Introduction
Renal fibrosis is the frequent final outcome of a
wide variety of progressive chronic kidney diseases (1). Epithelial-mesenchymal transition
(EMT) is a central mechanism in tubulointerstitial fibrosis, in
which tubular epithelial cell loss is accompanied by the deposition
of extracellular matrix (ECM) and accumulation of fibroblasts and
inflammatory cells in the intersitium (2–5).
The phenotypic conversion of epithelial cells to myofibroblast
(with expression of vimentin and less expression of E-cadherin) is
the main feature of this process (6). Increasing evidence suggests that
numerous genes are involved in tubular EMT (7–10).
The transforming growth factor (TGF)-β/Smad pathway is a key
promoter of this process (11,12). Increased glomerular expression of
TGF-β has been reported in experimental and human kidney disease
(1,5,13).
Mice with increased plasma TGF-β1 levels exhibited enhanced renal
fibrosis (14). Through the
induction of target genes, TGF-β signaling promotes fibroblast
survival and proliferation. The range of TGF-β-target genes include
microRNAs (miRNAs or miRs) (15).
miRNAs are small (21–23-nt) non-coding RNA molecules
that regulate gene expression by interacting with multiple mRNAs
and inducing translational suppression or degradation of mRNA
(16). miRNAs are involved in
regulating diverse physiological processes ranging from
embryogenesis, organ development, oncogenesis and the initial step
in EMT (17–19). Three miRNA families,
miR-21, miR-200 and miR-29, are regulated by
TGF-β and have been shown to modulate renal fibrosis either by
amplifying TGF-β signaling and promoting fibrosis (miR-21)
or by inhibiting EMT and reducing fibrosis (miR-29 and
miR-200) (20–22). Five members of the miR-200
family identified thus far are miR-200a, miR-200b,
miR-200c, miR-429 and miR-141. Published data
suggest that the miR-200 family inhibit EMT through directly
targeting zinc finger E-box-binding homeobox (ZEB)-1
and ZEB-2, which are E-cadherin transcriptional repressors
in kidney tubular cells (23).
Therefore, approaches to correct miRNA expression represent the
novel therapeutic strategies for these diseases.
Homeodomain interacting protein kinase 2 (HIPK2) is
a member of an evolutionary conserved family of serine/threonine
kinases (24), which is
considered as a tumor suppressor gene and mediates the activation
of Wnt, Notch, and TGF-β-induced signaling (25–27). HIPK2 is also considered as a
co-regulator of an increasing number of transcription factors
modulating numerous different basic cellular processes, including
apoptosis, proliferation, differentiation and development (24,28–30). Recently, HIPK2 has also been
identified as a key regulator in idiopathic pulmonary fibrosis
(IPF) and kidney fibrosis (31,32). In the kidney, HIPK2 mediates
apoptosis and EMT of renal tubular epithelial cells, contributing
to fibrosis. HIPK2 may be a potential target for anti-fibrosis
therapy (31). Given the ability
of the miR-200 family to inhibit EMT and the evidence of
HIPK2 in tissue fibrosis, whether miR-141 ameliorates
tubulointerstitial fibrosis was investigated by inhibition of EMT
through targeting HIPK2. In the present study, the first aim was to
define a role of miR-141 in regulating EMT in TGF-β-treated
human kidney 2 (HK-2) cells (normal renal tubular epithelial
cells). miR-141 hindered EMT by upregulating E-cadherin and
downregulating vimentin and fibroblast-specific protein 1 (FSP1)
expression through direct targeting of HIPK2, by binding to its
three prime untranslated region (3′-UTR).
Materials and methods
Cell lines and transfection
HK-2 cell lines were purchased from American Type
Culture Collection (Manassas, VA, USA) and maintained in RPMI-1640
medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal
bovine serum (Gibco, Carlsbad, CA, USA), 100 U/ml penicillin and
100 μg/ml streptomycin in a humidified 5% CO2
incubator at 37°C. Ectopic expression of miR-141 in HK-2
cells was achieved by transfection with miR-141 mimics
(Genepharma, Shanghai, China) using Lipofectamine 2000
(Invitrogen). Overexpression of HIPK2 was performed using
the HIPK2 ORF expression clone (GeneCopoecia, Guangzhou,
China).
RNA extraction and SYBR green
quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using the Trizol reagent as
recommended by the manufacturer (Invitrogen). RNA quality and
concentration were evaluated by spectrophotometry using a NanoDrop
2000c instrument (Thermo Scientific, Rockford, IL, USA). For miRNA
analysis, mature miR-141 was detected using a Hairpin-it™
miRNAs qPCR Quantitation kit (GenePharma, Shanghai, China).
U6 served as an internal reference. For HIPK2 mRNA
analysis, qPCR was performed using the Power SYBR-Green PCR master
mix (Takara, Dalian, China) on an ABI 7900HT PCR machine (Applied
Biosystems, Foster City, CA, USA), and data were normalized to
β-actin and further normalized to the negative control,
unless otherwise indicated. Data analysis was performed using the
2−ΔΔCt method. Human recombinant TGF-β1 was purchased
from Cell Signaling Technology (no. 8915; Danvers, MA, USA),
reconstituted in 20 mM citrate (pH 3.0) at a concentration of 100
μg/ml. Further dilution was made in PBS containing 2 mg/ml
albumin, stored at −20°C for future use.
Immunoblot analysis
Whole cell lysates were collected in
radioimmunoprecipitation assay buffer with protease inhibitor
cocktail (Roche, Indianapolis, IN, USA). Protein concentration was
measured using a bicinchoninic acid protein assay kit (Thermo
Scientific). Reduced protein (10–30 μg) in Laemmli sample
buffer was resolved using 6–12% sodium dodecyl sulfate
polyacrylamide gel and transferred to a nitrocellulose membrane
(Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5%
skimmed dry milk (Bio-Rad) in Tris-buffered saline (TBS) with 0.05%
Tween 20 (TBST) buffer for 1 h at room temperature, incubated with
primary antibody overnight at 4°C, followed by the appropriate
secondary immunoglobulin G antibody; anti-mouse, rabbit-horseradish
peroxidase (Bio-Rad). Membranes were washed thoroughly between
steps using TBST, and developed using the ECL Western blotting
detection kit (Bio-Rad). All the primary antibodies used were from
Cell Signaling Technology. The antibodies used were the following:
HIPK2 (no. 5091), vimentin (no. 5741), FSP1 (no. 13018), E-cadherin
(no. 3195) and β-actin (no. 4967). The blots were stripped using
stripping buffer (Thermo Scientific) prior to reprobing.
β-actin was used as an endogenous protein for normalization.
Images were analyzed by Quantity One software (Bio-Rad).
HIPK2 3′-UTR luciferase reporter
assays
The 3′-UTR for HIPK2 was PCR-amplified from
genomic DNA extracted from HK-2 cells. The PCR primers used to
amplify the Hipk2 3′-UTR were forward,
5′-CTCGAGACACTTTGCATAACGTATA-3′; and reverse,
5′-GCGGCCGCATTGGAACAGTAGTCATAT-3′, whereas the primers used to
amplify the mutant Hipk2 3′-UTR (with a mutant seed sequence
of miR-141) were forward, 5′-TTCAAATGAATATTTCGTCTAAATAAATAAAG-3′;
and reverse, 5′-TGTAGACGAAATATTCATTTGAATTTTCAAG-3′. The mutant
Hipk2 3′-UTR was identical to the wild-type sequence except
for the seed regions, in which the complementary sequence was used.
The mutant version was generated using the Stratagene QuikChange™
site-directed mutagenesis strategy (Stratagene, La Jolla, CA, USA).
Amplified 3′-UTRs were cloned downstream of the luciferase coding
region in the psiCECK™-2 vector (Promega, Madison, WI, USA). The
fidelity of all the constructs utilized in the study was confirmed
by sequencing (ABI PRISM 377 automated sequencer; Southern Medical
University, Guangzhou, Guangdong, China) and subsequent sequence
alignment (NCBI BLAST) using GenBank™ accession gene ID 28996. To
assess the effect of miR-141 on reporter activity, HK-2
cells were seeded in 96-well clusters 24 h prior to transfection.
The following day, medium was replaced with OptiMEM (Invitrogen),
and cells were co-transfected with 100 ng reporter plasmids along
with 100 ng control Rnilla-luciferase plasmid and either
scrambled control miRNA (Vector) or miR141 (miR-141
Sponge) using Lipofectamine 2000 (Invitrogen). The cells were
collected 48 h post-transfection and luciferase activity was
detected using a dual-luciferase reporter assay system (Promega).
All the experiments were performed in triplicate, and each
experiment was repeated at least three times.
Statistical analysis
Values are shown as mean ± standard error, unless
otherwise specified. SPSS 17.0 (Pearson; SPSS, Inc., Chicago, IL,
USA) was used to analyze data by unpaired student t test or
by analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-141 downregulates the expression of
EMT markers
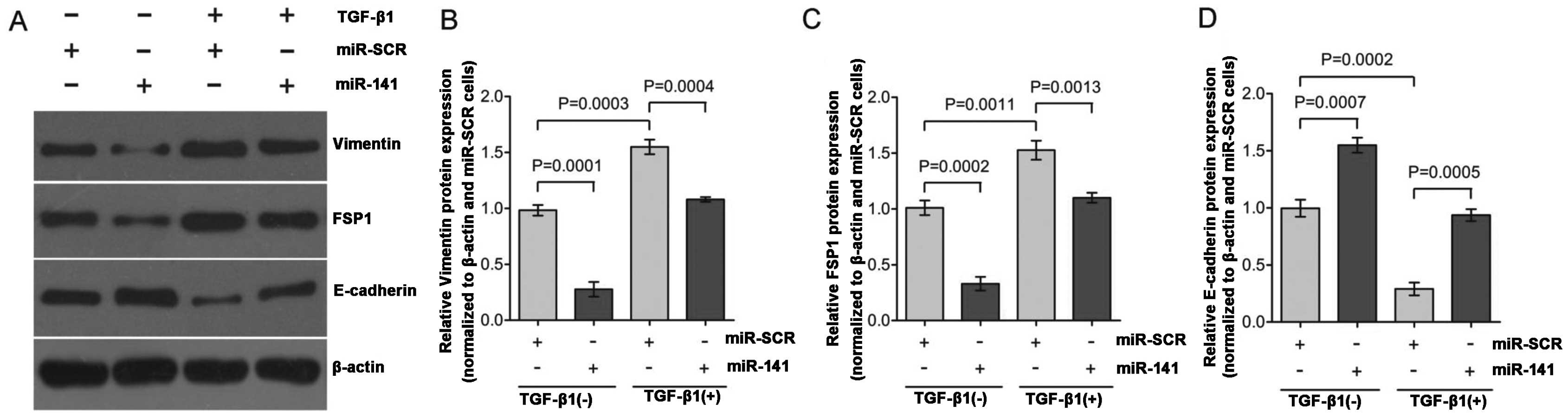
To study EMT, TGF-β1-induced EMT was used in the
HK-2 cells. Exposure of HK-2 cells to TGF-β1 (2 ng/ml) for 48 h
resulted in a significant decreased expression of the epithelial
marker, E-cadherin, and increased expression of mesenchymal
markers, vimentin and FSP1 (Fig.
1A; lane 1 compared to lane 3; lane 2 compared to lane 4).
These hallmark shifts at the molecular level indicate a successful
EMT program in HK-2 cells. miR-200 is enriched in the kidney
and lung, where it functions to maintain epithelial
differentiation. Among the five miR-200 family members,
miR-200a and miR-141 share the same seed sequence,
AACACU. Therefore, these two members may have the same mRNA
targets. A recent study found that miR-200a significantly
influenced the development and progression of TGF-β-dependent EMT
and fibrosis in vitro and in vivo (33). To study the actions of
miR-141 in the regulation of EMT, HK-2 cells were
transfected with miR-141 and changes of the gene expression
were compared with the negative scrambled control (miR-SCR). As
shown in Fig. 1A (lane 3 verses
lane 4), HK-2 cells transfected with miR-141 responded to
TGF-β1 treatment with >70% reduction of vimentin (Fig. 1B) and FSP1 expression (Fig. 1C), and >50% increase of
E-cadherin expression (Fig. 1D)
as compared to the control. Overall, these results indicate that
miR-141 is capable of functionally disrupting the EMT switch
in HK-2 cell by maintaining relative high levels of E-cadherin
expression, which is consistent with previous studies reporting
that the miR-200 family inhibits EMT by targeting E-cadherin
transcriptional repressors (23,34). TGF-β1 downregulates the
expression of miR-141 accompanied by upregulation of HIPK2.
Previous published data reported that treatment with TGF-β1 lead to
decreased expression of the miR-200 family, including
miR-141 (33). Therefore,
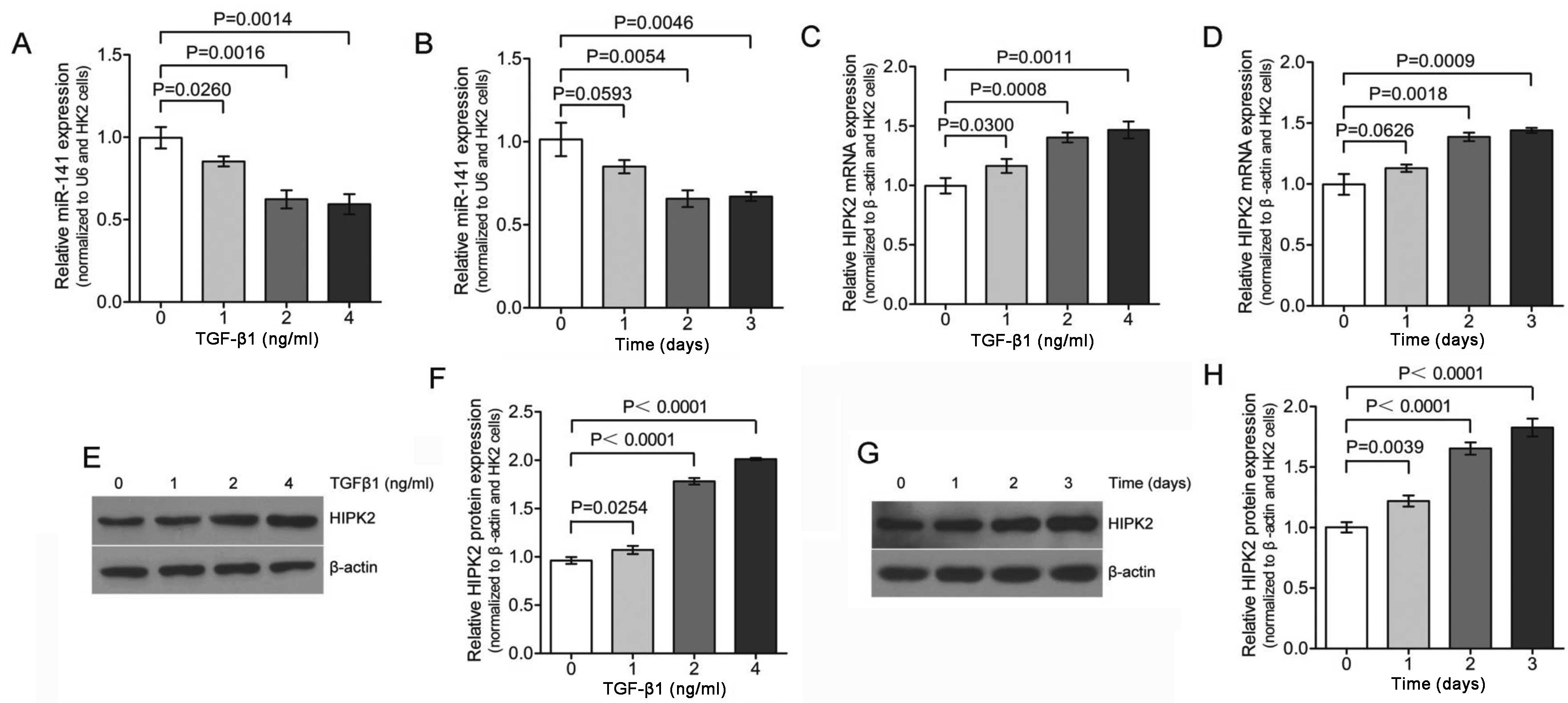
qPCR analysis was used to reveal that the expression of
miR-141 was significantly downregulated in HK-2 cell
following TGF-β1 treatment in a dose- and time-dependent manner
(Fig. 2A and B). The expression
of miR-141 was reduced to <30% of the pretreatment level
within 48 h of TGF-β1 exposure (Fig.
2B). These results confirm that TGF-β1 treatment lead to
decreased expression of miR-141.
A published systematic approach identified HIPK2 as
a key regulator of renal fibrosis (31). Another recent study implicated
dysregulated HIPK2 levels in idiopathic pulmonary fibrosis (IPF)
(32). Therefore, the role of
HIPK2 in TGF-β1 treated HK-2 cells was investigated. Initial qPCR
demonstrated TGF-β1-mediated induction of HIPK2 in HK-2 kidney
tubular cells (Fig. 2C and D) in
a dose- and time-dependent manner, which corresponded to the
decreased miR-141 expression. To verify the induction of
HIPK2, the expression was analyzed by western blotting in HK-2
cells. As shown in Fig. 2E and F,
HK-2 cells exposed to different dose of TGF-β1 for 2 days increased
the expression of HIPK2 significantly. The maximal HIPK2
mRNA and protein expression levels at 4 ng/ml TGF-β1 treatment were
~1.5-2-fold higher than the control. Furthermore, HK-2 cells
exposed to TGF-β1 (2 ng/ml) for 0–3 days also increased the
expression of HIPK2 significantly (Fig. 2G and H). This result is consistent
with the observation shown in Fig. 2C
and D, which means that the changes in HIPK2 expression were
also observed at the protein level. Collectively, these results
indicated that lower expression levels of miR-141 were
significantly associated with higher levels of HIPK2 mRNA
and protein expression in the same set of TGF-β1 treatment HK-2
cells.
Ectopic expression of HIPK2 promotes
EMT
Dysregulation of HIPK2 has been implicated in
increased proliferation, as it is typical in cancer or fibrosis
(35,36). A recently published study
identified HIPK2 as a key regulator of kidney fibrosis (31). Whether HIPK2 was sufficient to
induce EMT in the absence of TGF-β1 and what the potential role of
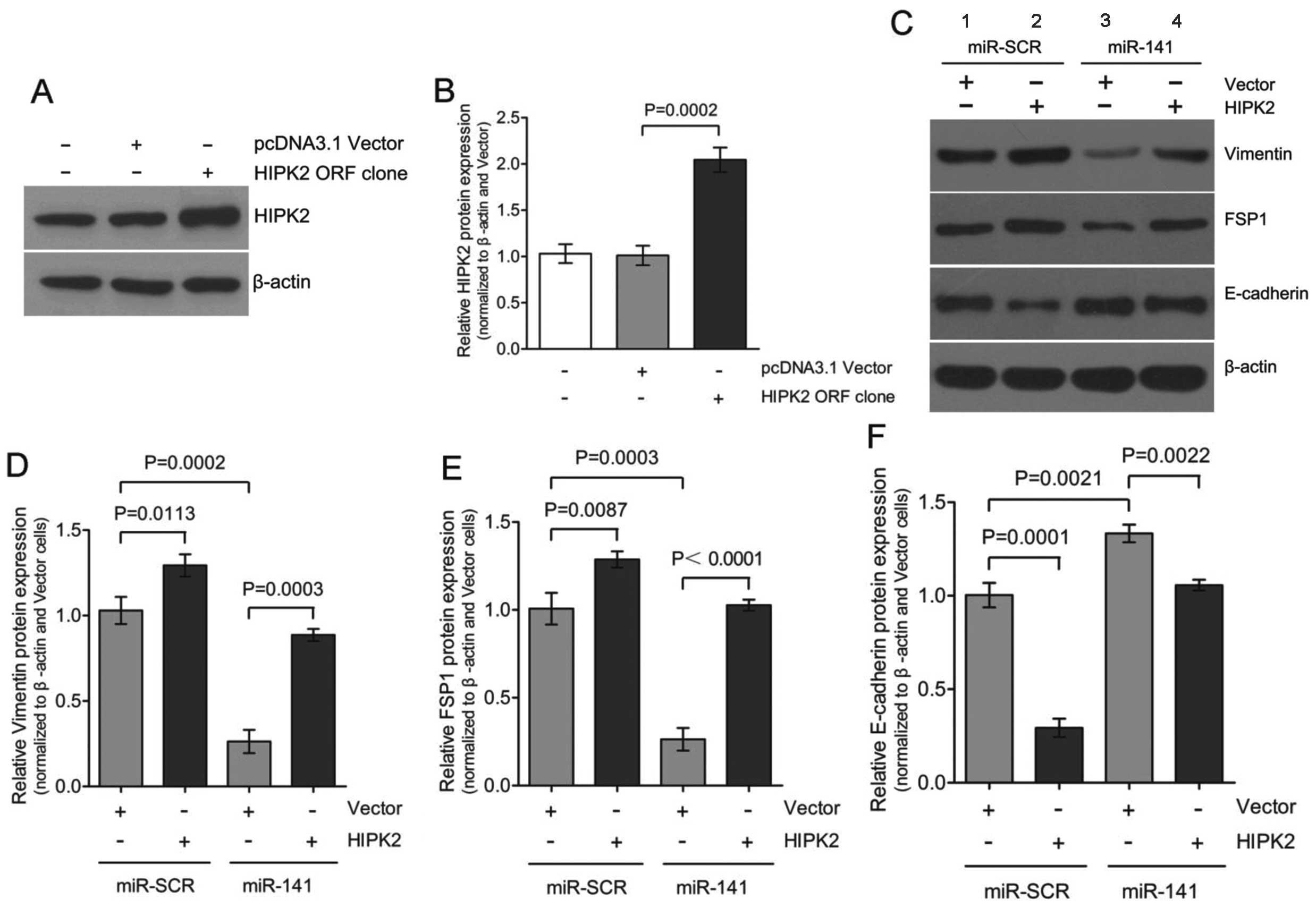
miR-141 was during this process were investigated. Transient
expression of HIPK2 ORF in HK-2 cells resulted in a
considerable upregulation of HIPK2 expression to 2.0±0.2-fold
(Fig. 3A and B) accompanied by
the gain of mesenchymal markers, vimentin and FSP1, and the loss
expression of epithelial marker E-cadherin (Fig. 3C; lanes 1 and 2). Co-transfection
of miR-141 with the HIPK2 ORF clone partially
restored E-cadherin expression and attenuated vimentin and FSP1
induction (Fig. 3C; lanes 2 and
4). These results are consistent with our previous results that
downregulation of miR-141 is accompanied by upregulation of
HIPK2 during TGF-β1-induced EMT in HK-2 cells. Therefore,
downregulation of miR-141 appears to be required for
mediating the upregulation of HIPK2 in the EMT process. HIPK2 can
mimic the TGF-β1-induced EMT process in HK-2 cells.
miR-141 directly targets HIPK2
The critical role of miR-141 in EMT prompted
the identification of the genes that were directly regulated by
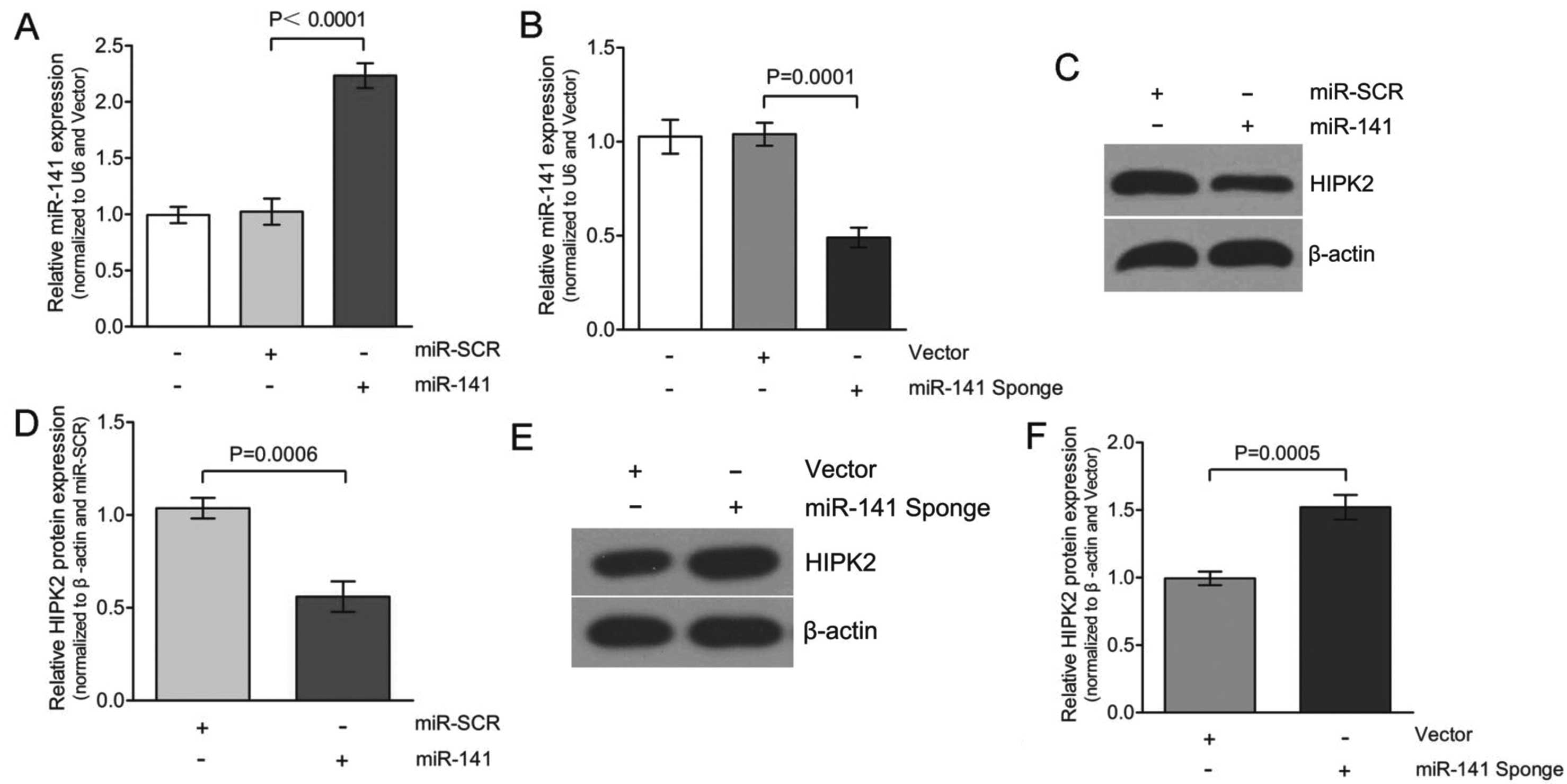
miR-141. Whether overexpression of miR-141 would
alter HIPK2 protein expression was initially determined. As shown
in Fig. 4A and C, transfecting
with miR-141 increased expression of miR-141 to
~2.3-fold, compared to the transfection of miR-SCR in the HK-2
cells. This increase in miR-141 expression decreased HIPK2
expression to an average of 50±5% in the three experiments
(Fig. 4D). Based on this
observation, we hypothesize that the miR-141 inhibitor
(miR-141 Sponge) should not be able to decrease HIPK2
expression. To test this hypothesis, HK-2 cells were transfected
with either vector or miR-141 Sponge and HIPK2 protein
expression was determined. The overexpression of miR-141
Sponge caused a downregulation of the miR-141 levels to
~0.5-fold compared to the transfection of the vector in the HK-2
cells (Fig. 4B and E).
Consequently, the decreased expression of miR-141 caused
upregulation of HIPK2 expression, with an average of 1.5±0.1-fold
in three experiments (Fig. 4F).
Therefore, the expression of HIPK2 was significantly decreased in
miR-141-transfected cells. Taken together, TGF-β1 (Fig. 2) and miR-141 inhibitor
(miR-141 Sponge) caused a similar effect on HIPK2 expression
in HK-2 cells.
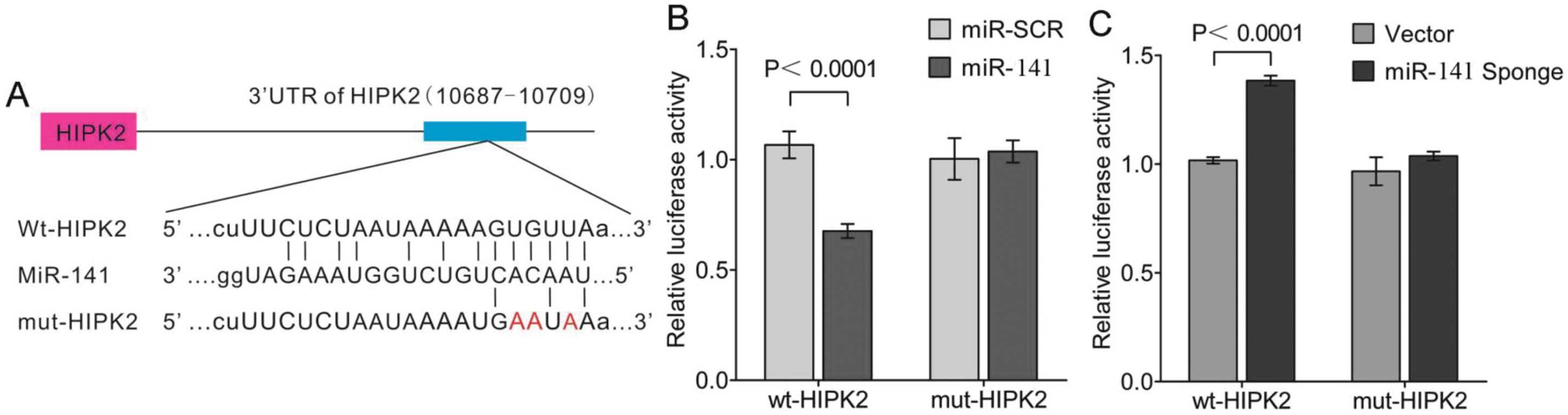
As miR-141 and HIPK2 3′-UTR share the
same seed sequence as shown (Fig.
5A), whether HIPK2 is the direct target of miR-141 on
translational repression was further investigated. In these
experiments, luciferase reporter constructs were used,
incorporating a wild-type or mutant 3′-UTR of HIPK2 in which
the sequence corresponding to the seed region was altered.
Co-transfection of wild-type HIPK2 luciferase reporter
construct or mutant 3′-UTR of HIPK2 with miR-141 or
miR-SCR in HK-2 cells resulted in a significantly reduced
HIPK2 3′-UTR luciferase activity expression. This
demonstrated that miR-141 directly repressed luciferase
activity with the wild-type 3′-UTR of HIPK2 (Fig. 5B), but not with the mutant 3′-UTR.
By contrast, miR-141 Sponge increased luciferase activity
with the wild-type HIPK2, but not with the mutant version of
HIPK2 (Fig. 5C).
Discussion
Tubular EMT is a series of highly-regulated
pathological events, which is one of the major mechanisms leading
to renal fibrosis (1,5,13,37). miRNAs are recognized to be
critical regulators of a number of important developmental,
homeostatic and pathogenic pathways. Recent findings suggest that
miRNAs are essential for kidney development and homeostasis. The
miR-200 family members were clearly downregulated in cells
that had undergone EMT in response to TGF-β, and the expression of
the miR-200 family alone was sufficient to prevent
TGF-β-induced EMT (38). The
study by Tamagawa et al (34) demonstrated the role of
miR-200c/miR-141 in the regulation of EMT and
migration in head and neck squamous cell carcinoma. However, the
functional involvement of miR-141 in EMT associated with
tubulointerstitial fibrosis, as well as direct targeting by
miR-141, has not been investigated. In the present study,
miR-141 influenced the progression of TGF-β1-induced EMT
in vitro. An EMT model was initially established using
TGF-β1-treated HK-2 cells by observing upregulation of vimentin and
FSP1 and downregulation of E-cadherin (Fig. 1A; lanes 1 and 3) at the protein
levels. Overexpression of miR-141 inhibited EMT progression
in TGF-β1-treated HK-2 cells by maintaining high E-cadherin
expression levels and low vimentin and FSP1 expression levels
(Fig. 1A; lanes 3 and 4).
Furthermore, miR-141 was found to be downregulated during
TGF-β1-induced EMT in HK-2 cells (Fig. 2). Given that reduced
miR-141 levels are associated with increased EMT expression,
and that restoring expression of miR-141 prevents EMT in
HK-2 cells, miR-141 appears to play an important role in
EMT, and therefore, renal fibrosis. Wang et al (33) also found that in early and more
advanced models of kidney disease, the downregulation of
miR-141 was also associated with increased TGF-β expression
and renal scarring. Additionally, downregulated expression of
miR-141 was accompanied by upregulated HIPK2
expression (Fig. 2). These
observations suggest a possible regulatory network that drives the
increased expression of HIPK2 during TGF-β1-induced EMT,
perspectives that warrant further investigation.
Although a number of different factors contribute to
renal fibrosis, the most known and studied profibrotic agent is
TGF-β1, which is increased in the diabetic kidney (39). Recently, HIPK2 has been also
identified as a main regulator of kidney fibrosis (31). In the kidney, HIPK2 mediates
apoptosis and EMT of renal tubular epithelial cells, contributing
to fibrosis (31). In the present
study, overexpression of HIPK2 resulted in the upregulation
of vimentin and FSP1, and downregulation of E-cadherin (Fig. 3), which indicates that HIPK2
mimicked TGF-β1-induced EMT in HK-2 cells. These initial studies
suggested that overexpression of miR-141 inhibited
TGF-β1-induced EMT (Fig. 1).
Given the possible link between miR-141 and HIPK2, whether
overexpression of miR-141 hindered HIPK2 expression in HK-2
cells was investigated further. To test this hypothesis, miR-SCR or
miR-141 were transfected with either vector or the
HIPK2 ORF clone and the EMT markers expression was
determined. The ability of miR-141 to attenuate HIPK2
expression has potential implications and may prove to be an
attractive option for targeting the EMT pathway.
Certain miRs are well known to be indiscriminate and
may bind to the UTR regions of a number of different genes. The
results of the present study showed that overexpression of
miR-141 significantly reduced the expression of HIPK2
(Fig. 4), and therefore, it is
possible that the 3′-UTR of HIPK2 may harbor a target site for
miR-141. The TargetScan database indicated that HIPK2 had a
target site for miR-141, as shown in Fig. 5A. In the study, miR-141 was
shown to be a direct repressor of HIPK2 as it targeted the 3′-UTR
of this gene, as shown by experiments using the wild-type and
mutant HIPK2 3′-UTR-luciferase constructs. HIPK2 was a
transcriptional cofactor in the downstream TGF-β/BMP signaling
pathway (27,31,40–42). Notably, loss of HIPK2 reduced
cellular responses to TGF-β during neuronal development and in
mouse models of renal fibrosis (27,40). In addition, the present
experimental results confirmed that HIPK2 was a functional target
of miR-141 in HK-2 cells. There are several lines of
evidence to support this. Firstly, protein expression of HIPK2 was
significantly decreased in the miR-141 group compared to the
miR-SCR group (Fig. 4).
Overexpression of miR-141 significantly downregulated HIPK2
by directly targeting the 3′UTR of HIPK2 mRNA, confirmed
using the luciferase reporter-gene assays (Fig. 5). This effect was largely
eliminated when the sites in HIPK2 3′UTR targeted by
miR-141 were mutated. Overexpression of HIPK2
mimicked TGF-β1-induced EMT, and overexpression of miR-141
rescued the expression of epithelial marker E-cadherin and
inhibited HIPK2-induced EMT (Fig. 3C
and F; lanes 3 and 4). These results strongly suggested that
miR-141, through directly targeting of HIPK2, plays
an essential role in renal proximal tubular EMT. miR-141
suppresses the expression of HIPK2 through directly
interacting with its wild-type 3′-UTR.
In the present study, ectopic expression of
miR-141 hindered the progression of EMT in TGF-β1-treated
HK-2 cells by downregulation of HIPK2 expression, maintaining a
high expression level of E-cadherin. Since HIPK2 has been indicated
to be involved in the progression of EMT by suppression of
E-cadherin expression, it may provide important targets for
therapeutic strategies. The findings in the study suggested that
miR-141 was downregulated in TGF-β1-treated HK-2 cells. This
not only reveals a close association between the miR-141
expression level and epithelial phenotypic conversion, but also
implicates a potential role of miR-141 in the regulation of
tubular dedifferentiation, which is a frequent pathogenesis in
tubular interstitial fibrosis in a number of types of chronic
kidney disease. Evidently, more studies are required to further
characterize the role of miR-141 in the pathogenesis of EMT,
as well as to depict detailed molecular pathways that are involved
in EMT and kidney fibrosis following chronic renal injury.
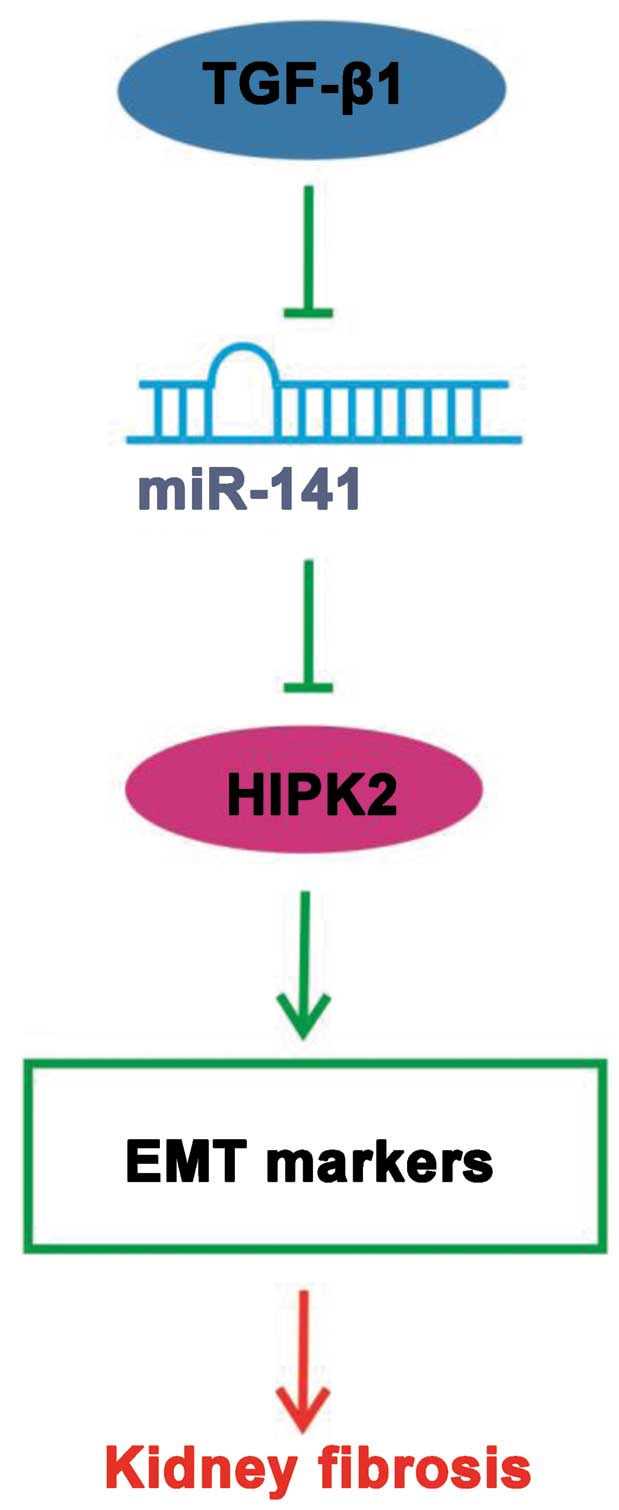
In conclusion, the present results indicate that
miR-141 coordinately regulates the TGF-β1-induced EMT
process through the HIPK2 signaling pathway (Fig. 6) and exogenous overexpression of
miR-141 may represent a promising approach for targeted
kidney fibriosis therapies.
Abbreviations:
|
EMT
|
epithelial mesenchymtion
transition
|
|
HIPK2
|
homeodomain-interacting protein
kinase 2
|
|
miRNA
|
microRNA
|
|
TGF-β
|
transforming growth factor-β
|
|
HK-2
|
human kidney 2
|
|
FSP1
|
fibroblast-specific protein 1
|
|
UTR
|
untranslated region
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
ORF
|
open reading frame
|
References
|
1
|
Liu Y: Epithelial to mesenchymal
transition in renal fibrogenesis: pathologic significance,
molecular mechanism, and therapeutic intervention. J Am Soc
Nephrol. 15:1–12. 2004. View Article : Google Scholar
|
|
2
|
Zeisberg M and Kalluri R: The role of
epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med
(Berl). 82:175–181. 2004. View Article : Google Scholar
|
|
3
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strutz F, Okada H, Lo CW, Danoff T, Carone
RL, Tomaszewski JE and Neilson EG: Identification and
characterization of a fibroblast marker: FSP1. J Cell Biol.
130:393–405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Strutz F and Müller GA: Renal fibrosis and
the origin of the renal fibroblast. Nephrol Dial Transplant.
21:3368–3370. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar
|
|
7
|
Li Y, Yang J, Luo JH, Dedhar S and Liu Y:
Tubular epithelial cell dedifferentiation is driven by the
helix-loop-helix transcriptional inhibitor id1. J Am Soc Nephrol.
18:449–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan R, Zhang J, Tan X, Zhang X, Yang J and
Liu Y: Downregulation of SnoN expression in obstructive nephropathy
is mediated by an enhanced ubiquitin-dependent degradation. J Am
Soc Nephrol. 17:2781–2791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang J, Shultz RW, Mars WM, Wegner RE, Li
Y, Dai C, Nejak K and Liu Y: Disruption of tissue-type plasminogen
activator gene in mice reduces renal interstitial fibrosis in
obstructive nephropathy. J Clin Invest. 110:1525–1538. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Zhang X, Li Y and Liu Y:
Downregulation of smad transcriptional corepressors snon and ski in
the fibrotic kidney: an amplification mechanism for TGF-beta1
signaling. J Am Soc Nephrol. 14:3167–3177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lan HY: Diverse roles of TGF-beta/smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067. 2011.
View Article : Google Scholar
|
|
12
|
Meng XM, Chung AC and Lan HY: Role of the
TGF-beta/BMP-7/smad pathways in renal diseases. Clin Sci (Lond).
124:243–254. 2013. View Article : Google Scholar
|
|
13
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Branton MH and Kopp JB: TGF-beta and
fibrosis. Microbes Infect. 1:1349–1365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patel V and Noureddine L: MicroRNAs and
fibrosis. Curr Opin Nephrol Hypertens. 21:410–416. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wienholds E, Kloosterman WP, Miska E,
Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen
S and Plasterk RH: MicroRNA expression in zebrafish embryonic
development. Science. 309:310–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yi R, O’Carroll D, Pasolli HA, Zhang Z,
Dietrich FS, Tarakhovsky A and Fuchs E: Morphogenesis in skin is
governed by discrete sets of differentially expressed microRNAs.
Nat Genet. 38:356–362. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev. Cancer. 6:259–269.
2006.
|
|
20
|
Zarjou A, Yang S, Abraham E, Agarwal A and
Liu G: Identification of a microRNA signature in renal fibrosis:
role of miR-21. Am J Physiol Renal Physiol. 301:F793–F801. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong M, Jiang L, Zhou Y, Qiu W, Fang L,
Tan R, Wen P and Yang J: The miR-200 family regulates
TGF-beta1-induced renal tubular epithelial to mesenchymal
transition through smad pathway by targeting ZEB1 and ZEB2
expression. Am J Physiol Renal Physiol. 302:F369–F379. 2012.
View Article : Google Scholar
|
|
22
|
Wang B, Komers R, Carew R, Winbanks CE, Xu
B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K,
Gregorevic P, Cooper ME and Kantharidis P: Suppression of
microRNA-29 expression by TGF-beta1 promotes collagen expression
and renal fibrosis. J Am Soc Nephrol. 23:252–265. 2012. View Article : Google Scholar :
|
|
23
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Calzado MA, Renner F, Roscic A and Schmitz
ML: HIPK2: a versatile switchboard regulating the transcription
machinery and cell death. Cell Cycle. 6:139–143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee W, rews BC, Faust M, Walldorf U and
Verheyen EM: Hipk is an essential protein that promotes notch
signal transduction in the drosophila eye by inhibition of the
global co-repressor groucho. Dev Biol. 325:263–272. 2009.
View Article : Google Scholar
|
|
26
|
Lee W, Swarup S, Chen J, Ishitani T and
Verheyen EM: Homeodomain-interacting protein kinases (Hipks)
promote Wnt/Wg signaling through stabilization of beta-catenin/Arm
and stimulation of target gene expression. Development.
136:241–251. 2009. View Article : Google Scholar
|
|
27
|
Zhang J, Pho V, Bonasera SJ, Holtzman J,
Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH and Huang EJ:
Essential function of HIPK2 in TGFbeta-dependent survival of
midbrain dopamine neurons. Nat Neurosci. 10:77–86. 2007. View Article : Google Scholar
|
|
28
|
D’Orazi G, Cecchinelli B, Bruno T, Manni
I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal
G, Piaggio G, Fanciulli M, Appella E and Soddu S:
Homeodomain-interacting protein kinase-2 phosphorylates p53 at ser
46 and mediates apoptosis. Nat Cell Biol. 4:11–19. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hofmann TG, Möller A, Sirma H, Zentgraf H,
Taya Y, Dröge W, Will H and Schmitz ML: Regulation of p53 activity
by its interaction with homeodomain-interacting protein kinase-2.
Nat Cell Biol. 4:1–10. 2002. View
Article : Google Scholar
|
|
30
|
Rinaldo C, Prodosmo A, Siepi F and Soddu
S: HIPK2: a multitalented partner for transcription factors in DNA
damage response and development. Biochem Cell Biol. 85:411–418.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin Y, Ratnam K, Chuang PY, Fan Y, Zhong
Y, Dai Y, Mazloom AR, Chen EY, D’Agati V, Xiong H, Ross MJ, Chen N,
Ma’ayan A and He JC: A systems approach identifies HIPK2 as a key
regulator of kidney fibrosis. Nat Med. 18:580–588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ricci A, Cherubini E, Ulivieri A, Lavra L,
Sciacchitano S, Scozzi D, Mancini R, Ciliberto G, Bartolazzi A,
Bruno P, Graziano P and Mariotta S: Homeodomain-interacting protein
kinase2 in human idiopathic pulmonary fibrosis. J Cell Physiol.
228:235–241. 2013. View Article : Google Scholar
|
|
33
|
Wang B, Koh P, Winbanks C, Coughlan MT,
McClelland A, Watson A, Jandeleit-Dahm K, Burns WC, Thomas MC,
Cooper ME and Kantharidis P: miR-200a prevents renal fibrogenesis
through repression of TGF-beta2 expression. Diabetes. 60:280–287.
2011. View Article : Google Scholar :
|
|
34
|
Tamagawa S, Beder LB, Hotomi M, Gunduz M,
Yata K, Grenman R and Yamanaka N: Role of miR-200c/mir-141 in the
regulation of epithelial-mesenchymal transition and migration in
head and neck squamous cell carcinoma. Int J Mol Med. 33:879–886.
2014.PubMed/NCBI
|
|
35
|
Wallace K, Burt AD and Wright MC: Liver
fibrosis. Biochem J. 411:1–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wynn TA: Integrating mechanisms of
pulmonary fibrosis. J Exp Med. 208:1339–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang J and Liu Y: Dissection of key events
in tubular epithelial to myofibroblast transition and its
implications in renal interstitial fibrosis. Am J Pathol.
159:1465–1475. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The mir-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hill C, Flyvbjerg A, Gronbaek H, Petrik J,
Hill DJ, Thomas CR, Sheppard MC and Logan A: The renal expression
of transforming growth factor-beta isoforms and their receptors in
acute and chronic experimental diabetes in rats. Endocrinology.
141:1196–1208. 2000.PubMed/NCBI
|
|
40
|
Chalazonitis A, Tang AA, Shang Y, Pham TD,
Hsieh I, Setlik W, Gershon MD and Huang EJ: Homeodomain interacting
protein kinase 2 regulates postnatal development of enteric
dopaminergic neurons and glia via BMP signaling. J Neurosci.
31:13746–13757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harada J, Kokura K, Kanei-Ishii C, Nomura
T, Khan MM, Kim Y and Ishii S: Requirement of the co-repressor
homeodomain-interacting protein kinase 2 for ski-mediated
inhibition of bone morphogenetic protein-induced transcriptional
activation. J Biol Chem. 278:38998–39005. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hofmann TG, Stollberg N, Schmit ML and
Will H: HIPK2 regulates transforming growth factor-beta-induced
c-jun NH (2)-terminal kinase activation and apoptosis in human
hepatoma cells. Cancer Res. 63:8271–8277. 2003.PubMed/NCBI
|