Introduction
Patients with ataxia telangiectasia (A-T), carrying
mutations at both ataxia telangiectasia mutated (ATM)
alleles (ATM−/−), present with progressive
cerebellar ataxia, susceptibility to cancer, immunodeficiency,
insulin resistance and hyperglycemia (1,2).
In addition, patients with A-T reportedly have increased plasma
cholesterol and triglyceride levels (3), which are the two major risk factors
for atherosclerosis. It is currently known that humans with the
heterozygous ATM mutation (ATM+/−),
which account for 0.5–2% of the total population, also have an
increase risk of developing atherosclerosis-related cardiovascular
diseases (4). Previous studies
have suggested that defective ATM function promotes atherosclerosis
through multiple systemic pro-atherosclerotic features, such as
metabolic syndrome, oxidative stress, DNA damage and mitochondrial
dysfunction (5,6). We have prevoiusly demonstrated that
ATM assists the clearance of plasma apolipoprotein
(Apo)E-deficient, ApoB48-containing (E−/B48)
lipoproteins in ApoE-deficient mice
(ApoE−/− mice) (7). However, to date, there is no exact
explanation available as to the mechanisms through which ATM is
involved in the endocytosis or removal of
E−/B48 lipoproteins in
ApoE−/− mice.
It is now known that a fraction of the ATM protein
is also present in the cytoplasm and is associated with vesicular
structures, such as peroxisomes, lysosomes and endosomes (8–10),
which indicates that ATM may be involved in the trafficking of
proteins and vesicles. Certain studies have reported that in the
absence of ATM, intracellular vesicle and/or protein transport may
be impaired, leading to abnormalities in endosomal function
(11). Therefore, we hypothesized
that ATM mutations in ApoE−/− mice
may lead to an overaccumulation of plasma ApoB-48-containing
lipoproteins, thus promoting the development of atherosclerosis,
which may be associated with the loss of or the impaired function
of cytosolic ATM protein. This may affect the process of
endocytosis of lipoproteins or it may influence lipid metabolism
enzymes involved in the endocytic scavenging process, and transport
of proteins to the peroxisomes and/or lysosomes.
Phosphatidylinositol-3-kinases (PI3Ks), are known to
play a key role in a wide range of cellular functions, including
cell growth, proliferation, differentiation, motility and survival
(12). It is also evident that
PI3Ks play an important role in endocytosis and vesicle transport,
including a role in the recruitment of regulatory proteins to the
plasma membrane, endocytic uptake and recycling of receptors
(13). Class III PI3Ks are
responsible for the production of phosphatidylinositol-3-phosphate
[PtdIns(3)P] (14), which is
enriched in the membranes of early endosomes and the internal
vesicles of multivesicular bodies (15,16). PtdIns(3)P recruits proteins
containing FYVE, PX or PH motifs, and is involved in the control of
vesicular transport and intracellular protein sorting (17,18). Certain studies have reported that
PI3Ks are involved in the metabolism of different lipoproteins. For
example, Shetty et al (19) demonstrated that PI3K plays an
important role in class B type I scavenger receptor subcellular
localization and selective lipid uptake in hepatocytes.
Kzhyshkowska et al (20)
reported that PI3K activity is required for the transfer of
stabilin-1 and its ligand, acetylated low-density lipoprotein, from
early endosomes to late endosomes. As ATM has been shown to possess
a carboxyl-terminal domain homologous to PI3Ks and that the ATM
protein regulates PI3K protein activity (21,22), we hypothesized that PI3K may
involved in the promotion of E−/B48
lipoprotein endocytosis by cytoplasmic ATM in the hepatocytes of
ApoE−/− mice.
In this study, we demonstrate that heterozygous
ATM mutation reduces the hepatocyte uptake of
E−/B48 lipoproteins in
ApoE−/− mice and that the ATM protein is
distributed in early and late endosomes. In addition, we reveal
that the activated ATM protein interacts with class III PI3K and
affects its activity and that a class III PI3K inhibitor attenuates
the intracellular total cholesterol accumulation induced by ATM
activation. The data presented in this study, provide insight into
the mechanisms behind the involvement of ATM in the process of
endocytosis of E−/B48 lipoproteins.
Materials and methods
Animals
ATM+/− mice were kindly
provided by Dr Anthony Wynshaw-Boris (University of California, San
Diego, CA, USA).
ApoB48/48/ApoE−/−
and 129vEv wild-type mice were obtained from the Jackson Laboratory
(Bar Harbor, ME, USA).
ApoB48/48/ApoE−/−
mice were obtained by crossbreeding
ApoE−/− mice with
ApoB48/48 mice.
ATM+/−/ApoE−/−
mice were obtained by crossbreeding ATM+/−
and ApoE−/− mice. In the present study,
ATM+/−/ApoE−/−mice
were used at 14 weeks of age. The mice appeared as healthy as their
ATM+/+/ApoE−/−
littermates. All procedures for handling the animals were approved
by the Institutional Animal Care and Use Committees of Meharry
Medical College (Nashville, TN, USA) and were performed in
accordance with the guidelines of the American Association for the
Accreditation of Laboratory Animal Care and the National Institutes
of Health and Animal Care Guidelines of the Animal Experimental
Committee of the College of Medicine, Wuhan University, Wuhan,
China.
Hepatocyte isolation and culture
Hepatocytes were isolated from
ApoE−/− mice. After the mice were
anesthetized and the livers exposed, the livers were first perfused
with calcium-free buffer for 1.5 min and then perfused with 0.25%
(w/w) collagenase type I (nitrogen) at 37°C in Williams E nutrient
medium for 4 min. Subsequently, the hepatocytes were isolated by
gently mincing the livers in Williams E nutrient medium containing
0.25% collagenase type I, filtered through a nylon gauze, and
centrifuged twice for 5 min at 50 × g at 4°C. The cell pellets
consisted of pure hepatocytes, as confirmed under a light
microscope, and the viability of the cells was 90% as determined by
trypan blue exclusion assay.
Preparation of
E−/B48 lipoproteins
E−/B48 lipoproteins were
prepared from the plasma of
ApoB48/48/ApoE−/−
mice as previously described in the study by Wu et al
(7). Briefly, mouse plasma was
overlaid with KBr gradient solution (d<1.006) and centrifuged at
120,000 rpm for 2 h with a Sorvall Discovery M150 ultracentrifuge
(Thermo Scientific, Waltham, MA, USA). The
E−/B48 lipoproteins were collected, dialyzed
in phosphate-buffered saline (PBS) (pH 7.4) containing 10 mM EDTA
for 48 h at 4°C, and filtered through a 0.45 μm filter. One
milliliter of E−/ B48 lipoproteins at a
concentration of 1 mg/ml was mixed with 0.2 ml of 1 M glycine
buffer in 0.25 M NaOH (pH 10) and then mixed with a solution
containing 7 μl of 100 mM iodine monochloride, 7 μl of 100 μCi/μl
125I, and 25 μl of 1 M glycine buffer in 0.25 M NaOH (pH
10). Subsequently, the reaction mixture was incubated at room
temperature for 10 min and then applied to a 10 DG chromatography
column (Bio-Rad Laboratories, Hercules, CA, USA) to remove free
iodine. The 125I-labeled E−/B48
lipoproteins were eluted with PBS (pH 7.4) and dialyzed extensively
against PBS (pH 7.4).
Binding and uptake assays
For the uptake experiments, the cells were washed
twice with 2 ml of serum-free medium, containing 0.2% BSA. The
cells were then incubated with 1 ml of the same medium, containing
various concentrations of labeled E−/B48
lipoproteins [very-low-density lipoprotein (VLDL)] for 2 h or
containing 20 μg/ml labeled E−/B48
lipoproteins for 30, 60, 120 and 240 min. For the determination of
surface bound proteins, the cells were pre-chilled on ice for 30
min before washing and incubating in medium containing
E−/B48 lipoproteins; the cells were then
incubated with 1 ml of the same medium, containing various
concentrations of labeled E−/B48 lipoproteins
for 2 h or containing 20 μg/ml labeled E−/B48
lipoproteins for 30, 60, 120 and 240 min. Following incubation with
E−/B48 lipoproteins, the medium was removed
and the cells were washed twice with 2 ml of ice-cold PBS
containing 0.2% BSA followed by 2 more washes with 2 ml of ice-cold
PBS. The cells were lysed by the addition of 1 ml of 0.5 M NaOH and
lysate was collected for the measurement of protein (10 μl of
aliquot) and radioactivity taken up by the cells.
Endosomal fraction isolation
Mouse liver endosomal fractions were isolated as
prevously described by Chen et al (23). Mouse livers were homogenized in
20% (w/v) homogenization buffer containing 0.25 M sucrose, 3 mM
imidazole (pH 7.4), 1.7 nM antipain, 2 nM leupeptin and 1 mM
phenylmethylsulfonyl fluoride. The homogenate was centrifuged at
460 × g for 10 min, the supernatant was saved and the pellet was
rehomogenized and centrifuged as described above. The pooled
supernatant was centrifuged at 24,000 × g for 10 min, and the
resulting supernatant (S2) was then centrifuged at 100,000 × g for
90 min. The resulting microsomal pellet (P3) was suspended in
homogenization buffer (1.0 g starting liver/2 ml homogenization
buffer) using 10 strokes at 1,000 rpm. The resuspended P3 was then
diluted with an equal volume of 2.0 M sucrose in 3 mM imidazole
buffer. Three milliliters aliquots of the resulting 1.15 M sucrose
fraction were successively overlaid with 1.0, 0.86, 0.6 and 0.25 M
sucrose solutions, all buffered with 3 mM imidazole. Following
centrifugation at 100,000 × g for 3.5 h, 3 distinct fractions of
average density 1.06, 1.09 and 1.12 g/ml were obtained. The 1.06
and 1.09 fractions were pooled to yield the late endosomal
fraction, and the 1.12 g/ml fraction contained early endosomes.
Western blot analysis
Endosomal fractions were isolated and prepared as
described above. Endosomal proteins were extracted by a dual
precipitation procedure. First, endosomal fractions were suspended
in 20% trichloroacetic acid (TCA) with 20 mM DTT (1 g of starting
liver/2 ml 20% TCA). Second, the suspension was allowed to
precipitate on ice for 2 h and centrifuged at 1000 × g for 10 min,
and the pellet was then suspended in acetone with 20 mM DTT (1 g of
starting liver/2 ml acetone). Proteins in the suspension were
precipitated at −20°C for 4 h and centrifuged at 1,000 × g for 5
min. Residual acetone was removed by lyophilization. The protein
pellet was solubilized in lysis buffer, sonicated at 100 W for 30
sec, and centrifuged at 25,000 × g for 1 h. Plasma membrane
proteins were extracted using the Plasma Membrane Protein
Extraction kit (Abcam, Cambridge, MA, USA). Nuclear and cytoplasmic
proteins from the cells were extracted using the Nuclear-Cytosol
Extraction kit (BioVision, Inc., Milpitas, USA). Protein expression
was determined by western blot analysis. Briefly, equal amounts of
protein were separated by 7.0 or 10% SDS-polyacrylamide gel
electrophoresis (SDS-PAGE) and elcetrophoretically transferred onto
nitrocellulose membranes. After being blocked with TBST containing
5% bovine serum albumin, the membranes were incubated with primary
antibodies against ATM (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) and phosphorylated ATM (p-ATM) protein (Cell Signaling
Technology, Danvers, MA, USA) at 4°C overnight, followed by
incubation with horseradish peroxidase-conjugated secondary
antibody for 1 h at room temperature. The immunostaining was
visualized by enhanced chemiluminescence, and the results were
normalized to β-actin expression.
Co-immunoprecipitation
The hepatocytes were pre-treated with chloroquine
(an ATM activator) for 1 h and then treated with 40 μg/ml
lipoproteins for 8 h. Cytoplasmic proteins (500 μg) from the cells
extracted using the Nuclear-Cytosol Extraction kit were used to
perform immunoprecipitation assays according to the manufacturer’s
instructions (Pierce, Rockford, IL, USA). Briefly, the cytoplasmic
lysates were incubated with ATM or class III PI3K antibody (Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature followed by
the addition of protein A/G-Sepharose beads and further overnight
incubation at 4°C with gentle rocking. The immunoprecipitates were
washed 3 times with lysis buffer. The samples were then subjected
to SDS-PAGE and immunoblotting.
Class III PI3K activity assay
Cytoplasmic proteins from cells were extracted using
the Nuclear-Cytosol Extraction kit (BioVision). The proteins were
immunoprecipitated with anti-class III PI3K antibody for 2 h at
room temperature, and subsequently incubated with A Sepharose beads
overnight at 4°C. The precipitates were then washed with lysis
buffer. The immunoprecipitated proteins were incubated with PIP
substrates in vitro, and the PI(3)P products were measured
with the use of class III PI3K ELISA kit (Echelon Biosciences, Salt
Lake City, UT, USA) according to the manufacturer’s
instructions.
Measurement of cellular cholesterol
Hepatocytes grown in 75-mm culture flasks were
pre-treated with 5 μM chloroquine, 10 μmol/l KU55933 (an
ATM-specific inhibitor), 10 μM LY290042 (a class III PI3K
inhibitor) or 5 mM 3-MA (a class III PI3K inhibitor) for 2 h and
then incubated with 40 μg/ml E−/B48
lipoproteins for 22 h followed by a 12-h equilibrium in
lipoprotein-free medium. Quantitative measurement of intracellular
total cholesterol (TC) in vitro was analyzed using the
method described in the study by Gamble et al (24). In brief, the hepatocytes were
collected and lipids were extracted by the addition of
chloroform:methanol (2:1). The lipid phase was collected, dried,
and then dissolved in isopropanol containing 10% Triton X-100. The
concentrations of TC were measured by enzymatic assays and
normalized to total cellular protein levels.
Statistical analysis
The data are expressed as the means ± SEM.
Comparisons among multiple groups were performed using one-way
ANOVA or two-way ANOVA followed by the Student-Newman-Keuls or
Dunnett’s test. Differences were considered significant at
P<0.05.
Results
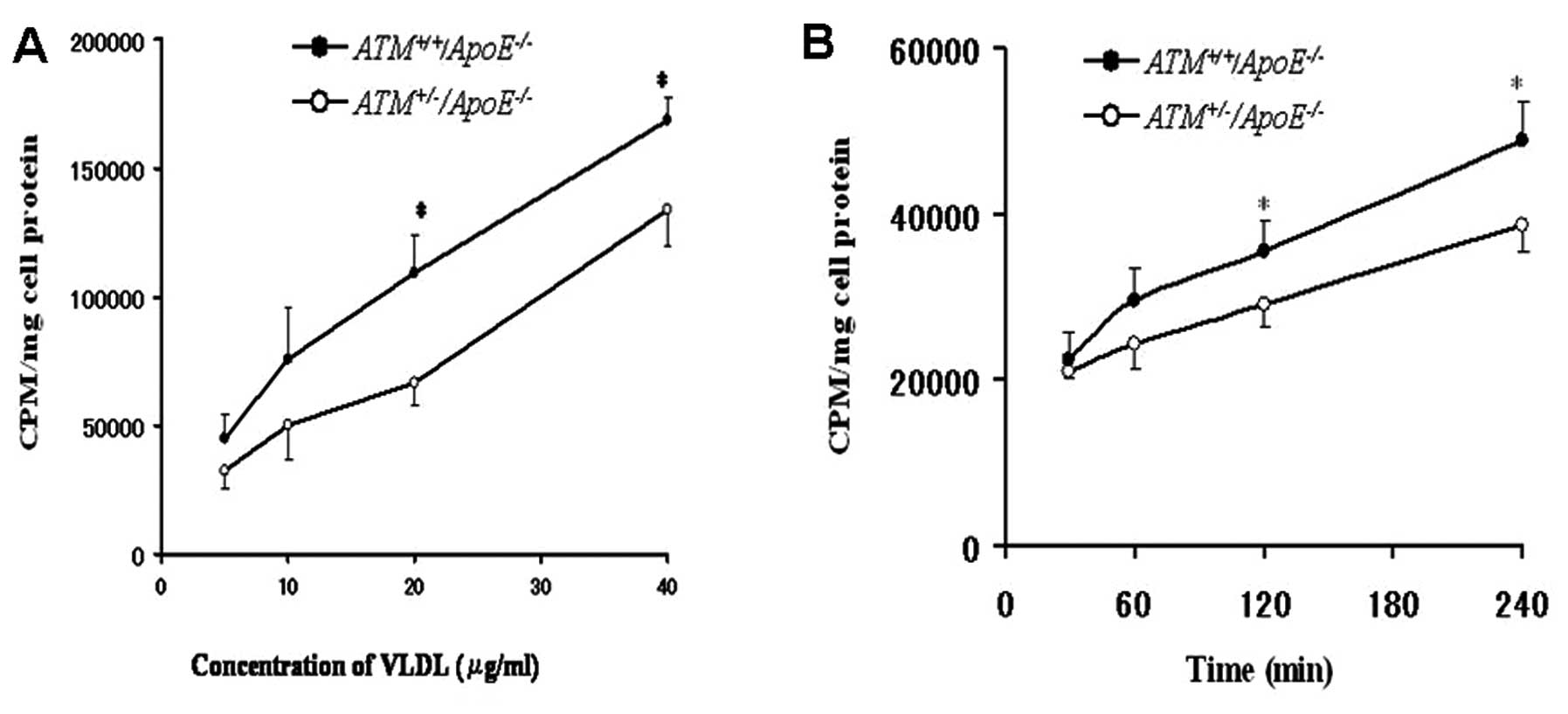
Uptake of E−/B48
lipoproteins
Both
ATM+/−/ApoE−/−
and
ATM+/+/ApoE−/−
hepatocytes absorbed E−/B48 lipoproteins in a
concentration and time-dependent manner. However, the uptake of
E−/B48 lipoproteins by the
ATM+/+/ApoE−/−
hepatocytes was greater than that of the
ATM+/−/ApoE−/−
hepatocytes. The uptake of E−/B48
lipoproteins by the
ATM+/+/ApoE−/−
hepatocytes was greater by 38–65% compared with the
ATM+/−/ApoE−/−
hepatocytes at the concentration range of 5–40 μg/ml; in
particular, a significant difference was observed between the
ATM+/−/ApoE−/−
and ATM+/+/
ApoE−/− hepatocytes at the concentration
range of 20–40 μg/ml and at the 120 and 240 min time points
(P<0.05) (Fig. 1).
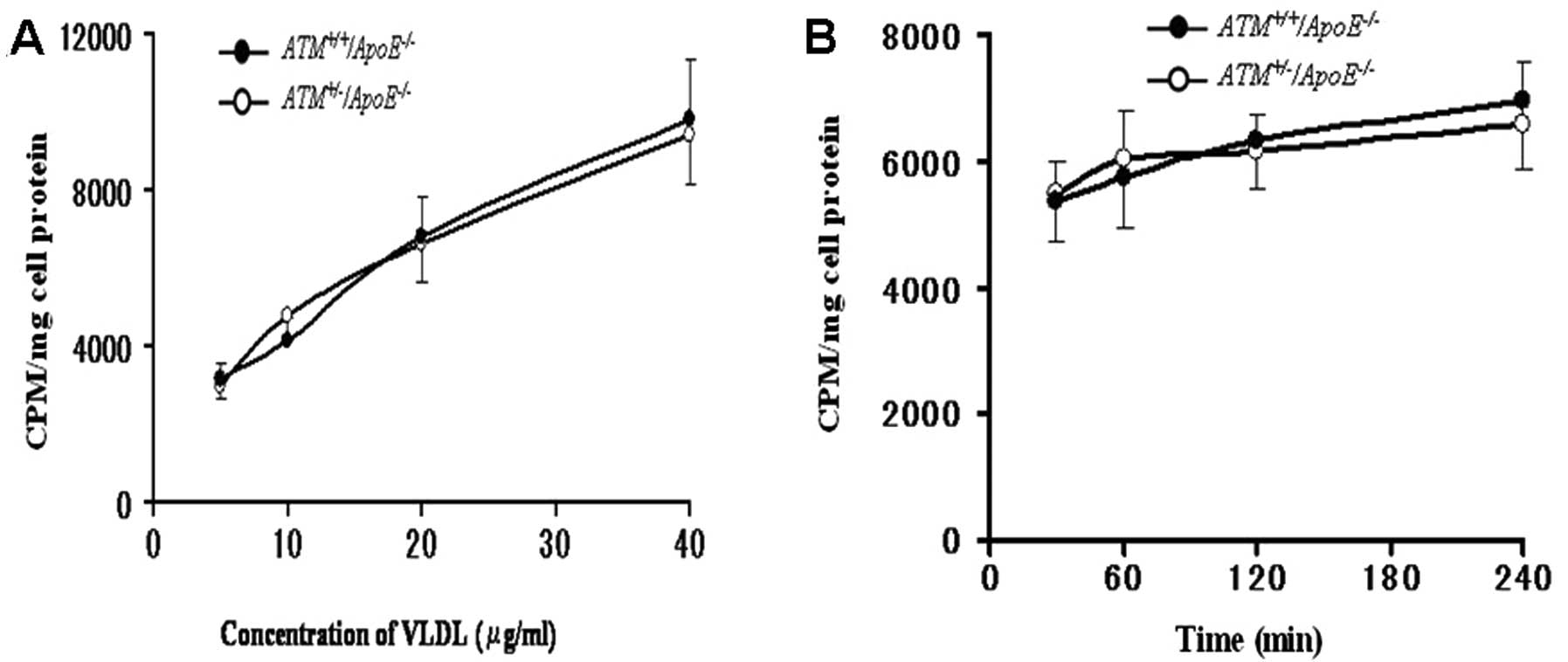
Binding of E−/B48
lipoproteins
Both
ATM+/−/ApoE−/−
and
ATM+/+/ApoE−/−
hepatocytes bound E−/B48 lipoproteins in a
concentration-dependent manner. However, no significant difference
was observed in the binding of E−/B48
lipoproteins to the
ATM+/−/ApoE−/−
and
ATM+/+/ApoE−/−
hepatocytes at the concentration range of 5–40 μg/ml (Fig. 2A). The binding of
E−/B48 lipoproteins to the
ATM+/−/ApoE−/−
and
ATM+/+/ApoE−/−
hepatocytes was not enhanced and no significant difference was
observed as time progressed (Fig.
2B).
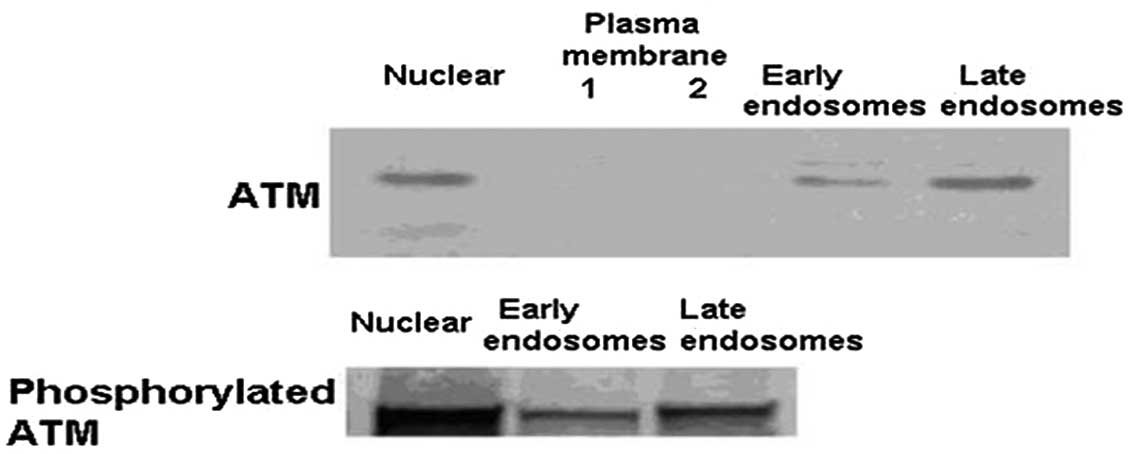
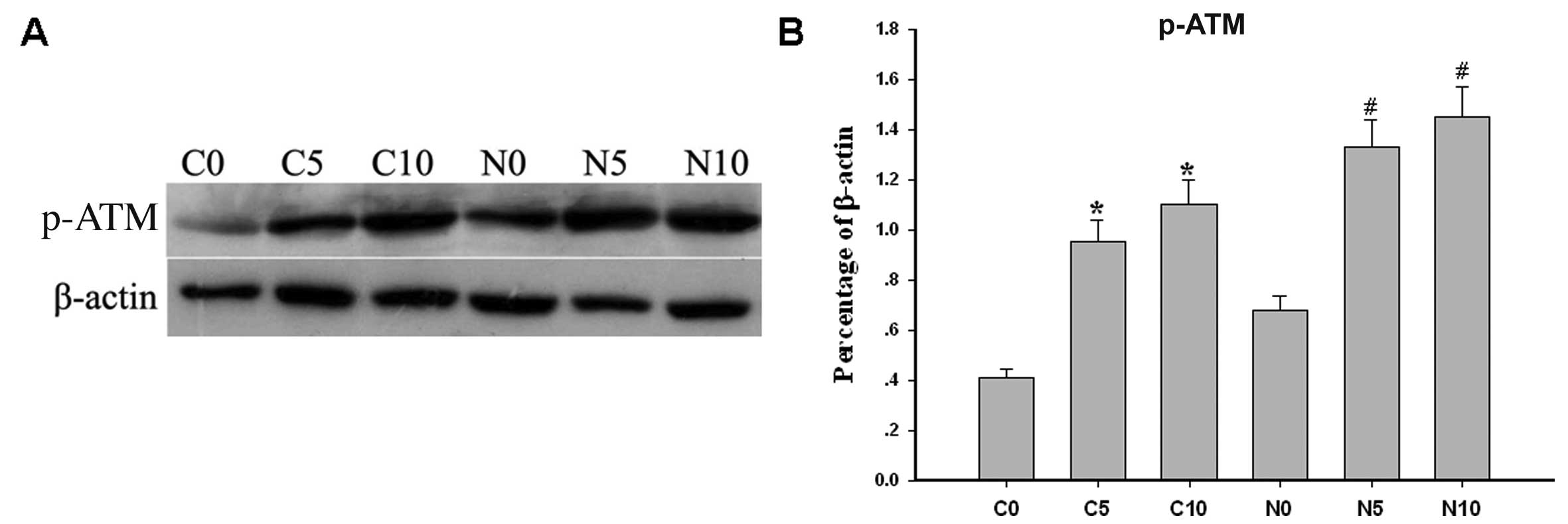
Distribution of ATM protein in endosomes
and ATM activation by chloroquine in the nucleus and cytoplasm
ATM protein and p-ATM protein were expressed in the
nucleus, early endosomes and late endosomes, but not in the plasma
membrane in the hepatocytes of ApoE−/−
mice (Fig. 3). The hepatocytes of
ApoE−/− mice were incubated with various
concentrations of chloroquine (0, 5 and 10 μmol/l) for 8 h at 37°C.
p-ATM levels in the nucleus and cytoplasm increased following
treatment with chloroquine in a dose-dependent manner (P<0.05)
(Fig. 4).
ATM protein interaction with class III
PI3K protein and its effect on class III PI3K activity
As ATM has been shown to possess a carboxyl-terminal
domain homologous to PI3Ks and that ATM protein regulate PI3K
activitys, we wished to determine whether ATM interacts with class
III PI3Ks and whether ATM activation affects the activity of class
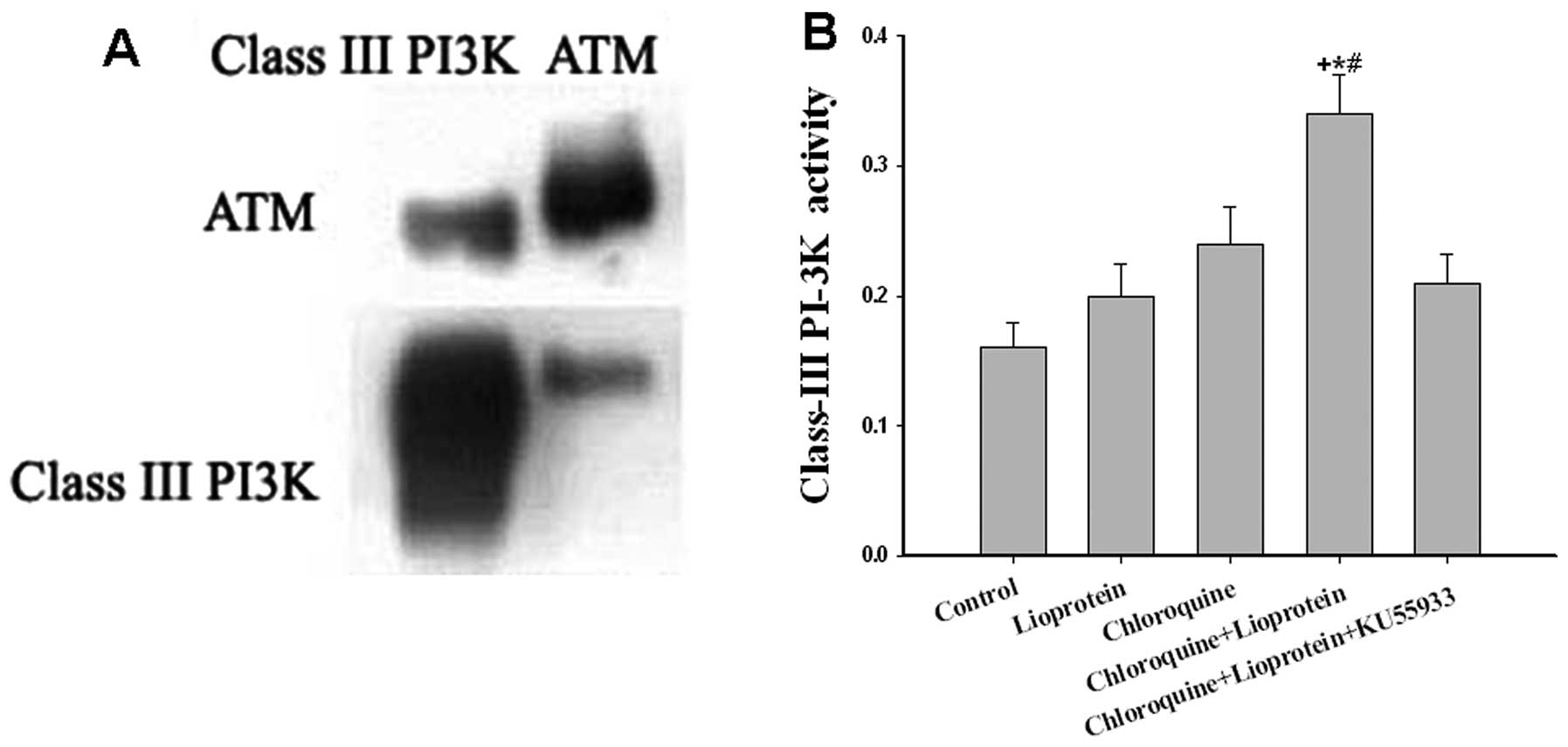
III PI3Ks. The results from co-immunoprecipitation analysis
indicated that cytoplasmic ATM protein interacted with cytoplasmic
class III PI3K protein (Fig. 5A).
In addition, when the hepatocytes were incubated with
E−/B48 lipoproteins alone, class III PI3K
protein activity increased; however, no significant difference was
observed between the control group (untreated group) and the group
treated with E−/B48 lipoproteins (P>0.05).
When the hepatocytes incubated with E−/B48
lipoproteins and chloroquine, class III PI3K activity markedly
increased (P<0.01); however, this effect was attenuated by the
ATM inhibitor, KU55933 (P<0.05) (Fig. 5B).
Class III PI3K inhibitor attenuates
intracellular total cholesterol accumulation induced by ATM
activation
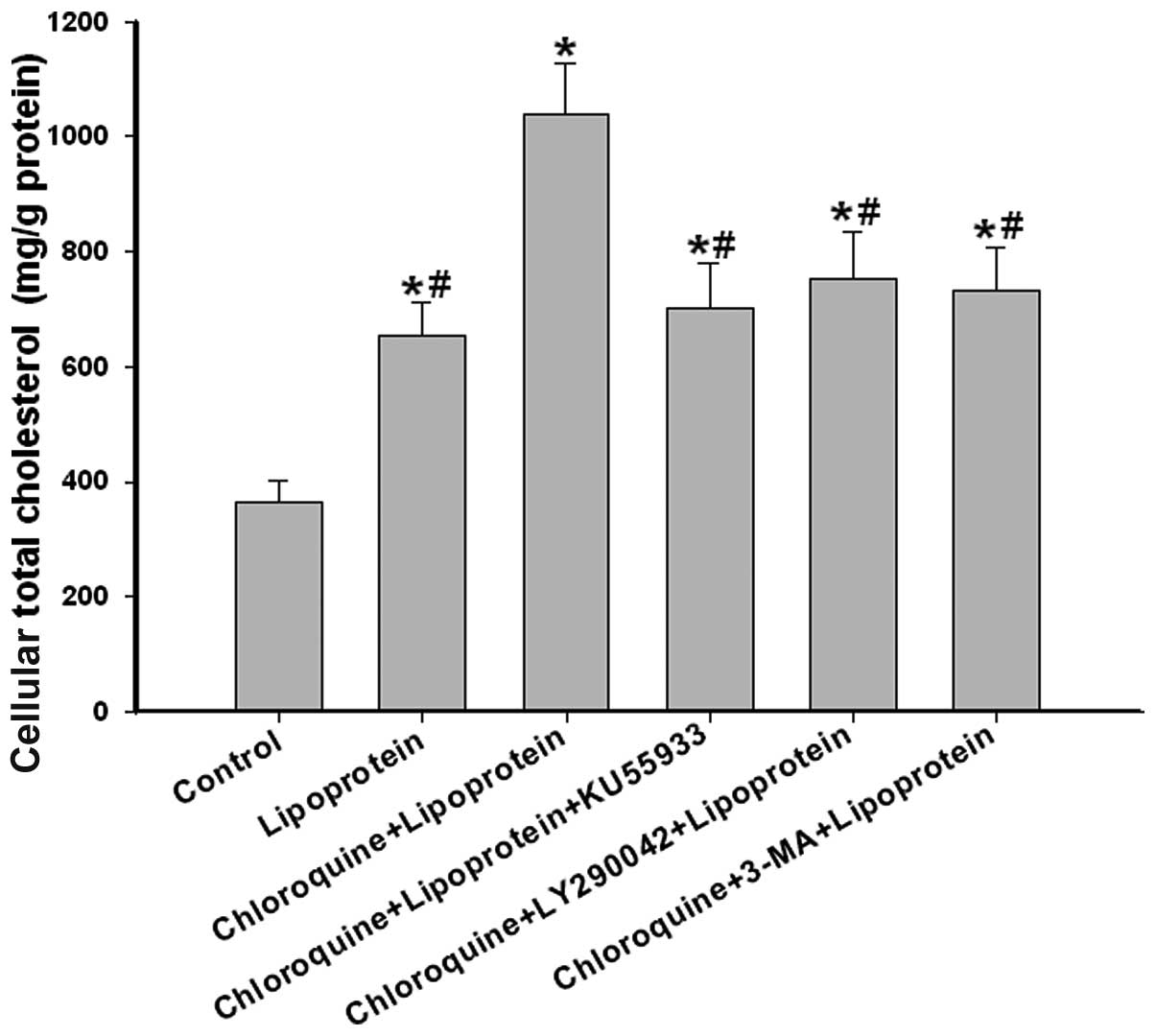
When the hepatocytes were incubated with
E−/B48 lipoproteins alone, the intracellular
total cholesterol of the hepatocytes increased, compared with the
control (untreated) group (653.8±58.2 vs. 362.5 ± 38.2 mg/g
protein). When the hepatocytes were incubated with
E−/B48 lipoproteins and chloroquine, the
intracellular total cholesterol of the hepatocytes markedly
increased compared with the lipoprotein-treated group (1038.5±88.3
vs. 653.8±58.2 mg/g protein). However, treatment with the ATM
inhibitor, KU55933, and the class III PI3K inhibitor, LY290042 and
3-MA, abolished the intracellular total cholesterol accumulation
induced by chloroquine (702.8±78.6 vs. 1038.5±88.3 mg/g protein),
(753.3±82.5 vs. 1038.5±88.3 mg/g protein) or (732.2±76.3 vs.
1038.5±88.3 mg/g protein), respectively (Fig. 6).
Discussion
The major findings of the present study are as
follows: i) the uptake of E−/B48 lipoproteins
by the
ATM+/+/ApoE−/−
hepatocytes was greater than that of the
ATM+/−/ApoE−/−
hepatocytes, although no significant difference was observed in the
binding of E−/B48 lipoproteins between the
ATM+/+/ApoE−/−
and
ATM+/−/ApoE−/−
hepatocytes; ii) a fraction of the ATM protein was expressed in
early endosomes and late endosomes, but not in the plasma membrane;
iii) ATM protein interacted with class III PI3K protein and the
activated ATM protein enhanced class III PI3K activity; iv) the
class III PI3K inhibitor, LY290042, abolished the intracellular
total cholesterol accumulation induced by ATM activation.
Under physiological conditions, ApoE activates
receptor-mediated endocytosis by binding to cell surface
low-density lipoprotein (LDL) receptor and LDL receptor-related
protein (LRP) (25). The deletion
of ApoE, lipoproteins containing ApoB100 (such as LDL) can still be
achieved by the interaction between ApoB100 and LDL receptor, but
lipoproteins containing ApoB48 can not enter cells through the LDL
receptor and LRP (26). We
previously reported that ATM heterozygous mutation in
ApoE−/− mice resulted in an
overaccumulation of plasma ApoB48-containing lipoproteins and
severe hypercholesterolemia occurred only with a combination of a
heterozygous ATM mutation and a null ApoE mutation,
but not wih the heterozygous ATM mutation alone or with the
combined heterozygous ATM and null LDL receptor
mutations (7). The present
experimental results revealed that despite the lack of ApoE,
lipoproteins containing ApoB48 can still be absorbed. Compared with
hepatocytes of
ATM+/+/ApoE−/−
mice, the uptake of E−/B48 lipoproteins by
hepatocytes of
ATM+/−/ApoE−/−
mice decreased significantly; however, no signficant difference was
observed in the binding of E−/B48
lipoproteins between hepatocytes from
ATM+/+/ApoE−/−
mice and those of
ATM+/−/ApoE−/−
mice. Based on these results, it can be concluded that there are
other pathways mediating the endocytosis of lipoproteins containing
ApoB48 without ApoE, and ATM facilitates the endocytosis of
lipoproteins containing ApoB48. In addition, we found that a
portion of the ATM protein was localized in early endosomes and
late endosomes, but not in the plasma membrane. These results
indicate that the ATM protein may participate in the endocytosis of
E−/B48 lipoproteins.
Since we observed that the ATM protein was
expressed in endosomes and it is known that class III PI3Ks are
responsible for the production of PtdIns(3)P in the membranes of
endosomes, and mediate vesicular transport, membrane trafficking
and intracellular protein sorting (14,27,28), our study focused on the effects of
ATM on the activity of class III PI3Ks. Certain studies have
reported that small doses of chloroquine activate nucleic ATM
proteins (29). In this study, we
observed that chloroquine activated ATM in the nucleus and
cytoplasm of hepatocytes in a dose-dependent manner. ATM has been
shown to possess a carboxyl-terminal domain homologous to PI3Ks and
ATM protein has been shown to interact with PI3K, regulating PI3K
activity (21,22). Similar to this result, in our
present study, we observed an interaction between cytoplasmic ATM
and class III PI3Ks; activated ATM increased class III PI3K protein
activity. Moreover, chloroquine, which activated ATM protein,
promoted intracellular total cholesterol accumulation, while the
class III PI3K inhibitor, LY290042 and 3-MA, inhibited this effect,
suggesting that class III PI3K protein plays an important role in
the ATM protein-mediated endocytosis of
E−/B48 lipoproteins in the hepatocytes of
ApoE−/− mice.
Previous studies have confirmed that there are
ApoE-independent mechanisms which mediate the uptake of
lipoproteins containing ApoB48. Magoori et al (30) reported that ApoE and
LRP-5 double knockout mice developed more severe
hypercholesterolemia than ApoE−/− mice and
that this hypercholesterolemia resulted mainly from an increased
level of ApoB-48-containing lipoproteins in the plasma, wheras
LRP-5 single knockout mice showed no significant difference
in plasma cholesterol levels. These results indicate that the LRP-5
mediates the ApoE-independent plasma lipoprotein metabolism
pathway. Therefore, it is possible that
E−/B48 lipoproteins interact with cell
membrane receptors, such as LRP-5 through unknown mechanisms, and
may activate cytosolic ATM; activated ATM in turn activates class
III PI3K, regulating the endocytosis of
E−/B48 lipoproteins. This may facilitate
E−/B48 lipoprotein degradation and
metabolism. Based on these results, we hypothesized that the
ATM/class III PI3K pathway may be involved in the endocytosis of
E−/B48 lipoproteins. In addition, it is
evident that chloroquine exerts an inhibitory effect on blood
lipids; however, the exact mechanisms involved are unclear. Our
results suggest that the decrease in blood lipid levels by
chloroquine may be associated with the activation of the ATM/class
III PI3K pathway, thus promoting the uptake of lipoproteins by
hepatocytes.
In conclusion, to the best of our knowledge, the
results presented in this study demonstrate for the first time that
a heterozygous mutation in ATM reduces the uptake of
E−/B48 lipoproteins by hepatocytes of
ApoE−/− mice. We also found that ATM was
distributed in early endosomes and late endosomes. Our results
demonstrated that the ATM protein interacted with class III PI3K
protein and the activated ATM protein enhanced class III PI3K
activity. In addition, the ATM activation promoted intracellular
total cholesterol accumulation; however, this was abolished by the
class III PI3K inhibitor, LY290042. These observations suggest that
ATM is involved in the endocytosis of E−/B48
lipoproteins through the class III PI3K protein. Our findings
provide further insight into the mechanisms through which the ATM
activation by chloroquine exerts beneficial effects, reducing blood
lipid levels.
Acknowledgements
This study was supported by a grant from the
National Nature Science Foundation of China (30971428 to D.W.).
References
|
1
|
Frappart PO and McKinnon PJ:
Ataxia-telangiectasia and related diseases. Neuromolecular Med.
8:495–511. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lavin MF: ATM: the product of the gene
mutated in ataxia-telangiectasia. Int J Biochem Cell Biol.
31:735–740. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Badalian LO and Kalinina LV: Lipid
metabolism disorder in ataxia-telangiectasia. Zh Nevropatol
Psikhiatr Im S S Korsakova. 76:665–669. 1976.(In Russian).
|
|
4
|
Su Y and Swift M: Mortality rates among
carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med.
133:770–778. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schneider JG, Finck BN, Ren J, et al:
ATM-dependent suppression of stress signaling reduces vascular
disease in metabolic syndrome. Cell Metab. 4:377–389. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercer JR, Cheng KK, Figg N, et al: DNA
damage links mitochondrial dysfunction to atherosclerosis and the
metabolic syndrome. Circ Res. 107:1021–1031. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu D, Yang H, Xiang W, et al: Heterozygous
mutation of ataxia-telangiectasia mutated gene aggravates
hypercholesterolemia in apoE-deficient mice. J Lipid Res.
46:1380–1387. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watters D, Kedar P, Spring K, et al:
Localization of a portion of extranuclear ATM to peroxisomes. J
Biol Chem. 274:34277–34282. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuljis RO, Chen G, Lee EY, Aguila MC and
Xu Y: ATM immunolocalization in mouse neuronal endosomes:
implications for ataxia-telangiectasia. Brain Res. 842:351–358.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barlow C, Ribaut-Barassin C, Zwingman TA,
et al: ATM is a cytoplasmic protein in mouse brain required to
prevent lysosomal accumulation. Proc Natl Acad Sci USA. 97:871–876.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lim DS, Kirsch DG, Canman CE, et al: ATM
binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci
USA. 95:10146–10151. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Backer JM: Phosphoinositide 3-kinases and
the regulation of vesicular trafficking. Mol Cell Biol Res Commun.
3:193–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hunyady L, Baukal AJ, Gaborik Z, et al:
Differential PI 3-kinase dependence of early and late phases of
recycling of the internalized AT1 angiotensin receptor. J Cell
Biol. 157:1211–1222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lemmon MA: Phosphoinositide recognition
domains. Traffic. 4:201–213. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gillooly DJ, Morrow IC, Lindsay M, et al:
Localization of phosphatidylinositol 3-phosphate in yeast and
mammalian cells. EMBO J. 19:4577–4588. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kutateladze TG: Phosphatidylinositol
3-phosphate recognition and membrane docking by the FYVE domain.
Biochim Biophys Acta. 1761:868–877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi JH, Hong WP, Kim MJ, Kim JH, Ryu SH
and Suh PG: Sorting nexin 16 regulates EGF receptor trafficking by
phosphatidylinositol-3-phosphate interaction with the Phox domain.
J Cell Sci. 117:4209–4218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shetty S, Eckhardt ER, Post SR and van der
Westhuyzen DR: Phosphatidylinositol-3-kinase regulates scavenger
receptor class B type I subcellular localization and selective
lipid uptake in hepatocytes. Arterioscler Thromb Vasc Biol.
26:2125–2131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kzhyshkowska J, Gratchev A, Brundiers H,
Mamidi S, Krusell L and Goerdt S: Phosphatidylinositide 3-kinase
activity is required for stabilin-1-mediated endosomal transport of
acLDL. Immunobiology. 210:161–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lavin MF, Khanna KK, Beamish H, Spring K,
Watters D and Shiloh Y: Relationship of the ataxia-telangiectasia
protein ATM to phosphoinositide 3-kinase. Trends Biochem Sci.
20:382–383. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khanna KK, Yan J, Watters D, et al:
Defective signaling through the B cell antigen receptor in
Epstein-Barr virus-transformed ataxia-telangiectasia cells. J Biol
Chem. 272:9489–9495. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen A, Guo Z, Zhou L and Yang H: Hepatic
endosome protein profiling in apolipoprotein E deficient mice
expressing apolipoprotein B48 but not B100. J Bioanal Biomed.
2:100–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gamble W, Vaughan M, Kruth HS and Avigan
J: Procedure for determination of free and total cholesterol in
micro- or nanogram amounts suitable for studies with cultured
cells. J Lipid Res. 19:1068–1070. 1978.PubMed/NCBI
|
|
25
|
Martins IJ, Hone E, Chi C, Seydel U,
Martins RN and Redgrave TG: Relative roles of LDLr and LRP in the
metabolism of chylomicron remnants in genetically manipulated mice.
J Lipid Res. 41:205–213. 2000.PubMed/NCBI
|
|
26
|
Hui DY, Innerarity TL, Milne RW, Marcel YL
and Mahley RW: Binding of chylomicron remnants and beta-very low
density lipoproteins to hepatic and extrahepatic lipoprotein
receptors. A process independent of apolipoprotein B48. J Biol
Chem. 259:15060–15068. 1984.PubMed/NCBI
|
|
27
|
Corvera S: Phosphatidylinositol 3-kinase
and the control of endosome dynamics: new players defined by
structural motifs. Traffic. 2:859–866. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Clague MJ, Urbé S and de Lartigue J:
Phosphoinositides and the endocytic pathway. Exp Cell Res.
315:1627–1631. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bakkenist CJ and Kastan MB: DNA damage
activates ATM through intermolecular autophosphorylation and dimer
dissociation. Nature. 421:499–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Magoori K, Kang MJ, Ito MR, et al: Severe
hypercholesterolemia, impaired fat tolerance, and advanced
atherosclerosis in mice lacking both low density lipoprotein
receptor-related protein 5 and apolipoprotein E. J Biol Chem.
278:11331–11336. 2003. View Article : Google Scholar
|