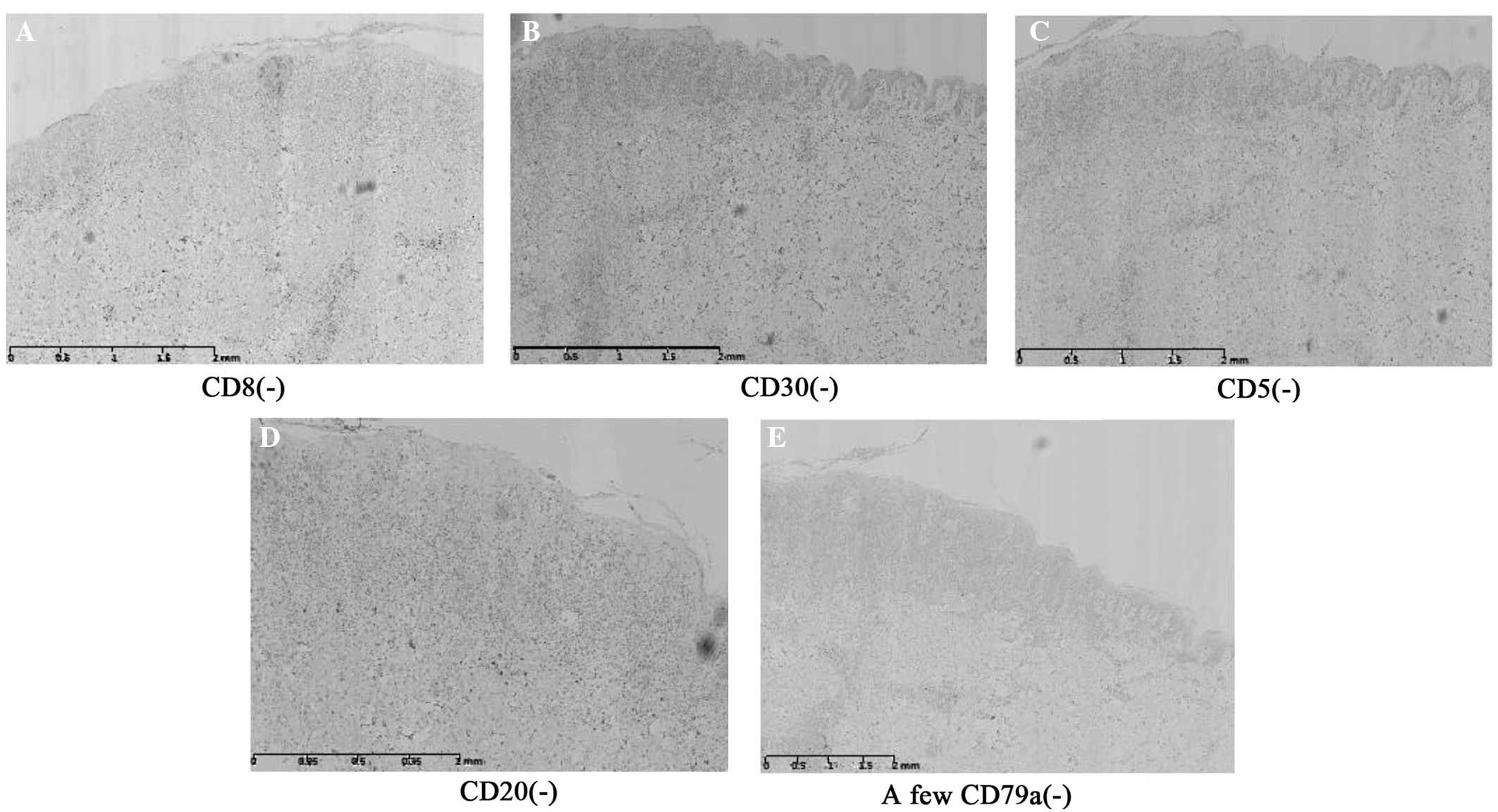
was 70% and CD45RO was partially positive (Fig. 3). In addition, the case was CD5-,
CD8-, CD20- and CD30-negative and a small amount of CD79a was
observed, which was considered negative (Fig. 4). Accordingly, the patient was
diagnosed with LyP of the type B/mycosis fungoides (MF) type in
histopathology. The patient was treated with Viaminate capsules (25
mg, three times a day for one month), Transfer Factor capsules (25
mg, three times a day for one month) and disodium glycyrrhizinate
(0.1 g, twice a day for one month) orally, and glucocorticoid cream
for external use (once a day for two weeks). Subsequent to the
treatment, the lesions gradually subsided and no new lesions formed
(Fig. 1).
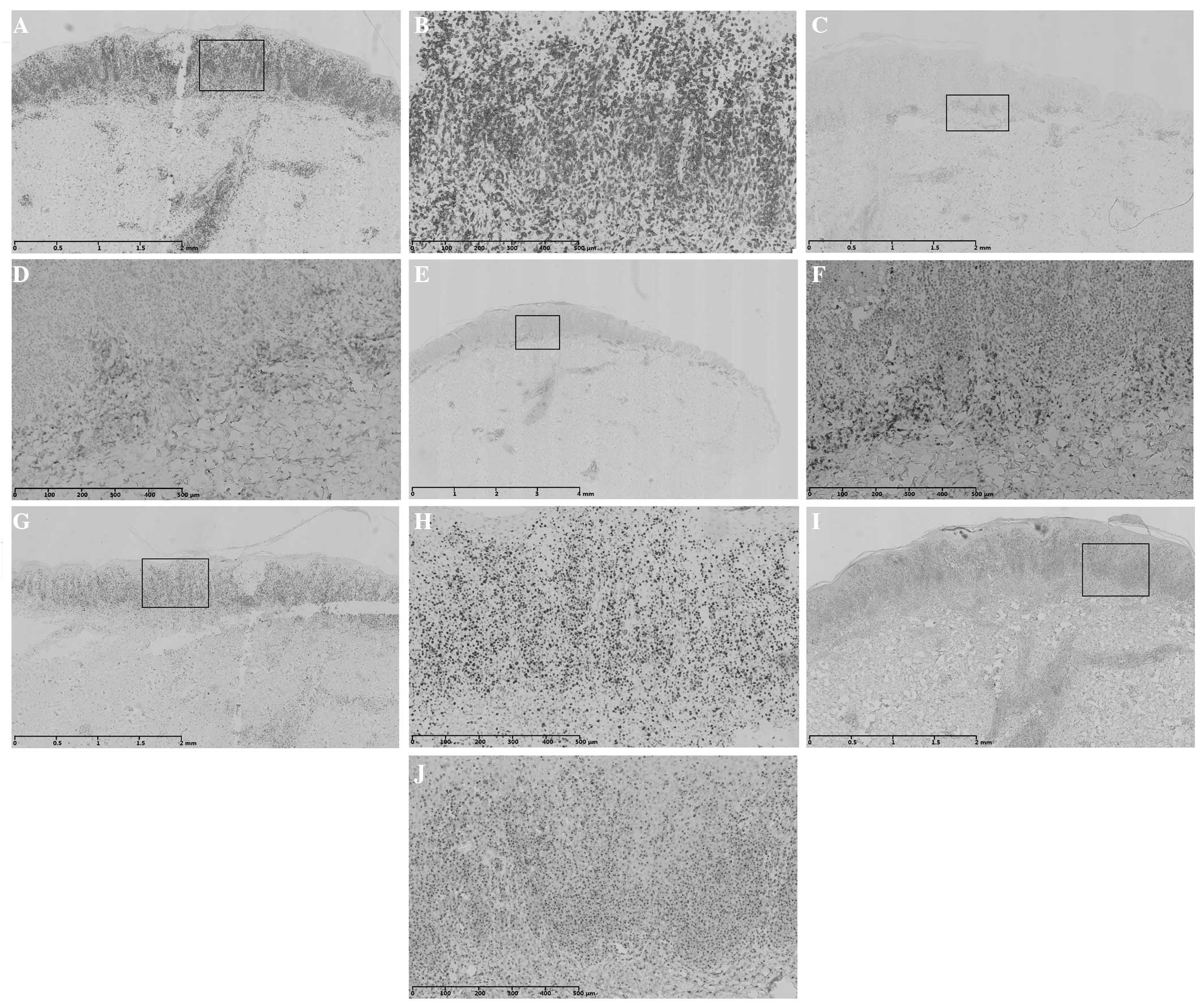 | Figure 3Immunohistochemical staining of case 1
shows positive or partly positive results for (A and B) CD3, (C and
D) CD4, (E and F) CD45RO, (G and H) Ki67 and (I and J) TIA-1. (B,
D, F, H and J) are magnified images (scale bar length = 500 μm) of
(A, C, E, G and I), respectively. Scale bar length: (A, C, G and I)
2 mm and (E) 4 mm. CD, cluster of differentiation; TIA-1, T-cell
intracellular antigen-1. |
Case 2
A 30-year-old male patient was admitted to the
Second Affiliated Hospital of Xi’an Jiaotong University exhibiting
nodules in the left waist that had been present for three months,
spreading over the whole body with ulceration and scab formation
for two months. Three months prior to admission, three purple-red
papules measuring 0.8×0.4 cm appeared in the left waist without
significant external stimuli, which did not bother the patient as
there were no clear symptoms. A month subsequent to this, similar
papules and nodules gradually appeared over the patient’s whole
body. They increased in number, ulcerated, generated an exudate,
and had a putrescent and scabbed central section. Some of the old
lesions spontaneously subsided, leaving blackish-brown
pigmentation. Concurrently, new lesions appeared that were painless
but mildly itchy. The local clinic diagnosed this case as herpes
and treated the patient with acyclovir for one month. However, the
curative effect was poor. Thus, in the hospital, this case was
diagnosed as pityriasis lichenoides et varioliformis acuta and the
patient was orally treated with clarithromcin (0.5 g, once a day),
levocetirizine hydrochloride tablets (5 mg, once a day) and
compound glycyrrhizin capsules (50 mg, three times a day) for one
week. Histopathological and immunohistochemical examination was
also conducted.
From the onset of the disease, the patient remained
normal in spirit, food intake, night rest and urinary function and
no significant changes in body weight occurred. The patient had no
history of severe disease and was not allergic to any food or
drugs. His father had succumbed to esophageal cancer and the
patient was unaware of any similar disease or other genetic
disorders in his family history. The results from a physical
examination revealed that the patient had stable vital signs and
superficial lymph nodes that were not palpably enlarged. No
abnormality of the heart, lungs or abdomen was identified. A number
of red papules and nodules appeared on the face, neck, trunk,
limbs, lip mucosa and scrotum of the patient. These were 0.2–2.0 cm
in diameter and contained sections that exhibited necrosis and
overlying central black crusts. Old lesions formed atrophic scars
of various sizes and with different degrees of pigmentation,
sections of which were slightly atrophied. When palpated, the
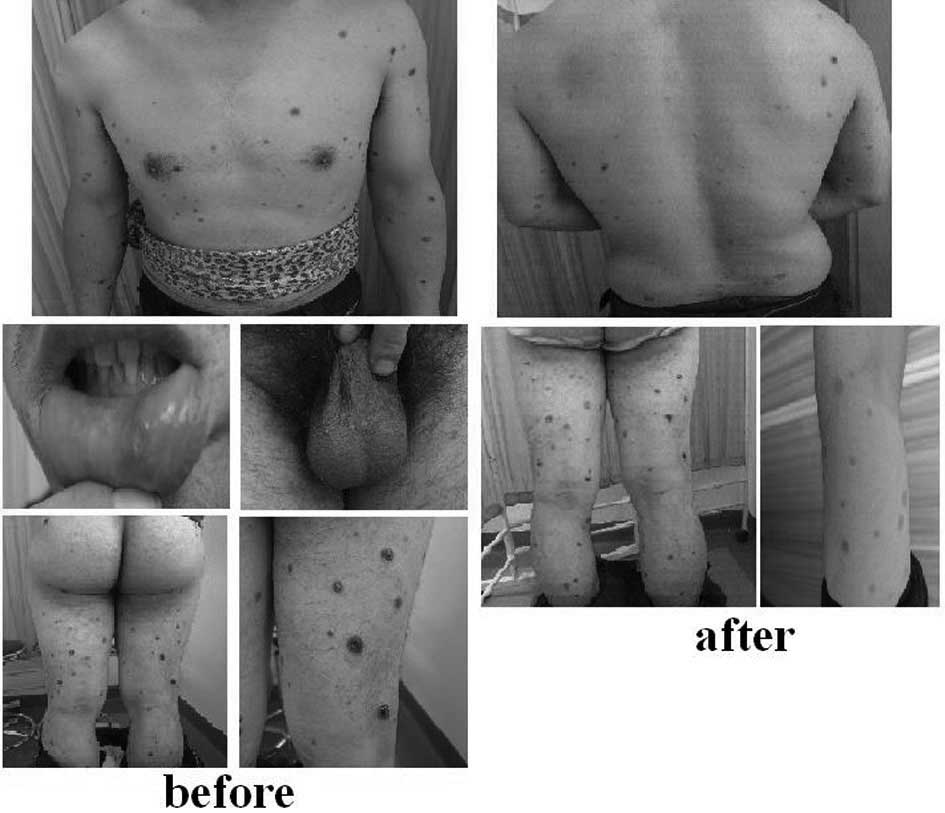
lesions felt hard and were not tender (Fig. 5).
Histopathological examination revealed
hyperkeratosis of the epidermis, dyskeratosis, acanthosis and
dermal edema. Dense lymphocytic infiltration was observed in bands,
and numerous atypical lymphocytes had infiltrated into the
epidermis. The papillary layer of the dermis exhibited evident
edema. Extravasation of the red blood cells was also apparent.
Scattered malignant lymph cells infiltrated into the mid-dermis and
some lymphocytes had invaginated, cerebriform nuclei (Fig. 6).
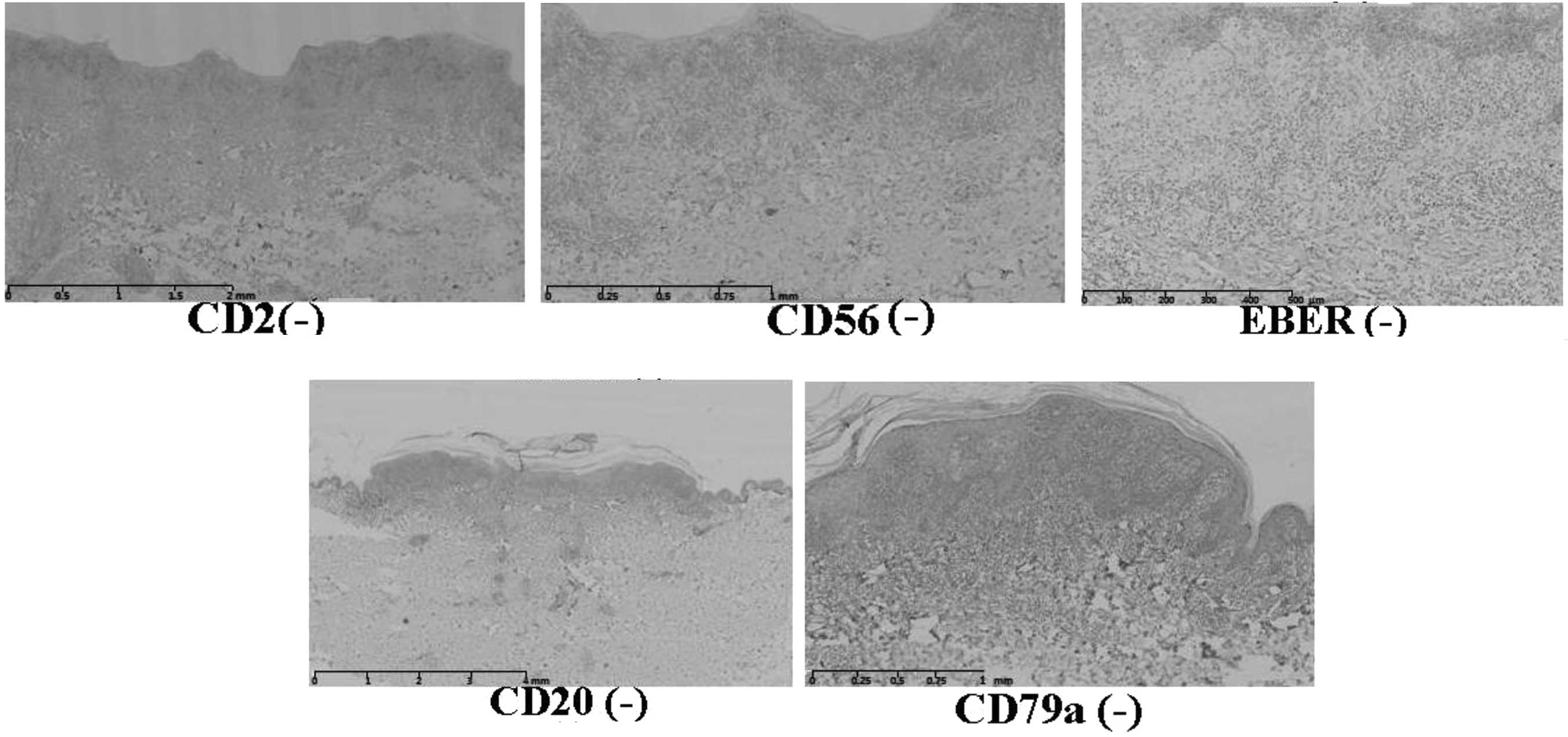
Immunohistochemical staining demonstrated that this
case was CD3-, CD5-, CD45RO- and anaplastic lymphoma kinase
(ALK)-positive, a few scattered large cells were CD30-positive and
40–50% of cells were Ki67-positive (Fig. 7). In addition, the case was CD2-,
CD20-, CD79a-, CD56- and Epstein-Barr (EB) virus-encoded RNA
(EBER)-negative (Fig. 8).
Accordingly, the patient was diagnosed with LyP type B. The patient
was treated with methylprednisolone tablets (4 mg, once every
morning) and Viaminate capsules (25 mg, three times a day) orally,
desonide cream (twice a day) and compound polymyxin B ointment for
external use (once a day) topically, and intramuscular mannatide
injection (5 mg, once every alternate day) for one month. Following
the treatment, the papules shrank and the ulcerated nodules became
crusted, flattened and gradually subsided. The original
pigmentation faded and no new lesions formed (Fig. 5).
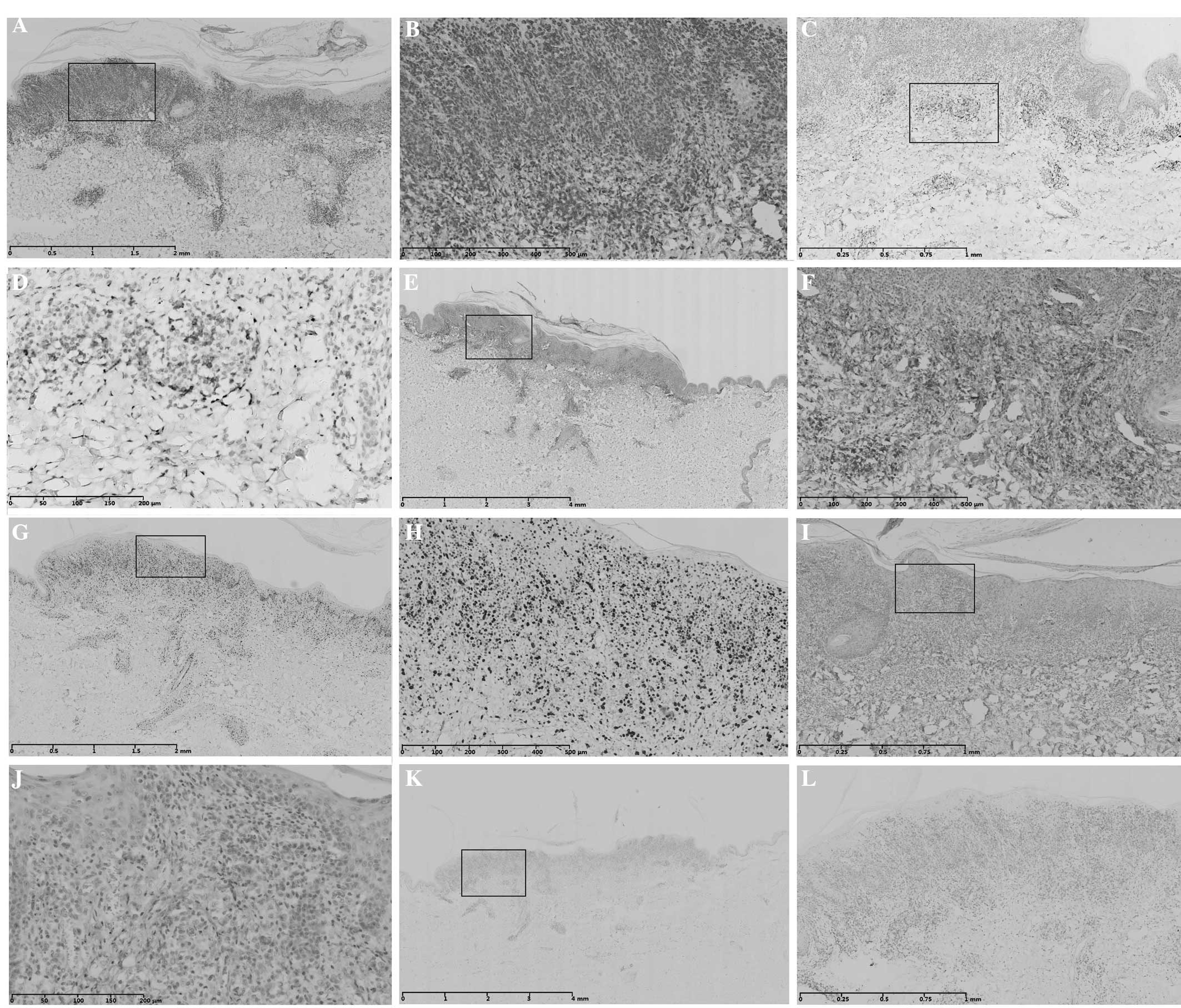 | Figure 7Immunohistochemical staining of case 2
shows positive or partly positive results for (A) CD3, (C) CD5, (E)
CD45RO, (G) Ki67, (I) CD30 and (K) anaplastic lymphoma kinase. (B,
D, F, H, J and L) are magnified images of (A, C, E, G, I and K),
respectively. Scale bar length: (A) 2 mm; (B) 500 μm; (C) 1 mm; (D)
200 μm; (E) 4 mm; (F) 500 μm; (G) 2 mm; (H) 500 μm; (I) 1 mm; (J)
200 μm; (K) 4 mm; (L) 1 mm. CD, cluster of differentiation. |
Discussion
The term LyP was originally used by Macaulay
(5) in 1968 to describe ‘a
self-healing rhythmical paradoxical eruption, histologically
malignant but clinically benign’. However, the classification
system for cutaneous lymphomas has evolved rapidly. During
consensus meetings in 2003–2004, the World Health
Organization-European Organization for Research and Treatment of
Cancer (WHO-EORTC) classification grouped LyP among the indolent
cutaneous T-cell lymphomas (6,7). LyP
is classified as a cutaneous lymphoma due to its association with
other malignant lymphoproliferative disorders. LyP is part of a
spectrum of CD30-positive cutaneous lymphoproliferative diseases
that also includes primary cutaneous anaplastic large cell lymphoma
(ALCL) and borderline CD30+ lesions (8,9). The
etiology and pathogenesis of LyP remain unclear, but have been
speculated to have an association with infections by paramyxovirus,
human leukemia/lymphoma virus or EB virus (10–13).
LyP can be found in individuals of any age, but most
commonly occurs in adults, particularly those aged >40 years,
with no significant difference between genders (14,15).
It is mainly divided into three types in the clinical setting.
These are the classic type (type A), an MF-like type in which
plaque parapsoriasis or MF-like plaques are present (type B), and a
limited granuloma type, which is similar to Hodgkin’s disease (type
C). Since the 1980s, several subtypes have been reported, including
LyP of hair follicles, eosinophilic histiocytosis and
hypereosinophilic syndrome (16,17).
The lesions frequently occur on the trunk and proximal extremities
and may also be found on the palms and soles, scalp and the genital
and oral mucosa. LyP is similar to pityriasis lichenoides et
varioliformis auta clinically and its characteristics include skin
lesions that appear in groups, reddish, purple or red-brown papules
and recurring nodules that appear in small or large numbers (up to
several hundreds) and distribute symmetrically with unequal size
(typically <2 cm). A few papules develop into vesicles and
pustules, and they can also develop into masses with diameters of
5–15 cm, followed by gradual necrosis, ulceration, or the formation
of a scab or surface scaliness (18). In general, single lesions regress
in 3–12 weeks, leaving hyperpigmentation or atrophic scars. Since
LyP is chronic and skin lesions persist in recurring and fading,
lesions of various types may be present during the same period of
time. Lesions confined to a region are known as limited type LyP,
which is rare in clinical practice (19). Lymphadenopathy, thyroiditis, fever,
weight loss and fatigue may also be associated with LyP (20).
The 2005 WHO-EORTC cutaneous lymphoma classification
divided LyP into types A, B and C, but there are no strict
boundaries separating these types from each other. Occasionally,
individual lesions may combine A, B or C types, which is termed a
hybrid. In type A, or the histiocytic type, specimens display dense
mixed infiltrates characterized by large atypical lymphocytes and
neutrophils, eosinophils, histiocytes and small lymphocytes. The
large atypical cells do not form sheets or constitute >50% of
the infiltrating cells. In type B, or the lymphocytic type,
specimens display a monomorphous infiltrate of small to
medium-sized lymphocytes with cerebriform nuclei similar to those
observed in MF. This variant has also been referred to as MF-like
LyP in the literature (21). In
type C, specimens display cytological features similar to those of
type A and ALCL, and the atypical lymphoid cells either form sheets
or large nodules and these represent >50% of the infiltrating
cells. This type has been referred to as borderline LyP-ALCL
(22).
Atypical cells of the three types described above
can express proteins associated with cytotoxic T cells, including
TIA-1, perforin and granzyme B (GrB). These cells are mostly
ALK-negative, but few of them are positive. They are usually
epithelial membrane antigen- and CD15-negative. The percentage of
the total cells that are Ki67-positive is 50–95% (23).
In addition to these three previously known types, a
novel type of LyP, designated type D, was recently proposed
(24,25). Although the clinical presentation
of type D LyP is similar to that of CD8-positive cytotoxic T-cell
lymphoma, type D LyP has a more favorable prognosis. These
conditions differ in that CD30 expression in LyP type D is
positive. Thus, the clinical diagnosis must be conducted carefully
to identify the condition correctly.
Since LyP is self-limiting and the prognosis is
promising, with the exception of a few lesions that do not heal
well, and lesions in the face, hand, foot and genitals causing
aesthetic, function and scarring problems, the majority of patients
do not require active treatment. The current reported treatment
methods are the systemic or local application of glucocorticoids,
phototherapy (psoralen and ultraviolet A light, ultraviolet B
treatment and photodynamic therapy), retinoic acid, methotrexate,
interferon, radiotherapy, nitrogen mustard and hormone replacement
therapies (26–31). The five-year survival rate is 100%
(32,33). However, a number of studies have
found that 10–20% of patients may exhibit diseases prior to,
coexisting with or subsequent to LyP, including MF, Hodgkin’s
lymphoma, ALCL or other tumors. These diseases have a high chance
of developing into malignant lymphoma; therefore, it is recommended
that all patients with LyP should be followed-up throughout their
lifetime (34–38).
The two cases described in this paper involve
typical skin lesions, characterized by multiple papules and nodules
with gradually broken and scabbed centers. Since the clinical
manifestation is similar to that of pityriasis lichenoides et
varioliformis acuta, it is easily misdiagnosed. Therefore,
histopathological and immunohistochemical examinations were
conducted and finally the two patients were diagnosed with type B
LyP. The treatments prescribed to the two patients included
retinoic acid and immunomodulatory drugs taken orally and
corticosteroid ointment for external use. In view of the increased
severity of case 2, a small dose of hormone orally and antibiotic
for external use was included. The two patients obtained a
satisfactory curative effect. As Lyp may be secondary to other
malignant tumors, and due to the short duration of follow-up, close
observation of the patients remains necessary in the future.
References
|
1
|
Zic JA: Lymphomatoid papulosis treatment
& management. Medscape Website. Available at: http://emedicine.medscape.com/article/1098954-treatmenturi.
Accessed January 24, 2012
|
|
2
|
Fernández-Guarino M, Carrillo-Gijón R and
Jaén-Olasolo P: Lymphomatoid papulosis: clinical and pathological
findings in 18 patients. Actas Dermosifiliogr. 103:388–393.
2012.(In Spanish).
|
|
3
|
Martorell-Calatayud A, Hernández-Martín A,
Colmenero I, et al: Lymphomatoid papulosis in children: report of 9
cases and review of the literature. Actas Dermosifiliogr.
101:693–701. 2010.(In Spanish).
|
|
4
|
Magro CM and Crowson AN: CD30-positive
lymphoproliferative disorders including lymphomatoid papulosis,
borderline CD3-positive lymphoproliferative disease, anaplastic
large cell lymphoma, and T-cell-rich CD30-positive large B cell
lymphoma. The Cutaneous Lymphoid Proliferations: A Comprehensive
Textbook of Lymphocytic Infiltrates of the Skin. Magro CM, Crowson
AN and Mihm MC: John Wiley & Sons, Inc; Hoboken, NJ: pp.
440–444. 2007
|
|
5
|
Macaulay WL: Lymphomatoid papulosis. A
continuing self-healing eruption, clinically benign -
histologically malignant. Arch Dermatol. 97:23–30. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Willemze R, Jaffe ES, Burg G, et al:
WHO-EORTC classification for cutaneous lymphomas. Blood.
105:3768–3785. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Willemze R and Meijer CJ: Classification
of cutaneous T-cell lymphoma: from Alibert to WHO-EORTC. J Cutan
Pathol. 33(Suppl 1): 18–26. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Slater DN: The new World Health
Organization-European Organization for Research and Treatment of
Cancer classification for cutaneous lymphomas: a practical marriage
of two giants. Br J Dermatol. 153:874–880. 2005. View Article : Google Scholar
|
|
9
|
de Souza A, el-Azhary RA, Camilleri MJ,
Wada DA, Appert DL and Gibson LE: In search of prognostic
indicators for lymphomatoid papulosis: a retrospective study of 123
patients. J Am Acad Dermatol. 66:928–937. 2012.PubMed/NCBI
|
|
10
|
Laube S, Stephens M, Smith AG, et al:
Lymphomatoid papulosis in a patient with atopic eczema on long-term
ciclosporin therapy. Br J Dermatol. 152:1346–1348. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leo AM and Ermolovich T: Lymphomatoid
papulosis while on efalizumab. J Am Acad Dermatol. 61:540–541.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JH, Lee J, Lee JH, et al:
Lymphomatoid papulosis in a patient treated with adalimumab for
juvenile rheumatoid arthritis. Dermatology. 225:259–263. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SK and Kim YC: Lymphomatoid papulosis
after allogenic stem cell transplantation. Eur J Dermatol.
19:520–521. 2009.PubMed/NCBI
|
|
14
|
Atkins KA, Dahlem MM and Kohler S: A case
of lymphomatoid papulosis with prominent myxoid change resembling a
mesenchymal neoplasm. Am J Dermatopathol. 25:62–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Neer FJ, Toonstra J, Van Voorst Vader
PC, et al: Lymphomatoid papulosis in children: a study of 10
children registered by the Dutch Cutaneous Lymphoma Working Group.
Br J Dermatol. 144:351–354. 2001.PubMed/NCBI
|
|
16
|
Chen L, Jin HZ, Yin HL, et al:
Lymphomatoid papulosis. Study on the prevention and treatment of
tumor. Zhong Liu Fang Zhi Yan Jiu. 27:130–132. 2000.(In
Chinese).
|
|
17
|
Willemze R, Meyer CJ, Van Vloten WA and
Scheffer E: The clinical and histological spectrum of lymphomatoid
papulosis. Br J Dermatol. 107:131–144. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Querfeld C, Kuzel TM, Guitart J and Rosen
ST: Primary cutaneous CD30+ lymphoproliferative
disorders: new insights into biology and therapy. Oncology
(Willston Park). 21:689–696; discussion 699–700. 2007.
|
|
19
|
Heald P, Subtil A, Breneman D and Wilson
LD: Persistent agmination of lymphomatoid papulosis: an equivalent
of limited plaque mycosis fungoides type of cutaneous T-cell
lymphoma. J Am Acad Dermatol. 57:1005–1011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shao Z, Zuo C, Ge L, et al: Lymphomatoid
papulosis: Case report and literature review. Wei Xun Huan Xue Za
Zhi. 22:34–36. 2012.(In Chinese).
|
|
21
|
Saggini A, Gulia A, Argenyi Z, et al: A
variant of lymphomatoid papulosis simulating primary cutaneous
aggressive epidermotropic CD8+ cytotoxic T-cell
lymphoma: Description of 9 cases. Am J Surg Pathol. 34:1168–1175.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
El Shabrawi-Caelen L, Kerl H and Cerroni
L: Lymphomatoid papulosis: reappraisal of clinicopathologic
presentation and classification into subtypes A, B, and C. Arch
Dermatol. 140:441–447. 2004.PubMed/NCBI
|
|
23
|
Liao QL, Chen XD, Wang W, et al: The
clinical pathological analysis of primary cutaneous
CD30+ lymphoproliferative disease. Zhong Hua Pi Fu Ke Za
Zhi. 44:151–154. 2011.(In Chinese).
|
|
24
|
Andersen RM, Larsen MS, Poulsen TS, et al:
Lymphomatoid papulosis type D or an aggressive epidermotropic
CD8(+) cytotoxic T-cell lymphoma? Acta Derm Venereol. 94:474–475.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sim JH and Kim YC: CD8+ lymphomatoid
lapulosis. Ann Dermatol. 23:104–107. 2011. View Article : Google Scholar
|
|
26
|
Hoetzenecker W, Guenova E, Hoetzenecker K,
et al: Successful treatment of recalcitrant lymphomatoid papulosis
in a child with PUVA-bath photochemotherapy. Eur J Dermatol.
19:646–647. 2009.PubMed/NCBI
|
|
27
|
Coelho JD, Afonso A and Feio AB: Regional
lymphomatoid papulosis in a child - treatment with a UVB
phototherapy handpiece. J Cosmet Laser Ther. 12:155–156. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rodrigues M, McCormack C, Yap LM, et al:
Successful treatment of lymphomatoid papulosis with photodynamic
therapy. Australas J Dermatol. 50:129–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yip L, Darling S and Orchard D:
Lymphomatoid papulosis in children: experience of five cases and
the treatment efficacy of methotrexate. Australas J Dermatol.
52:279–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shang SX, Chen H, Sun JF and Xu XL:
Regional lymphomatoid papulosis successfully controlled by
interferon α-2b and nitrogen mustard solution. Chin Med J (Engl).
126:3194–3195. 2013.PubMed/NCBI
|
|
31
|
Spires N and McGibbon D: Lymphomatoid
papulosis improving on hormone-replacement therapy. Clin Exp
Dermatol. 34:635–636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grange F and Bagot M: Prognosis of primary
cutaneous lymphomas. Ann Dermatol Venereol. 129:30–40. 2002.(In
French).
|
|
33
|
Nijsten T, Curiel-Lewandrowski C and Kadin
ME: Lymphomatoid papulosis in children: a retrospective cohort
study of 35 cases. Arch Dermatol. 140:306–312. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kunishige JH, McDonald H, Alvarez G,
Johnson M, Prieto V and Duvic M: Lymphomatoid papulosis and
associated lymphomas: a retrospective case series of 84 patients.
Clin Exp Dermatol. 34:576–581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bekkenk MW, Geelen FA, van Voorst Vader
PC, et al: Primary and secondary cutaneous CD30(+)
lymphoproliferative disorders: a report from the Dutch Cutaneous
Lymphoma Group on the long-term follow-up data of 219 patients and
guidelines for diagnosis and treatment. Blood. 95:3653–3661.
2000.PubMed/NCBI
|
|
36
|
Beljaards RC and Willemze R: The prognosis
of patients with lymphomatoid papulosis associated with malignant
lymphomas. Br J Dermatol. 126:596–602. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tan AW and Giam YC: Lymphomatoid papulosis
associated with recurrent cutaneous T-cell lymphoma. Ann Acad Med
Singapore. 33:110–112. 2004.PubMed/NCBI
|
|
38
|
Rosen ST and Querfild C: Primary cutaneous
T-cell lymphomas. Hematology Am Soc Hematol Educ Program.
2006:323–330. 2006. View Article : Google Scholar
|