Introduction
Mantle cell lymphoma (MCL) is a rare agressive type
of B-cell non-Hodgkin’s lymphoma with an estimated annual incidence
in Europe of 0.45/100,000 individuals (1). MCL is a biologically and clinically
heterogeneous disease; the immunophenotype of neoplastic cells
reflects the phenotype of cells similar to lymphocytes in the
mantle zone of normal germinal follicles (2). The genetic hallmark of MCL cells is
a translocation between chromosomes 11 and 14, t(11;14)(q13;q32),
juxtaposing the gene for immunoglobulin heavy chain and the gene
encoding cyclin D1. This results in cyclin D1 overexpression
(3,4).
The standard of care for newly diagnosed MCL
patients is combined immunochemotherapy alternating rituximab-CHOP
(R-CHOP; cyclophosphamide, vincristine, doxorubicin and prednisone)
and R-HDAC (high-dose cytarabine). The addition of rituximab and
HDAC to CHOP has improved the survival of MCL patients in the last
2 decades from 3 to 5 years. However, the response to therapy tends
to be short and virtually all patients sooner or later relapse.
There is no standard of care for relapsed or refractory MCL
patients. Salvage therapy usually comprises diverse regimens based
on fludarabine, gemcitabine, cisplatin, bendamustine, bortezomib
(inhibitor of 26S proteasome) or temsirolimus (inhibitor of mTOR).
Recently, several new experimental molecules have shown promise in
the therapy of relapsed or resistant MCL, including lenalidomide
(immunomodulatory agent), ibrutinib (PCI-32765, inhibitor of
Bruton’s tyrosine-kinase), new monoclonal antibodies (e.g.,
anti-CD20 ofatumumab), as well as other agents (5). Combination therapies are currently
being evaluated in clinical trials; however, novel drugs are
required.
The tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is considered one of the novel experimental
molecules with strong antitumor effects. TRAIL is a type II
transmembrane protein from the tumor necrosis factor superfamily
(6,7) expressed mostly by cells of the
immune system (natural killer cells, cytotoxic T-cells, macrophages
and dendritic cells). The main function of this molecule is thought
to be in tumor immunosurveillance, but its actual molecular role
remains to be elucidated.
TRAIL can trigger extrinsic apoptotis in target
cells by binding to TRAIL death receptors located on the cell
surface (8). This interaction is
performed by a long extracellular C-terminal region of the TRAIL
molecule. There are 4 distinct cell surface TRAIL receptors in
humans (DcR1, DcR2, DR4 and DR5) encoded by separate genes
(9,10). However, only DR4 and DR5 contain a
functional death domain (structurally conserved protein interaction
domain) and are capable of signaling apoptosis. Two decoy receptors
(DcR1 and DcR2) lack a functional death domain and inhibit TRAIL
signaling by competing with death receptors for TRAIL (9,10).
The binding of TRAIL to DR4 or DR5 leads to receptor
homotrimerization and formation of the death-inducing signaling
complex (DISC) (11). Through the
DISC a caspase machinery is activated, which results in apoptosis
(12). TRAIL death receptors DR4
and DR5 are ubiquitously expressed, indicating that most tissues
and cell types are potential targets of TRAIL signaling (13). Nevertheless, TRAIL seems to induce
apoptosis only in tumor cells but not in healthy tissues. Due to
its selective pro-apoptotic effect, TRAIL has attracted much
attention for its possible use in cancer therapy. In vitro,
a recombinant soluble TRAIL molecule has shown cytostatic or
cytotoxic effects in a wide variety of tumor cell lines, including
leukemia and lymphoma cells, but not in normal cells (6,7,10,11,14–19). The administration of a recombinant
soluble TRAIL molecule has been shown to induce the regression or
complete remission of tumors in tumor xenograft models (11,20–26). The efficacy of recombinant TRAIL
and agonistic antibodies recognizing either receptor DR4 or DR5 has
been investigated in numerous clinical trials, as recently reviewed
(27).
TRAIL has also shown promising pro-apoptotic effects
in a variety of lymphoma cell lines including MCL (15). However, as with other drugs,
cancer cells can develop resistance to TRAIL following prolonged
exposure to sublethal doses of TRAIL (14,28). Resistance to TRAIL-mediated
apoptosis can arise due to changes at the cell membrane level
(typically by loss of expression or mutation of functional DR4
and/or DR5 at the cell surface) or on the intracellular level (such
as incorrect formation of DISC and abberant expression of caspases)
(29). The successful therapy of
malignancies in general, and particularly those with very poor
prognosis, such as MCL, depends on the effective management of drug
resistance. An in-depth understanding of the processes involved in
the development of drug resistance and a detailed description of
secondary molecular changes associated with resistance are
essential for successful cancer therapy. Specific molecular changes
which occur in drug-resistant cells can confer a potential
selective disadvatage to such cells and may be used as targets for
the effective elimination of drug-resistant lymphoma cells.
The aim of this study was to elucidate the molecular
mechanisms responsible for TRAIL resistance in MCL cells, as well
as the secondary molecular alterations associated with this
process. We also aimed to identify the phenotypic features specific
for TRAIL-resistant MCL cells. If identified, these molecular
features can be, at least theoretically, used as molecular targets
for the effective elimination of TRAIL-resistant lymphoma cells in
experimental therapies.
Materials and methods
Cell growth and cellular toxicity
assay
HBL-2 cells were grown in Iscove’s modified
Dulbecco’s medium in the presence of 10% foetal bovine serum, 1%
penicillin-streptomycin solution in a 37°C humidified atmosphere
with 5% CO2. TRAIL-resistant HBL-2/R cells were derived
by selective pressure of increasing concentrations of human
recombinant TRAIL (Apronex Biotechnologies, Czech Republic) up to
1,000 ng/ml in medium from the wild-type HBL-2 cells in 5 weeks.
The toxicity of TRAIL to HBL-2 and HBL-2/R was measured using the
colorimetric WST-8-based Quick Cell proliferation Assay kit II
(BioVision, San Francisco, CA, USA) according to the manufacturer’s
instructions. Briefly, 40,000 cells were seeded in a 96-well plate
in 300 μl of medium supplemented with increased concentrations of
TRAIL up to 1,000 ng/ml in medium for 1–4 days. After the addition
of WST reagent, absorbance was measured on a Sunrise microplate
absorbance reader (Tecan Group Ltd., Männedorf, Switzerland) with a
450 nm reading filter and 630 nm reference filter. The absorbance
of free medium was used as the background level, triplicate samples
were grown and measured for each cell type and TRAIL concentration.
Mean values were calculated. All chemicals were purchased from
Sigma-Aldrich (St. Louis, MO, USA) unless specified otherwise.
Flow cytometric analysis
HBL-2 and HBL-2/R cells (2×105 cells for
each assay) were washed in PBS buffer (0.5% foetal bovine serum in
PBS), stained with phycoerythrin-conjugated antibodies against
TRAIL receptors DR4, DR5, DcR1 and DcR2 (anti-hTRAIL R1,
anti-hTRAIL R2, anti-hTRAIL R3 and anti-hTRAIL R4; R&D Systems,
Minneapolis, MN, USA) and analyzed by flow cytometry in triplicate
(FASCCanto II, BD Biosciences, San Jose, CA, USA). Unstained cells
and cells incubated with isotype controls served as the background
fluorescence controls.
Sample preparation for two-dimensional
electrophoresis
HBL-2 and HBL-2/R cells (6×107) were
harvested, washed twice with PBS and cell pellets were frozen and
stored at −80°C. Samples were thawed and homogenized in lysis
buffer [7 M urea, 2 M thiourea, 4% CHAPS, 60 mM dithiothreitol
(DTT) and 1% ampholytes (Bio-Lyte 3–10 Buffer, Bio-Rad, Hercules,
CA, USA)] and protease inhibitor cocktail (Roche Diagnostics GmbH,
Mannheim, Germany) for 20 min at room temperature with occasional
vortexing. Samples were sedimented at 18,000 × g for 20 min at room
temperature, supernatants were collected and protein concentration
was determined by the Bradford assay (Bio-Rad). Protein
concentrations in all samples were equalized to 3.3 mg/ml by
dilution with lysis buffer.
Two-dimensional electrophoresis
IPG strips (pH 4.0–7.0, 24 cm; ReadyStrip, Bio-Rad)
were rehydrated overnight in 450 μl of sample, representing 1.5 mg
of protein. Isoelectric focusing was performed for 70 kVh, with
maximum voltage not exceeding 5 kV, current limited to 50 μA per
strip and temperature set to 20°C (Protean IEF Cell, Bio-Rad). Six
replicates were run for each cell type. Focused strips were briefly
rinsed in deionized water, equilibrated and reduced in
equilibration buffer supplemented with DTT (6 M urea, 50 mM Tris pH
8.8, 30% glycerol, 2% SDS and 450 mg DTT per 50 ml) for 15 min and
then alkylated in equilibration buffer with iodacetamide (1.125 mg
iodacetamide per 50 ml of the buffer). Equilibrated strips were
then secured on 10% SDS-PAGE and secured in place by molten
agarose. SDS-PAGE electrophoresis was performed in a
Tris-glycine-SDS system using a 12-gel Protean Dodeca Cell
apparatus (Bio-Rad) with buffer circulation and external cooling
(20°C). Gels were run at a constant voltage of 45 V per gel for 30
min and then at a constant voltage of 200 V for 6 h. Gels were
washed 3 times for 15 min in deionized water to remove redundant
SDS. Gels were then stained with colloidal Coomassie Brilliant Blue
(SimplyBlue™ Safestain, Invitrogen, Carlsbad, CA, USA) overnight
and briefly de-stained in deionized water.
Gel image analysis and extraction of
peptides
Stained gels were scanned with GS 800 calibrated
densitometer (Bio-Rad) and image analysis was performed with
Progenesis™ software (Nonlinear Dynamics, Ltd., Newcastle upon
Tyne, UK) in semi-manual mode with 6 gel replicates for each cell
type. Normalization of gel images was based on total spot density,
and integrated spot density values (spot volumes) were then
calculated after background subtraction. Average spot volume values
(averages from the all 6 gels in the group) for each spot were
compared between the groups. Protein spots were considered
differentially expressed if their average normalized spot volume
difference was >1.5-fold. As determined by the Student’s t-test,
a p-value <0.05 was considered to indicate a statistically
significant difference.
Protein digestion and peptide
extraction
Spots containing differentially expressed proteins
were excised from the gels, cut into small pieces and washed 3
times with 25 mM ammonium bicarbonate in 50% acetonitrile (ACN).
The gels were then dried in a SpeedVac Concentrator (Eppendorf,
Hamburg, Germany). Sequencing grade modified trypsin (Promega,
Madison, WI, USA) (6 ng/μl of trypsin in 25 mM ammonium bicarbonate
in 5% ACN) was added. Following overnight incubation at 37°C, the
resulting peptides were extracted with 50% ACN.
Matrix-assisted laser
desorption/ionization-time of flight mass spectrometry (MALDI-TOF
MS) and identification of selected proteins
Peptide samples were spotted on a polished steel
target plate (Bruker Daltonics, Bremen, Germany) and allowed to dry
at room temperature. Matrix solution (3 mg
α-cyano-4-hydroxycinnamic acid in 1 ml of 50% ACN containing 0.1%
trifluoroacetic acid) was then added. MS was performed on an
Autoflex II MALDI-TOF/TOF mass spectrometer (Bruker Daltonics)
using a solid nitrogen laser (337 nm) and FlexControl software
(Bruker Daltonics) in reflectron mode with positive ion mass
spectra detection. The mass spectrometer was externally calibrated
with Peptide Calibration Standard II (Bruker Daltonics). Spectra
were acquired in the mass range 800–4,000 Da. The peak lists were
generated using FlexAnalysis and searched against Swiss-Prot (2011
version, 524420 sequences) using Mascot software. The peptide mass
tolerance was set to 50 ppm, taxonomy Homo sapiens, missed
cleavage was set to 2, fixed modification for cysteine
carbamidomethylation, and variable modifications for methionine
oxidation and protein N-terminal acetylation.
Western blot analysis
Cells were lysed in NHT buffer (140 mM NaCl, 10 mM
HEPES, 1.5% Triton X-100, pH 7.4). Protein concentration in the
collected supernatants was determined by the Bradford assay
(Bio-Rad). Lysate samples (25–70 μg) were combined with SDS loading
buffer containing 2-mercaptoethanol and boiled for 5 min.
Triplicate samples were separated on 12% SDS-PAGE minigels in
Tris-glycine buffer (Bio-Rad). Electrophoresis was performed at a
constant voltage for 30 min at 45 V per gel, and then at 90 V per
gel until the dye front reached the gel bottom. Proteins were
transferred onto 0.45 μm PVDF membranes (Milipore, Billerica, MA,
USA) in a semi-dry blotter (Hoefer, San Francisco, CA, USA) at 0.8
mA/cm2 of membrane. Membranes were incubated with
blocking buffer containing PBS (Invitrogen), 0.1% Tween-20 and 5%
non-fat dried milk for 1 h. As primary antibodies anti-adenine
phosphoribosyltransferase (APRT; 1:1,000, rabbit polyclonal
antibody), anti-purine nucleoside phosphorylase (PNP; 1:1,000,
mouse monoclonal antibody) and anti-GAPDH (1:10,000, rabbit
polyclonal antibody) were used. After thoroughly washing in
blocking buffer, a secondary horseradish peroxidase-conjugated
anti-mouse or anti-rabbit antibody was added (1:10,000). GAPDH was
used as the loading control. The signal was detected using Western
Blotting Luminol Reagent (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) and membranes were exposed to X-ray films (Kodak,
Rochester, NY, USA). All used antibodies were from Santa Cruz
Biotechnology.
Results
Molecular changes associated with the generation of
drug-resistant cells can confer potential selective disadvantage.
Such a ‘weakness’ may be used as druggable target for effective
elimination of drug-resistant lymphoma cells. Our aim was to
elucidate the molecular changes associated with the development of
TRAIL resistance in (originally TRAIL-sensitive) MCL cells in order
to identify such a cellular ‘weakness’ of TRAIL-resistant MCL
cells. To identify the specific protein expression changes in the
TRAIL-resistant cells, we derived a TRAIL-resistant HBL-2 subclone
(HBL-2/R) from the originally TRAIL-sensitive HBL-2 cell line, and
performed differential analysis of the surface expression of TRAIL
receptors and comparative proteomic analysis of the HBL-2/R and
HBL-2 cells.
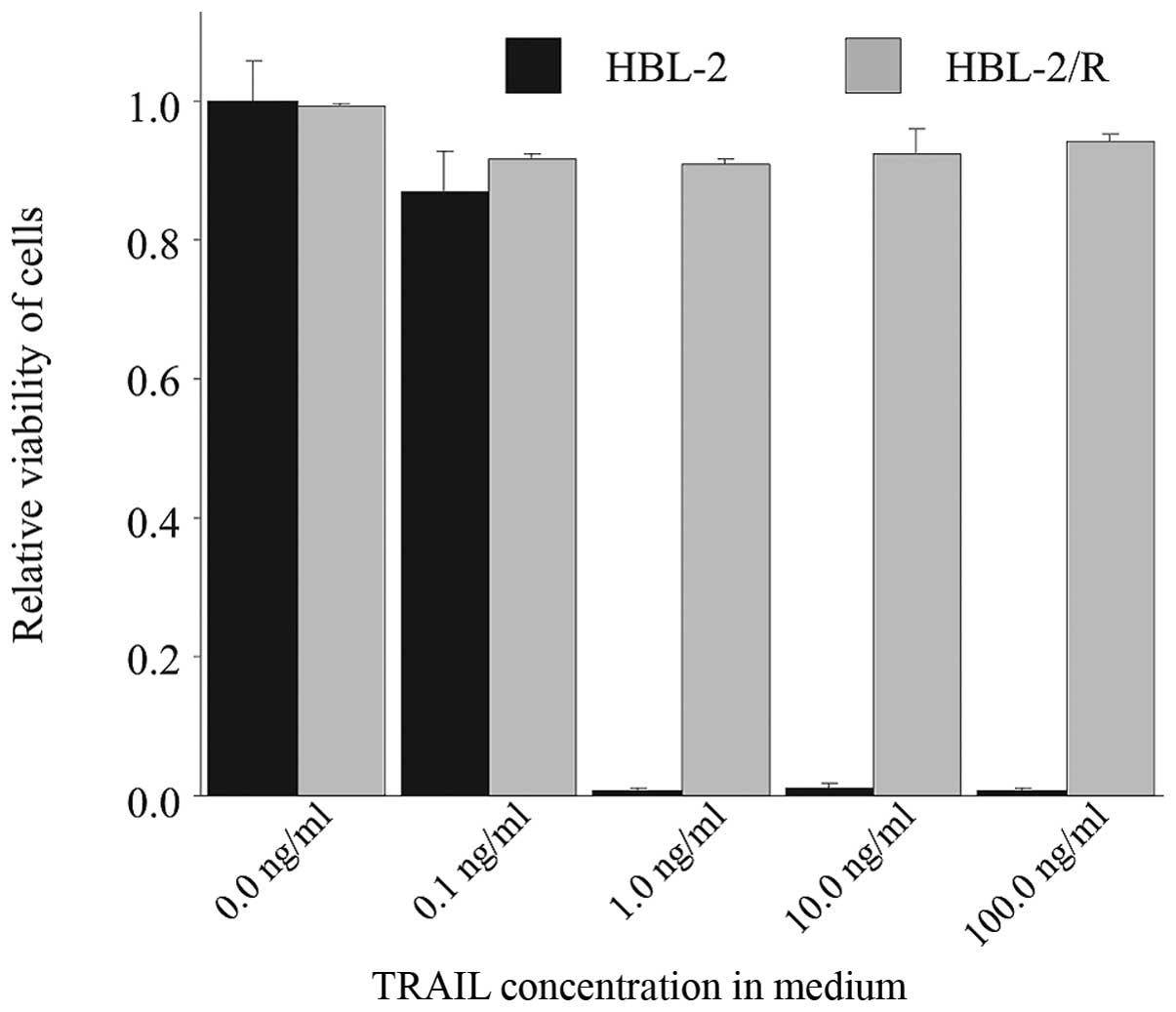
TRAIL-resistant cell line
The TRAIL-resistant HBL-2 subclone (HBL-2/R) was
derived from the originally TRAIL-sensitive HBL-2 cell line by
selective pressure of increasing TRAIL concentration in medium over
5 weeks. While the IC50 for TRAIL in the originally
sensitive HBL-2 cells was 1 ng/ml at 48 h (data not shown), the
resulting HBL-2/R subclone proliferated in up to 1,000 ng/ml TRAIL
concentration in medium and was therefore >1,000-fold more
resistant to TRAIL than the HBL-2 cells (Fig. 1).
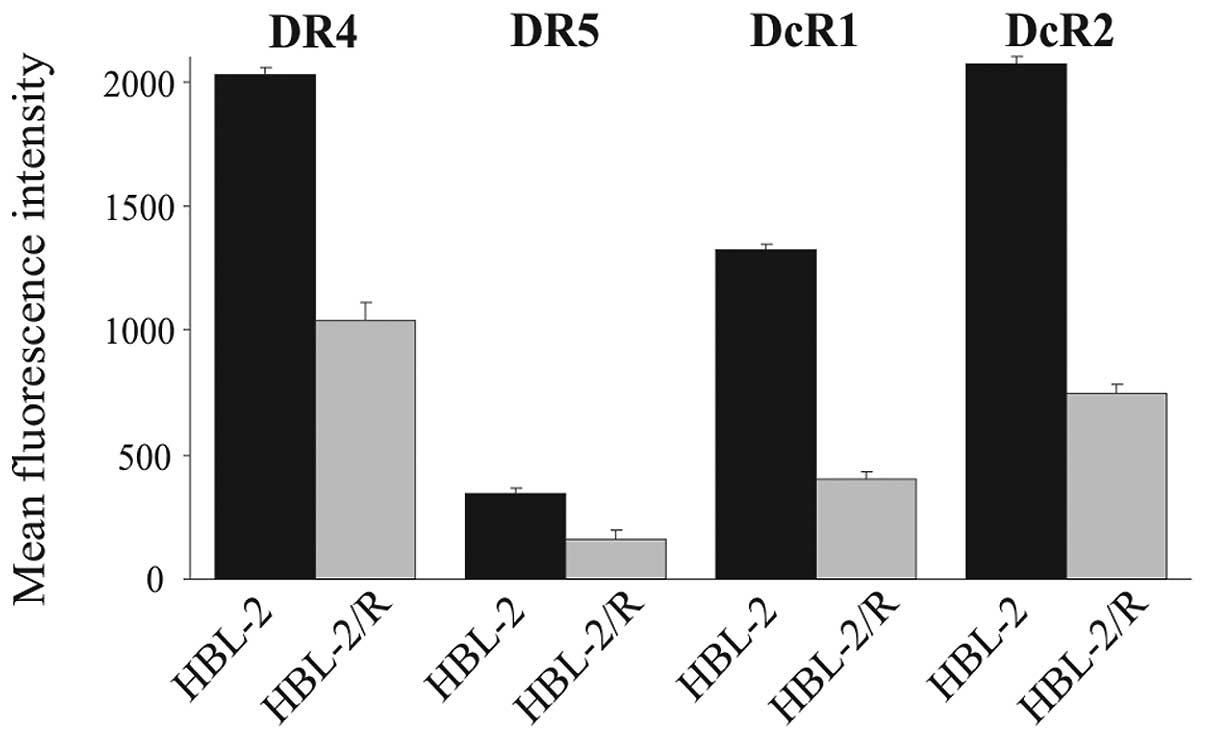
TRAIL receptors - flow cytometric
analysis of cell surface expression
The attenuated expression of TRAIL death receptors
DR4 and DR5 has been previously described as a cause of TRAIL
resistance. We therefore determined the relative expression of
TRAIL receptors in HBL-2 and HBL-2/R cells by flow cytometry
(Fig. 2). The expression of DR4,
DR5, DcR1 and DcR2 in the HBL-2/R cells was markedly decreased
compared to the HBL-2 cells. The marked downregulation of death
receptors DR4 and DR5 explains the resistance of the HBL-2/R cells
to TRAIL, while the downregulation of decoy receptors DcR1 and DcR2
may indicate further, more complex phenotypic changes in the
HBL-2/R cells.
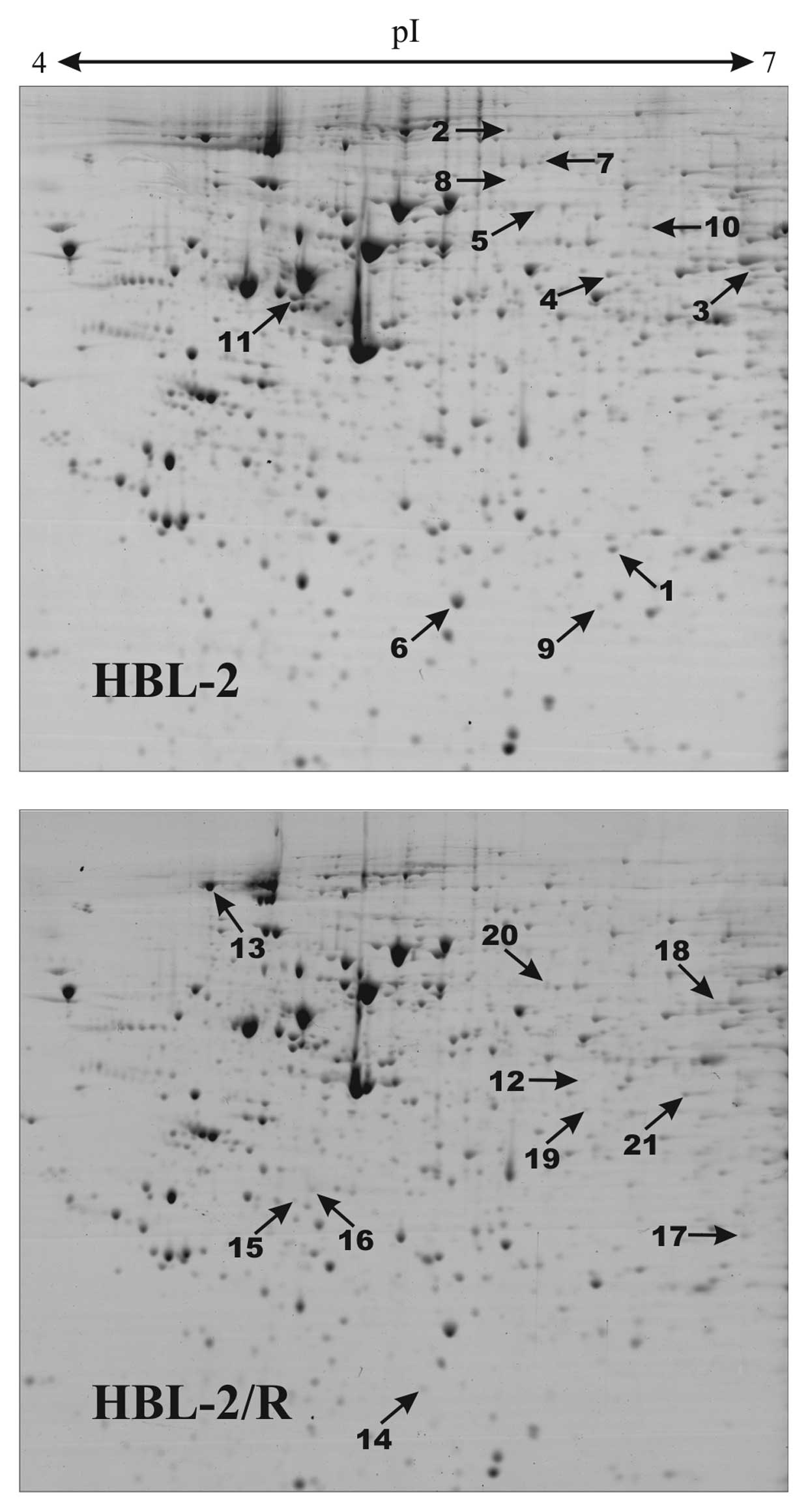
Proteomic analysis
In order to identify specific changes in protein
expression associated with TRAIL resistance in HBL-2/R cells, we
performed comparative proteomic analysis of cellular homogenates of
HBL-2/R and TRAIL-sensitive HBL-2 cells. Using two-dimensional
electrophoresis of total cell lysates, we reproducibly detected 820
protein spots on Coomassie Brilliant Blue-stained gels. We found 21
protein spots to be significantly quantitatively changed
(upregulated or downregulated, change >1.5-fold; p<0.05) in
HBL-2/R cells (Fig. 3). Using
MALDI-TOF/TOF mass spectrometry we identified all 21 proteins
differentially expressed in HBL-2/R cells (Table I).
 | Table IList of proteins differentially
expressed in HBL-2/R cells (difference at least 1.5-fold and
statistical significance p<0.05). |
Table I
List of proteins differentially
expressed in HBL-2/R cells (difference at least 1.5-fold and
statistical significance p<0.05).
| Spot no. | Swiss-Prot
no.a | Protein name | Fold change | Mascot
scoreb | Sequence cov.
(%)c | Mr |
|---|
| Proteins
upregulated in HBL-2/R cells |
| 1 | P04792 | Heat shock protein
β-1 | 3.9 | 84 | 51 | 22826 |
| 2 | P42704 | Leucine-rich PPR
motif-containing protein, mitochondrial | 2.6 | 100 | 23 | 159003 |
| 3 | O75351 | Vacuolar protein
sorting-associated protein 4B | 2.6 | 171 | 32 | 49443 |
| 4 | P23381 | Tryptophanyl-tRNA
synthetase, cytoplasmic | 2.4 | 240 | 54 | 53474 |
| 5 | P20591 | Interferon-induced
GTP-binding protein Mx1 | 2.2 | 176 | 42 | 75872 |
| 6 | P09211 | Glutathione
S-transferase P | 1.9 | 110 | 56 | 23569 |
| 7 | P06396 | Gelsolin | 1.9 | 115 | 22 | 86043 |
| 8 | P13010 | X-ray repair
cross-complementing protein 5 | 1.7 | 262 | 46 | 83222 |
| 9 | Q9HAV7 | GrpE protein
homolog 1, mitochondrial | 1.6 | 99 | 44 | 24492 |
| 10 | O43776 | Asparaginyl-tRNA
synthetase, cytoplasmic | 1.5 | 250 | 41 | 63758 |
| 11 | Q15084 | Protein
disulfide-isomerase A6 | 1.5 | 76 | 29 | 48490 |
| Proteins
downregulated in HBL-2/R cells |
| 12 | P08559 | Pyruvate
dehydrogenase E1 component subunit α | 3.2 | 111 | 32 | 43952 |
| 13 | P19338 | Nucleolin | 2.4 | 146 | 29 | 76625 |
| 14 | P07741 | Adenine
phosphoribosyltransferase | 2.2 | 227 | 79 | 19766 |
| 15 | O75792 | Ribonuclease H2
subunit A | 1.7 | 348 | 72 | 33716 |
| 16 | Q07955 |
Serine/arginine-rich splicing factor
1 | 1.7 | 82 | 35 | 27842 |
| 17 | P00491 | Purine nucleoside
phosphorylase | 1.6 | 182 | 68 | 32325 |
| 18 | P12268 |
Inosine-5′-monophosphate dehydrogenase
2 | 1.6 | 230 | 44 | 56226 |
| 19 | P40121 | Macrophage-capping
protein | 1.6 | 102 | 41 | 38760 |
| 20 | P13674 | Prolyl
4-hydroxylase subunit α-1 | 1.5 | 234 | 48 | 61296 |
| 21 | Q15019 | Septin-2 | 1.5 | 62 | 26 | 41689 |
Functional annotations of the identified
differentially expressed proteins were analyzed using the Kyoto
Encyclopedia of Genes and Genomes (KEGG) database. Among the 21
identified proteins we found molecules involved in diverse
functions, including cytoskeleton regulation, ribosome synthesis
and maturation, RNA metabolism, chromosome translocation, DNA
repair and replication, as well as protein folding. However, one
pathway was markedly enriched in our set (hsa00230 - purine
metabolism) represented by 3 differentially expressed proteins.
These 3 molecules are key enzymes of the purine nucleotide
metabolism (Fig. 5) and all 3 are
downregulated in TRAIL-resistant HBL-2/R cells [PNP (downregulated
1.6-fold in HBL-2/R cells), APRT (downregulated 2.2-fold in HBL-2/R
cells) and inosine-5′-monophosphate dehydrogenase 2 (IMPDH2,
downregulated 1.6-fold in HBL-2/R cells)].
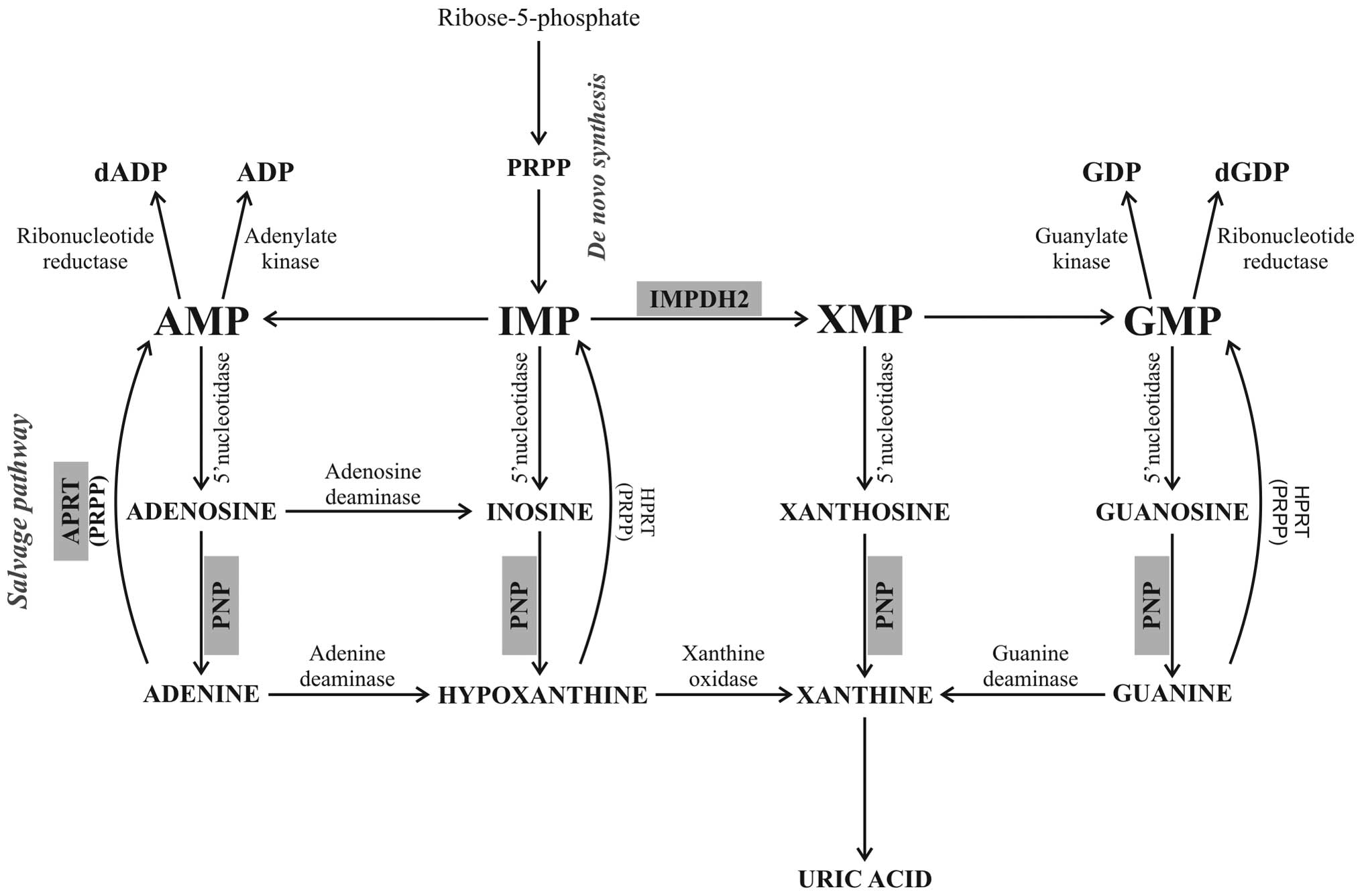 | Figure 5Scheme of the purine metabolism
pathways, showing the position of IMPDH2, APRT and PNP in purine
nucleotide biosynthesis, adopted from a previous study (35). The de novo synthesis of
purine nucleotides begins with the phosphorylation of
ribose-5-phosphate to form PRPP. In a number of reactions, PRPP
creates the first fully formed nucleotide, IMP. IMP is converted by
IMPDH2 to GMP. PNP catalyzes the reversible cleavage of purine
nucleosides, releasing purine nucleobases (adenine, hypoxanthine,
xanthine and guanine). In the salvage pathway the free nucleobases
can be reconverted back to nucleoside-5′-monophosphates in a
reaction with activated sugar (PRPP) catalyzed by APRT. IMPDH2,
inosine-5′-monophosphate dehydrogenase 2; APRT, adenine
phosphoribosyltransferase; PNP, purine nucleoside phosphorylase;
PRPP, 5-phosphoribosyl-1-pyrophosphate; IMP,
inosine-5′-monophosphate; GMP, guanosine-5′-monophosphate; dADP,
deoxyadenosine diphosphate; ADP, adenosine diphosphate; GDP,
guanosine diphosphate; dGDP, deoxyguanosine diphosphate; AMP,
adenosine monophosphate; XMP, xanthosine monophosphate. |
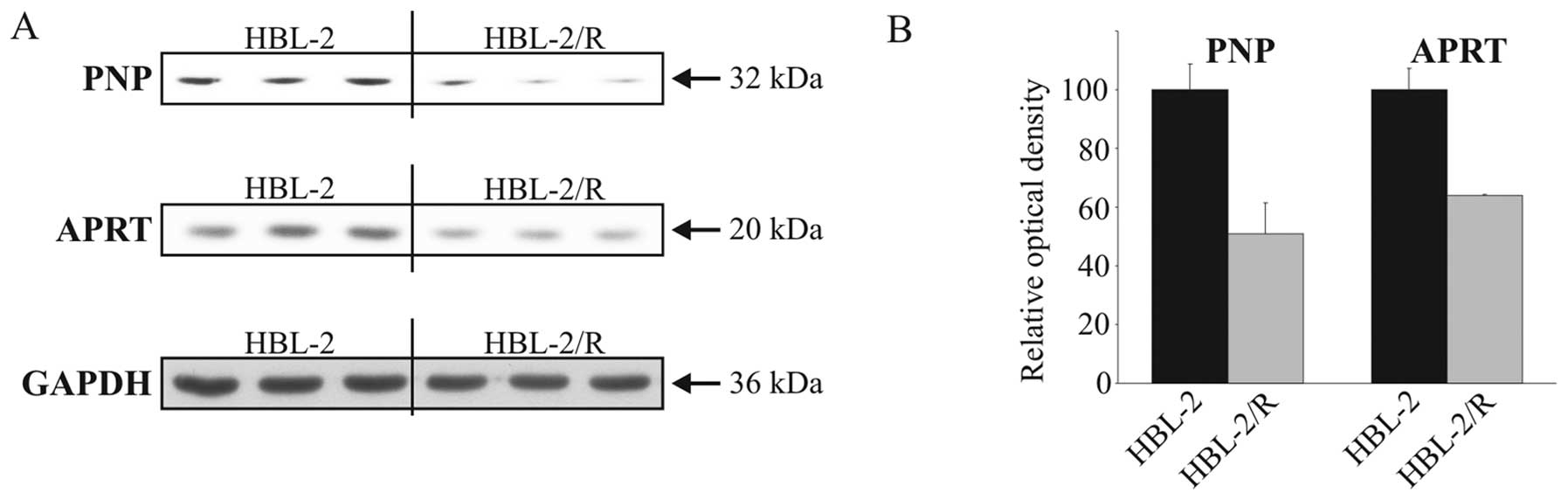
Verification of proteomic analysis
To confirm the results of proteomic analysis by an
independent method we verified the decreased expression of the 2
proteins involved in purine metabolism, namely PNP and APRT, by
western blot analysis in HBL-2 and HBL-2/R cell lysates (Fig. 4).
Discussion
The downregulation of the 3 key enzymes of purine
metabolism can have a profound effect on nucleotide homeostasis in
TRAIL-resistant lymphoma cells. Purine nucleotides, the building
blocks for synthesis of DNA, RNA and enzyme co-factors, are
recruited either from de novo purine synthesis from low
molecular weight precursors or by recycling of free nucleobases in
the so-called salvage pathway. Both pathways lead to the production
of nucleoside-5′-phosphates (Fig.
5). Both pathways can supply cellular demand independently;
however, their importance in different tissues is variable. In
leukemic and lymphoma cells the salvage pathway is considered the
major source of purine nucleotides (30,31).
The de novo synthesis of purine nucleotides
requires 5-phosphoribosyl-1-pyrophosphate (PRPP), ATP, glutamine,
glycine, CO2, aspartate and formate to create the first
fully formed nucleotide, inosine-5′-monophosphate (IMP). IMP
represents a branch point for purine biosynthesis, since it can be
converted either to guanosine-5′-monophosphate (GMP) by IMPDH2
(downregulated in HBL-2/R cells) or to adenosine-5′-monophosphate
(Fig. 5).
The catabolism of purine nucleotides leads to the
liberation of free purine bases by PNP (downregulated in HBL-2/R
cells). In the salvage pathway the free bases are reconverted back
to nucleoside-5′-monophosphates in a reaction with activated sugar
(PRPP) catalyzed by APRT (downregulated in HBL-2/R cells) or
hypoxanthine-guanine phosphoribosyltransferase (32) (Fig.
5). Ribonucleotides are converted by ribonucleotide reductase
into the corresponding deoxyribonucleotides.
The delicate balance of enzyme activities and
concentrations of products and intermediates are critical for
purine (nucleotide) homeostasis. The inhibition of PNP results in
the accumulation of its substrate, 2′-deoxyguanosine which is
further phosphorylated to deoxyguanosine triphosphate (dGTP). A
high intracellular concentration of dGTP inhibits cell
proliferation and induces apoptosis (33–35). If APRT is inhibited, accumulated
adenine is oxidized to insoluble 2,8-dihydroxyadenine. Accumulation
of this precipitate results in cell death (32). Similarly, the inhibition of IMPDH2
leads to depletion of guanosine nucleotides, which blocks DNA
synthesis and cell division (36,37).
Disruption of the purine nucleotide metabolism
generally results in an accumulation and/or a lack of
ribonucleotides or deoxyribonucleotides or metabolic intermediates
with potentially cytotoxic consequences. The observed decreased
expression of the 3 purine metabolism enzymes affects both de
novo synthesis and the salvage pathway of purine metabolism and
may also affect purine nucleotide homeostasis in TRAIL-resistant
HBL-2/R cells. Such an imbalance may represent a selective
disadvantage for the affected cells. Such a ‘weakness’ may not be
apparent under normal circumstances but may become critical under
stress or unfavorable conditions. As the proliferation rates of
HBL-2/R and HBL-2 cells are comparable, the proposed imbalance in
purine nucleotide metabolism in TRAIL-resistant cells is possibly
mild and/or well compensated in vitro. However, this
‘weakness’ may become apparent due to lack of building blocks for
DNA and RNA synthesis in the environment or upon further disruption
of purine metabolism. Since both pathways of purine metabolism are
compromised in TRAIL-resistant MCL cells, these cells should be
vulnerable to further inactivation of purine nucleotide metabolism
enzymes. Therefore, drugs that target (already disbalanced) purine
metabolism should be highly cytotoxic to TRAIL-resistant cells
(compared to non-malignant cells) and may therefore be selectively
effective in the elimination of TRAIL-resistant MCL cells in
experimental therapy. There are several approved inhibitors of
purine metabolism, such as methotrexate (inhibits purine de
novo synthesis via dihydrofolate reductase) (38), ribavirin and mycophenolic acid
(inhibitors of IMPDH2) (39,40) or forodesine (a novel inhibitor of
PNP) (41,42), available for clinical use.
The adaptation of cancer cells to cytostatic and
cytotoxic drugs is associated to a certain degree with extensive
changes in the cell phenotype. Some of the molecular changes,
although seemingly unrelated to the mechanism of resistance, can
provide a selective disadvantage to the cells and such a ‘weakness’
may be used as a potential therapeutic target. By the presented
proteomic analysis of the changes associated with resistance to
TRAIL in MCL HBL-2 cells, we demonstrated the downregulation of all
types of TRAIL receptors and identified the altered expression of
several proteins including 3 enzymes of the purine metabolism
pathway. This downregulated pathway potentially represents a
‘weakness’ of the TRAIL-resistant MCL cells and has potential as a
therapeutic target for the selective elimination of such cells in
the future.
Acknowledgements
This study was supported by the Grant Agency of
Charles University (GAUK 251180 111210 and 253284 700712), by the
Grant Agency of the Czech Republic (305/09/1390), by the Ministry
of Education, Youth and Sports (PRVOUK P24/LF1/3 and SVV
2012-264507), UNCE 204021 and the Ministry of Health of the Czech
Republic (IGA MZ NT12248-5, IGA-MZ NT13201-4).
References
|
1
|
Sant M, Allemani C, Tereanu C, et al:
Incidence of hematologic malignancies in Europe by morphologic
subtype: results of the HAEMACARE project. Blood. 116:3724–3734.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perez-Galan P, Dreyling M and Wiestner A:
Mantle cell lymphoma: biology, pathogenesis, and the molecular
basis of treatment in the genomic era. Blood. 117:26–38. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsujimoto Y, Yunis J, Onorato-Showe L,
Erikson J, Nowell PC and Croce CM: Molecular cloning of the
chromosomal breakpoint of B-cell lymphomas and leukemias with the
t(11;14) chromosome translocation. Science. 224:1403–1406. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Williams ME, Swerdlow SH, Rosenberg CL and
Arnold A: Characterization of chromosome 11 translocation
breakpoints at the bcl-1 and PRAD1 loci in centrocytic lymphoma.
Cancer Res. 52:5541S–5544S. 1992.PubMed/NCBI
|
|
5
|
Humala K and Younes A: Current and
emerging new treatment strategies for mantle cell lymphoma. Leuk
Lymphoma. Feb 19–2013.(Epub ahead of print).
|
|
6
|
Wiley SR, Schooley K, Smolak PJ, et al:
Identification and characterization of a new member of the TNF
family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sheridan JP, Marsters SA, Pitti RM, et al:
Control of TRAIL-induced apoptosis by a family of signaling and
decoy receptors. Science. 277:818–821. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ashkenazi A: Targeting death and decoy
receptors of the tumour-necrosis factor superfamily. Nat Rev
Cancer. 2:420–430. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Castro Alves C, Terziyska N, Grunert M, et
al: Leukemia-initiating cells of patient-derived acute
lymphoblastic leukemia xenografts are sensitive toward TRAIL.
Blood. 119:4224–4227. 2012.PubMed/NCBI
|
|
12
|
Peter ME and Krammer PH: The
CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Spierings DC: Tissue distribution of the
death ligand TRAIL and its receptors. J Histochem Cytochem.
52:821–831. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Petrak J, Toman O, Simonova T, et al:
Identification of molecular targets for selective elimination of
TRAIL-resistant leukemia cells. From spots to in vitro assays using
TOP15 charts. Proteomics. 9:5006–5015. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Molinsky J, Klanova M, Koc M, et al:
Roscovitine sensitizes leukemia and lymphoma cells to tumor
necrosis factor-related apoptosis-inducing ligand-induced
apoptosis. Leuk Lymphoma. 54:372–380. 2013. View Article : Google Scholar
|
|
16
|
Klener P, Leahomschi S, Molinsky J, et al:
TRAIL-induced apoptosis of HL60 leukemia cells: two distinct
phenotypes of acquired TRAIL resistance that are accompanied with
resistance to TNFalpha but not to idarubicin and cytarabine. Blood
Cells Mol Dis. 42:77–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leahomschi S, Molinsky J, Klanova M, et
al: Multi-level disruption of the extrinsic apoptotic pathway
mediates resistance of leukemia cells to TNF-related
apoptosis-inducing ligand (TRAIL). Neoplasma. 60:223–231. 2013.
View Article : Google Scholar
|
|
18
|
Wen J, Ramadevi N, Nguyen D, Perkins C,
Worthington E and Bhalla K: Antileukemic drugs increase death
receptor 5 levels and enhance Apo-2L-induced apoptosis of human
acute leukemia cells. Blood. 96:3900–3906. 2000.PubMed/NCBI
|
|
19
|
Plasilova M, Zivny J, Jelinek J, et al:
TRAIL (Apo2L) suppresses growth of primary human leukemia and
myelodysplasia progenitors. Leukemia. 16:67–73. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ashkenazi A, Pai RC, Fong S, et al: Safety
and antitumor activity of recombinant soluble Apo2 ligand. J Clin
Invest. 104:155–162. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Di Pietro R and Zauli G: Emerging
non-apoptotic functions of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)/Apo2L. J Cell Physiol.
201:331–340. 2004.
|
|
22
|
Kelley SK, Harris LA, Xie D, et al:
Preclinical studies to predict the disposition of Apo2L/tumor
necrosis factor-related apoptosis-inducing ligand in humans:
characterization of in vivo efficacy, pharmacokinetics, and safety.
J Pharmacol Exp Ther. 299:31–38. 2001.
|
|
23
|
Lawrence D, Shahrokh Z, Marsters S, et al:
Differential hepatocyte toxicity of recombinant Apo2L/TRAIL
versions. Nat Med. 7:383–385. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roth W, Isenmann S, Naumann U, et al:
Locoregional Apo2L/TRAIL eradicates intracranial human malignant
glioma xenografts in athymic mice in the absence of neurotoxicity.
Biochem Biophys Res Commun. 265:479–483. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Walczak H, Miller RE, Ariail K, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hylander BL, Pitoniak R, Penetrante RB, et
al: The anti-tumor effect of Apo2L/TRAIL on patient pancreatic
adenocarcinomas grown as xenografts in SCID mice. J Transl Med.
3:222005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 14–May;2012.(Epub ahead of
print). View
Article : Google Scholar : 1642012.PubMed/NCBI
|
|
28
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maksimovic-Ivanic D, Stosic-Grujicic S,
Nicoletti F and Mijatovic S: Resistance to TRAIL and how to
surmount it. Immunol Res. 52:157–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scavennec J, Maraninchi D, Gastaut JA,
Carcassonne Y and Cailla HL: Purine and pyrimidine ribonucleoside
monophosphate patterns of peripheral blood and bone marrow cells in
human acute leukemias. Cancer Res. 42:1326–1330. 1982.PubMed/NCBI
|
|
31
|
Natsumeda Y, Prajda N, Donohue JP, Glover
JL and Weber G: Enzymic capacities of purine de novo and salvage
pathways for nucleotide synthesis in normal and neoplastic tissues.
Cancer Res. 44:2475–2479. 1984.PubMed/NCBI
|
|
32
|
Bollee G, Harambat J, Bensman A,
Knebelmann B, Daudon M and Ceballos-Picot I: Adenine
phosphoribosyltransferase deficiency. Clin J Am Soc Nephrol.
7:1521–1527. 2012. View Article : Google Scholar
|
|
33
|
Bantia S, Miller PJ, Parker CD, et al:
Purine nucleoside phosphorylase inhibitor BCX-1777 (Immucillin-H) -
a novel potent and orally active immunosuppressive agent. Int
Immunopharmacol. 1:1199–1210. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bantia S, Montgomery JA, Johnson HG and
Walsh GM: In vivo and in vitro pharmacologic activity of the purine
nucleoside phosphorylase inhibitor BCX-34: the role of GTP and
dGTP. Immunopharmacology. 35:53–63. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Galmarini CM, Popowycz F and Joseph B:
Cytotoxic nucleoside analogues: different strategies to improve
their clinical efficacy. Curr Med Chem. 15:1072–1082. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Allison AC and Eugui EM: Mycophenolate
mofetil and its mechanisms of action. Immunopharmacology.
47:85–118. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hedstrom L: IMP dehydrogenase: structure,
mechanism, and inhibition. Chem Rev. 109:2903–2928. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fairbanks LD, Ruckemann K, Qiu Y, et al:
Methotrexate inhibits the first committed step of purine
biosynthesis in mitogen-stimulated human T-lymphocytes: a metabolic
basis for efficacy in rheumatoid arthritis? Biochem J. 342:143–152.
1999. View Article : Google Scholar
|
|
39
|
Allison AC and Eugui EM: The design and
development of an immunosuppressive drug, mycophenolate mofetil.
Springer Semin Immunopathol. 14:353–380. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou S, Liu R, Baroudy BM, Malcolm BA and
Reyes GR: The effect of ribavirin and IMPDH inhibitors on hepatitis
C virus subgenomic replicon RNA. Virology. 310:333–342. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gandhi V, Kilpatrick JM, Plunkett W, et
al: A proof-of-principle pharmacokinetic, pharmacodynamic, and
clinical study with purine nucleoside phosphorylase inhibitor
immucillin-H (BCX-1777, forodesine). Blood. 106:4253–4260. 2005.
View Article : Google Scholar
|
|
42
|
Miles RW, Tyler PC, Furneaux RH,
Bagdassarian CK and Schramm VL: One-third-the-sites
transition-state inhibitors for purine nucleoside phosphorylase.
Biochemistry. 37:8615–8621. 1998. View Article : Google Scholar : PubMed/NCBI
|