Introduction
The majority of colorectal cancer (CRC) cases
annually diagnosed occur due to sporadic events; however, up to 6%
are attributed to known monogenic disorders. To date, an
etiological association with CRC has been demonstrated for three
hereditary syndromes: Familial adenomatous polyposis (FAP) syndrome
(1), MYH-associated
polyposis (MAP) syndrome (2) and
hereditary nonpolyposis colorectal cancer (HNPCC) syndrome or Lynch
syndrome (LS). LS is the most common inherited CRC type and is
associated with mutations in DNA mismatch repair (MMR) genes
(3), mainly MLH1 and
MSH2 but also MSH6 and PMS2. A germline point
mutation in MSH3 was found to be associated with the LS
phenotype (4). Besides CRC, the
spectrum of LS encompasses other primary tumor types (5). The Amsterdam criteria were the first
diagnostic guidelines designed to identify families affected by LS
(6). As the Amsterdam criteria
were rated as being too stringent and not sufficiently sensitive,
the Bethesda guidelines were subsequently developed to improve the
identification of patients eligible for genetic testing (7).
Loss of MMR gene function manifests as high levels
of microsatellite instability (MSI-H) that occurs in >90% of all
LS carcinomas (7). The MSI-H
status has also been described in sporadic CRC associated with
BRAF gene mutation, namely the c.1799T>A (p.V600E)
mutation. This mutation is not present in LS-associated cancers
(8). Therefore, BRAF
mutation testing has been proposed as a means to exclude sporadic
MSI CRC cases from germline MMR gene testing (9).
The present study assessed the microsatellite
instability (MSI) status and BRAF V600E mutations in DNA
extracted from tumor tissues of patients selected according to
revised Bethesda guidelines. Hence, MLH1 and MSH2
genes were screened for germline mutations in patients at risk for
LS. By using approaches of previous studies, the present study
identified LS patients carrying germline mutations in these genes,
of which two mutations were novel. Using a combination of
computational approaches, co-segregation analysis and RNA assay, a
likely pathogenicity of these novel MLH1 mutations was
identified in the present study.
Patients and methods
Patients
The patients were recruited from several hospitals
(AOU Federico II and IRCS Pascale of Naples, AOU SUN of Caserta) in
Campania (southern Italy). Thirty-nine subjects with CRC were
selected according to Bethesda guidelines (7). All patients selected for the present
study belonged to families that did not completely fulfill the
Amsterdam criteria but in which multiple members were affected by
LS-associated cancer. Moreover, colon cancer was diagnosed in
almost all patients at <50 years of age with preferential
localization at the ascending (right) colon. Furthermore, as
negative controls, 100 samples from healthy patients were collected
from the Clinical Department of Laboratory Medicine of the hospital
affiliated to ‘Federico II’ university (Naples, Italy).
Samples from all subjects were collected after being
granted authorization from the local ethics committee ‘Comitato
etico per le attività Biomediche-Carlo Romano’ of the University of
Naples ‘Federico II’ (protocol no. 120/10). Once the authorization
was obtained, the study received ethical approval, and
participants’ informed and written consent was obtained. For each
patient, experiments were performed on DNA extracted from
peripheral blood lymphocytes and from paraffin-embedded tumor
tissues. For the healthy samples, the DNA was extracted only from
peripheral blood lymphocytes.
Isolation of genomic DNA
Total genomic DNA was extracted from 4 ml peripheral
blood lymphocytes using a BACC2 Nucleon kit (Amersham Pharmacia
Biotech, Amersham, UK). For each paraffin block, five 20-µm
sections were cut and collected in a 1.5-ml microtube. Briefly, 1
ml xylene was added to each tube followed by incubation at room
temperature for 20 min to completely remove the paraffin. The tubes
were then centrifuged at 15,000 rpm for 2 min and the supernatant
was discarded. The pellet was re-hydrated with a descendent
gradient series of ethanol (500 µl pure ethanol, 500
µl 90% ethanol, 500 µl 80% ethanol and 10% ethanol).
The tissue pellet was re-suspended in 1 ml distilled water for 30
min at room temperature. Subsequently, the DNA was extracted using
a BACC2 Nucleon kit (Amersham Pharmacia Biotech).
DNA amplification and microsatellite
analysis
MSI was tested on paired samples of lymphocyte DNA
and in paraffin-embedded tumor tissues of the colon. The MSI status
was evaluated with a fluorescent multiplex system comprising five
mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and NR-27),
three dinucleotide repeats (D2S123, D5S346 and D17S250) and two
tetranucleotide repeats using the CC-MSI kit (AB ANALITICA, Padova,
Italy) and subsequent capillary electrophoresis analysis using an
ABI 3130 Prism (Applied Biosystems, Fisher Thermo Scientific,
Waltham, MA, USA). Tumors were classified as ‘highly unstable’
(MSI-H), if at least 30% of the markers showed instabilities and
‘with low levels of instability’ (MSI-L), if at least 10% of the
markers showed instabilities; if no allele difference between DNA
extracted from normal and tumorous tissues was observed, tumors
were classified as microsatellite stable (MSS) (7).
BRAF V600E mutation analysis
For BRAF V600E genotyping, genomic DNA
extracted from paraffin-embedded and blood lymphocytes from
patients with MSI-H and MSI-L tumors were amplified using a
customized primer pair (forward primer, exon 15,
5′-TGCTTGCTCTGATAGGAAAATG AGA-3′ and reverse primer, exon 15,
5′-CTCAGCAGCA TCTCAGGGCC-3′). PCR reactions were performed in a
total volume of 50 µl containing 5 µl of 10X PCR
buffer, 200 µM of each dNTP, 25 pM of each primer, 1.5 mM of
MgCl2, 2 U of FastStart Taq DNA polymerase (Roche,
Basel, Switzerland) and 100 ng of genomic DNA. PCR conditions were
as follows: 95°C for 4 min, 35 cycles with 95°C for 30 sec, 60°C
for 30 sec and 72°C for 45 sec, followed by a final extension step
at 72°C for 7 min. PCR prodoucts were sequenced in forward and
reverse directions using an ABI 3100 Genetic Analyser (Applied
Biosystems Inc., Foster City, CA, USA).
Mutation analysis: Amplification,
denaturing high-performance liquid chrmoatography (dHPLC) and
sequencing
All MLH1 and MSH2 exons were
amplified, including intronexon boundaries, on DNA extracted from
blood lymphocytes of patients with MSI-H or MSI-L tumors, using
customized primer sets. Prior to dHPLC analysis, the polymerase
chain reaction (PCR) products were separated on a 1–2% agarose gel
to check for unspecific amplicons. A Transgenomic Wave DNA Fragment
Analysis system (3500 HT; Transgenomic, Inc., Omaha, NE, USA) was
used to perform dHPLC analysis. Abnormal HPLC chromatograms were
identified by visual inspection on the basis of the appearance of
one or more additional peaks with a lower retention time. For all
samples exhibiting abnormal dHPLC profiles, genomic DNA was
re-amplified and sequenced in the forward and reverse directions
using an ABI 3100 Genetic Analyser (Applied Biosystems).
In silico analysis
Structural analysis of missense point mutations is
important to understand the functional activity of the mutated
protein. The present study used three complementary algorithms for
functional impact prediction of novel missense variants: Sorting
Intolerant From Tolerant (SIFT) (http://blocks.fhcrc.org/sift/SIFT.html) (10), Polymorphism Phenotyping (PolyPhen)
(http://genetics.bwh.harvard.edu/pph/)
(11) and PredictProtein server
(http://www.predict-protein.org)
(12). Predictions were based on
a combination of phylogenetic, structural and sequence annotation
information characterizing a substitution with its position in the
protein. In addition, the silent novel variant discovered in the
present study was analyzed using the Human Splicing Finder (HSF)
software (http://www.umd.be/HSF/) (13), a tool designed to predict the
effects of mutations on splicing signals or to identify splicing
motifs in human sequences. It contains all available matrices for
auxiliary sequence prediction and also presents a novel position
weight matrix to assess the strength of 5′ and 3′ splice sites and
branch points.
Reverse transcription PCR and
quantitative (real-time) PCR (qPCR) of MLH1 cDNA
Total RNA was extracted from lymphocytes of the
patient carrying the c.438A>G mutation in the MLH1 gene
and from five normal controls using TRIzol reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA). cDNA was synthesized using 1
µg total RNA, 500 ng random hexamers and 1 µl
SuperScript III reverse transcriptase (Invitrogen Life
Technologies), in the presence of 4 µl 5X RT buffer, 1
µl dithiothreitol (0.1 M) and 1 mM deoxynucleotide
triphosphates (Invitrogen Life Technologies). The reaction was run
on a PCR thermocycler for 50 min at 42°C in a 20-µl reaction
volume, heated to 70°C for 15 min and subsequently chilled on ice.
PCR amplification reactions of the entire MLH1 cDNA were
performed using a customized primer pair (1F
5′-ACGTTTCCTTGGCTCTTCTG-3′ and 19R 5′-AATC
AATCCACTGTGTATAAAGGAA-3′). Amplified fragments were visualized on
an 8% polyacrylamide gel. Each band was excised from the gel and
re-suspended in 30 µl water overnight. Then, 1 µl was
re-amplified and subsequently sequenced using the same primer pair.
Next, the relative expression of the cDNA of the patient vs. that
of the wild-type controls (10 healthy samples) was evaluated by
qPCR based on SYBR-Green fluorescence on a CFX96 Real Time System
instrument from Bio-Rad Laboratories, Inc. (Hercules, CA, USA).
Three pairs of forward and reverse primers for MLH1 cDNA
quantification were used which amplified fragments spanning between
exons 3–5, 4–5 and 13–14 (Table
I). The β-glucuronidase gene (GUS) was used as
housekeeping gene for normalization. The PCR cycling conditions
were as follows: 3 min at 95°C followed by 40 cycles at 95°C for 15
sec, 60°C for 30 sec and 72°C for 20 sec without final elongation.
The specificity of qPCR products was evaluated by melting curve
analysis and by visualization on 2.5% agarose gels containing
ethidium bromide on a shortwave UV radiation transilluminator. To
evaluate qPCR efficiencies, a 10-fold serially diluted cDNA was
used for each amplicon, and the slope values given by the
instrument were used in the following formula: Efficiency =
[10(1/slope)]−1. All primer sets had efficiencies of
100±10%. Each experiment was performed in triplicate.
 | Table IPrimer sequences and sizes of
amplification fragments for MLH1 mRNA quantification. |
Table I
Primer sequences and sizes of
amplification fragments for MLH1 mRNA quantification.
| Primer
name/specificity | Primer sequences
(5′→3′) | Amplification
fragment size (bp) |
|---|
| cMLH1
forward primer, exon 3 |
CCAGTATTTCTACCTATGGCTTTCGACGTG | 198 |
| cMLH1
reverse primer, exon 5I |
GGTTTAGGAGGGGCTTTCAG | |
| cMLH1
forward primer, exon 4 |
AACGAAAACAGCTGATGGAA | 103 |
| cMLH1
reverse primer, exon 5II |
GATCTGGGTCCCTTGATTGC | |
| cMLH1
forward primer exon 13 |
GCAGGGACATGAGGTTCTCC | 169 |
| cMLH1
reverse primer, exon 14 |
GCTTGGTGGTGTTGAGAAGG | |
| GUS forward
primer |
GAAAATATGTGGGTTGGAGAGCTCATT | 120 |
| GUS reverse
primer |
CCGAGTGAAGATCCCCTTTTTA | |
Relative expression was calculated using the
comparative Ct method and normalized against the Ct of GUS
mRNA to acquire and analyze data, as previously described (14). The qPCR assays were performed
using the CFX Manager Software (version 2.1; Bio-Rad Laboratories,
Inc.) and were compared with the corresponding values from an
average of 10 samples of healthy controls to calculate the relative
expression.
Results
MSI analysis and BRAF V600E mutation
detection
The present study performed MSI analysis on 39
unrelated index cases with CRC that fulfilled the revised Bethesda
guidelines (5). The MSI-H status
was identified in 15/39 DNA samples extracted from tumor tissues of
patients, while the MSI-L status was identified in 11/39 patients;
tumors of 13 patients were free of MSI (MSS). V600E genotyping was
performed in 26 patients classified as MSI-H and MSI-L, and no
heterozygous or homozygous patients were observed.
Mutation analysis
All MLH1 and MSH2 exons were analyzed
in DNA extracted from 26 patients with MSI-H and MSI-L tumors. As
shown in Table II, six germline
mutations were identified in the MLH1 gene and five in the
MSH2 gene. Overall, 11 germline mutations were identified in
these genes in 12/26 patients; two of which were novel mutations
that have not previously been reported in the NCBI SNP
database, the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), the
International Society for Gastrointestinal Hereditary Tumours group
(InSight; http://www.insight-group.org/) or the MMR variants
database (15). The two novel DNA
variants (c.438A>G and c.1844T>C in the MLH1 gene)
were not detected in the 100 healthy controls. To verify the
pathogenicity of these novel variants, in silico analysis
was performed with software used in previous studies (10–13). The results are shown in Table III. In silico analysis
performed using the HSF software showed that the silent mutation,
c.438A>G of the MLH1 gene, occurs in a region involved in
the splicing process. PCR analysis of the entire MLH1 cDNA
showed an absence of amplification product corresponding to the
wild-type MLH1 cDNA (Fig.
1A). No abnormal aberrant splicing was identified in this
patient; however, PCR analysis showed different amplification
products between the patient and the healthy controls. Each
amplification product visualized on the gel was extracted and
sequenced, and the bands revealed the several splicing isoforms of
MLH1 mRNA, as described in a previous study (16). A qPCR experiment was then
performed in order to quantitatively assess the MLH1 mRNA
expression. Three regions of the MLH1 cDNA (fragments
spanning between exons 3–5, 4–5 and 13–14) were amplified using the
GUS gene as a reference (Fig. 1B and
C). In the patient examined, a quantitative alteration of the
MLH1 cDNA was found. In particular, transcripts including
exons 3–5 and 4–5, where the mutation occurred, were less
quantitatively expressed compared to those in the healthy control
samples (Fig. 1C). These results
indicated that this mutation prevents the formation of the
full-length MLH1 cDNA but not that of the alternative
splicing isoforms that are missing in certain exons.
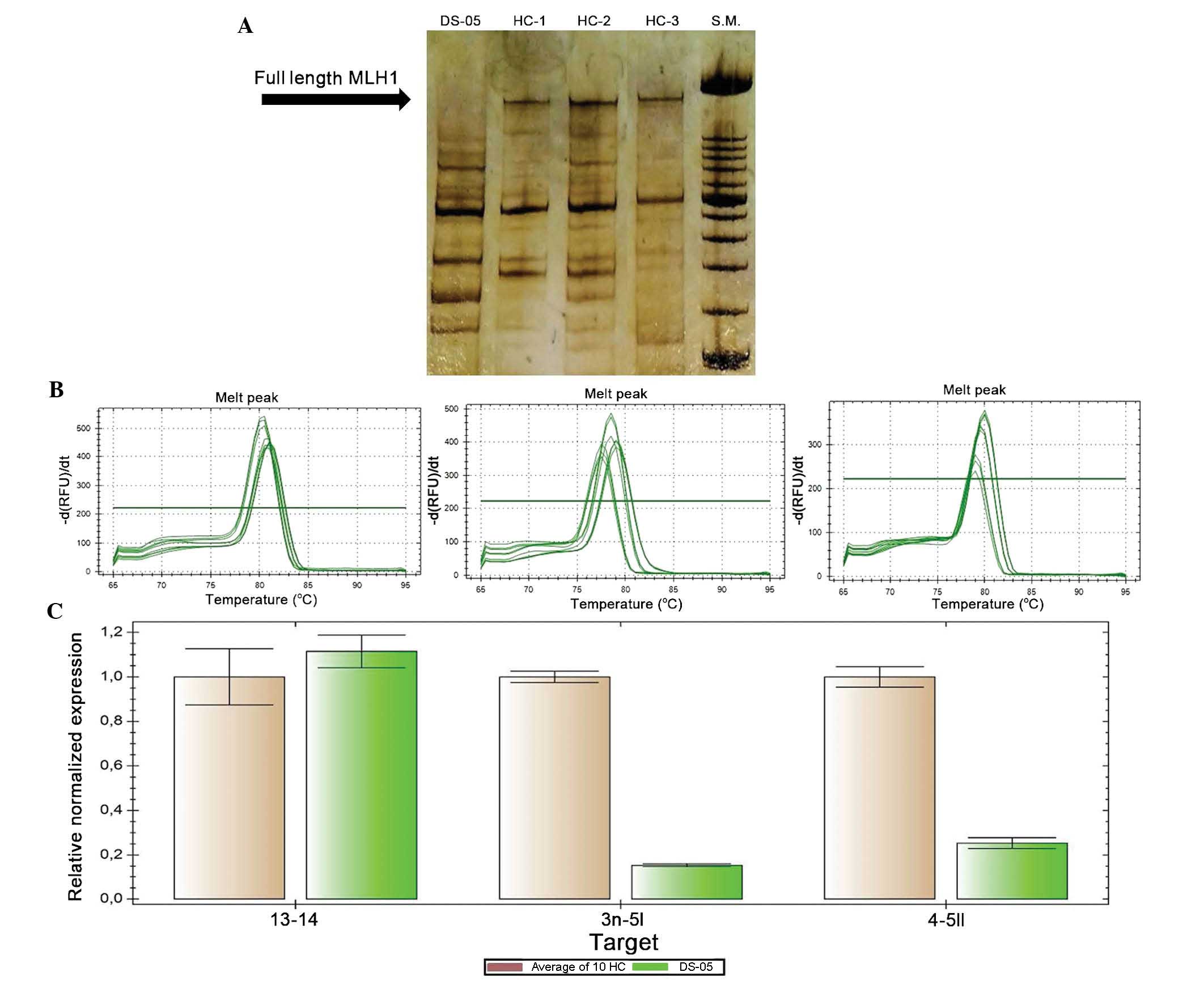 | Figure 1MLH1 cDNA analysis of a
patient carrying novel silent variant, c.438A>G and of three
healthy controls. (A) Detection of the PCR MLH1 cDNA results
on 8% polyacrylamide gel; no abnormal aberrant splicing is shown;
arrow indicates amplification product corresponding to full-length
MLH1 cDNA; the other amplification products corresponding to
alternative splicing isoforms are visible on the gel. (B) Melting
curve analysis of quantitative real-time polymerase chain reaction
amplification products corresponding to exons 13–14, 3–5 and 4–5,
respectively, of MLH1 cDNA. (C) Relative expression,
calculated using the comparative Ct method, of MLH1 cDNA,
including fragments 13–14, 3n–5, and 4–5 II normalized to
β-glucuronidase levels, in an average of 10 HC and in a patient.
Values are expressed as the mean ± standard deviation. S.M., size
marker XIV; HC, healthy control; DS-05, patient. |
| Table IISequence variations evaluated by
DHPLC and sequencing analysis in patients with MSI-H and MSI-L. |
Table II
Sequence variations evaluated by
DHPLC and sequencing analysis in patients with MSI-H and MSI-L.
| Patient ID | Exon of gene | Nucleotide
change | Amino acid
change |
Authors/(Refs.)a | MSI phenotype | Clinical
phenotype |
|---|
| 00–13 | 3 of
hMLH1 | c.304 G>A |
p.[Glu101Valfs*14,
Glu102Lys] | Ellison et
al 2001 | MSI-H | Cancer of small
intestine diagnosed at age 43; mother presented a colon polyp at
age 73, MUT+. |
| DS-05 | 5 of
hMLH1 | c.438 A>G | p=Gln146 | Present
studyb | MSI-H | Cancer of the
ascending colon at age 44 and kidney cancer at age 48; sister died
of colon cancer at age 53. |
| 01–04 | 12 of
hMLH1 | c.1321 G>A | Ala441Thr | Tannergard et
al 1995 | MSI-H | Cancer of the
ascending colon at age 44; maternal uncle died of rectal cancer at
age 57, MUT+. |
| 08–01 | 16 of
hMLH1 | c.1844 T>C | Leu615Pro | Present
studyb | MSI-H | Rectal cancer
diagnosed at age 44; mother with stomach cancer diagnosed at age
75, MUT+; daughter and son with adenocarcinoma and adenoma with
severe dysplasia of the ascending colon diagnosed at age 32 and 35,
respectively (both are MUT+). |
| 09–08 | 19 of
hMLH1 | 30_32 delTTC,
(3′UTR) | | Viel et al
1997 | MSI-H | Cancer of the
ascending colon at age 45; father with kidney cancer diagnosed at
age 60, MUT+. |
| 14–35 | 14 of
hMSH2 | c.2251G>C | p.Gly751Arg | De Lellis et
al 2013 | MSI-H | Cancer of the
ascending colon diagnosed at age 36; no family history (adopted
subject). |
| 10–04 | 3 of
hMSH2 | c.435T>G | p.Ile145Met | Kariola et
al 2003 | MSI-H | Cancer of the
ascending colon at age 35; maternal grandmother died of rectal
cancer at age 77. |
| 03–13 | 5 of
hMSH2 | c.942+3 A>T | p.Val265_Gln31
4del | Wijnen et al
1997 | MSI-H | First subject:
cancer of the ascending colon diagnosed at age 23; no reported
family history. Second subject: rectal cancer and polyp on the
ascending colon diagnosed at age 29; maternal uncle stomach
cancer. |
| 11–25 | 6 of
hMSH2 | c.984C>T | p=Ala328 | Curia et al
1999 | MSI-L | Tubular-adenoma
with severe dysplasia of the ascending colon diagnosed at age 58
and prostate cancer diagnosed at age 68; sister with adenocarcinoma
of the ascending colon diagnosed at age 49; daughter with
endometrial cancer diagnosed at age 35. |
| Table IIIIn silico analysis of the
exonic variants in the MLH1 gene. |
Table III
In silico analysis of the
exonic variants in the MLH1 gene.
| Mutation | PolyPhen
prediction | SIFT
prediction | PredictProtein
prediction | HSF prediction |
|---|
| c.438A>G | ND | ND | ND | 5′ss ΔCV
(c.437_445)=−322.52
3′ss ΔCV ×2 (c.419_423)=−157.19
(c.420_424)=−1654.17
+SF2/ASFa (c.436_442)
−ESEb (c.434_439)
−EIEc ×3 (c.434_439)
(c.435_440)
(c.437_442)
+9G8d (c.438_443)
−ESSe (c.436_443)
+IIEf (c.438_443)
−hnRNPA1d (c.437_443)
−ESRg (c.434_439) |
| c.1844T>C | Probably
damaged | Damaging | Strong signal for
mutation effect | ND |
Computational analysis performed for the novel
missense mutation, c.1844T>C in the MLH1 gene using
PolyPhen, SIFT and PredictProtein software (Table III) showed that the consequent
change in the amino acid (Leu615Pro) probably had a damaging effect
on protein function. Moreover, a clear familial segregation of this
mutation was observed for the disease.
The other nine mutations identified in the present
study have been previously reported in a mutation database
(InSight) (14) as pathogenetic
or unclassified variants (UVs) of the MLH1 and MSH2
genes.
Germline mutations in MLH1 and MSH2
genes were detected in 11/15 patients with MSI-H tumors (73%) and
in 1/11 patients with MSI-L tumors (9%).
In Table II, the
identified germline mutations, the MSI status of patients’ tumors
and clinical phenotypes of each subject carrying mutations in
MLH1 or MSH2 genes are listed.
Discussion
The present study was performed on a cohort of 39
subjects with a diagnosis of CRC at an early age and with a
familial background of LS. For all patients that fulfilled the
revised Bethesda guidelines, an MSI analysis was performed using
DNA extracted from tumorous tissues. Twenty-six of these patients
had an MSI-H or MSI-L status, while the remaining 13 patients
showed no MSI. Thus, the 26 patients with MSI-H and MSI-L underwent
MMR germline testing. The 13 remaining subjects with negative LS
diagnosis were excluded from these experiments; however, given the
selection criteria for enrolment in the present study, these cases
are not to be considered sporadic CRC cases, as they were likely to
have genetic causes. Recently, it has been described that other
Mendelian syndromes with autosomal-dominant inheritance patterns,
including the phosphatase and tensin homolog (PTEN) Hamartoma Tumor
Syndrome (PHTS), show an overlapping clinical presentation with LS,
but tumors do not show any MSI (17). In line with this, in previous
studies by our group, one patient with MSS status of tumorous
tissue, who underwent germline testing for the PTEN gene, showed a
germline mutation in this gene (18), which was associated with the
disease in the family (19).
Alternatively to the PHTS syndrome, an alteration of inflammatory
pathways associated with a dysregulation of cell proliferation
pathways (such as WNT/β-catenin) in colon mucosa and which may also
be inherited in a Mendelian manner (20,21), may have been the underlying cause
in the MSS-status CRC cases in the present study. Therefore, for
CRC cases without MSI but with a family history of LS, other
genetic factors should be considered for making an accurate
differential diagnosis of LS.
In the present study, V600E genotyping was performed
on DNA extracted from tumorous tissues with MSI-H or MSI-L status
(26/39); as expected, in none of these, the mutation of the
BRAF gene was detected. An MMR gene mutation was
identified in 12/26 selected cases, namely in 11/15 patients with
MSI-H tumors and in 1/11 patients with MSI-L tumors; therefore, the
mutation detection rate was 46%. The mutation detection rate was
significantly higher (73%) if only MSI-H cases were considered.
Although no point mutations were detected in the main MMR
genes (MLH1/MSH2), in the remaining 14 patients with MSI-H
or MSI-L tumors, the causes of the disease were likely to be other
types of mutation, including re-arrangements, deletions or
duplications in these same genes (22) or mutations in other MMR
genes (PMS2, MSH6 and MLH3) (23,24), which were not detectable in the
present study.
The present study identified 11 germline mutations
in 12 patients; whenever possible, the familial segregation of the
mutation with the disease was confirmed (Table II). Two of these germline
variants were novel mutations in the MLH1 gene that were not
found in the control population panel of 100 healthy blood donors.
Computational analysis was used to evaluate the putative functional
effects of these two novel sequence variants. PolyPhen, SIFT and
PredictProtein software were used for the missense variant and HSF
software for the silent variant identified in the MLH1 gene.
This software is commonly used to study unclassified variants (UVs)
found in patients with LS (25).
The novel mutation c.438A>G in exon 5 of the
MLH1 gene was identified in a patient who had developed two
primitive malignancies and showed an MSI-H status. This was a
silent variant for which the HSF analysis showed a possible
negative effect on the splicing process. In human disease genes,
there are several mutations in exonic splicing enhancer control
sequences that have been shown to cause aberrant exon skipping
(2,26). However, no abnormal aberrant
splicing of MLH1 mRNA was found in this patient (no. DS-05),
but PCR analysis of the entire MLH1 cDNA showed an absence
of amplification product corresponding to wild-type cDNA compared
to healthy controls. Furthermore, qPCR analysis detected a
significant reduction in MLH1 mRNA expression in tissue from
patient no. DS-05, who carried the novel mutations, or rather in
transcript fragments that included the exon 5. Although the
mechanism of splicing site selection may also significantly differ
depending on individual or tissue-specific differences (27), the silent mutation may have
altered the normal splicing process, preventing the formation of
full-length MLH1 cDNA. This may explain why the PCR analysis
of the entire MLH1 cDNA showed an apparent increase of the
alternative splicing isoforms compared to those in the wild-type
cDNA. Therefore, the sole formation of alternative splicing
isoforms of the MLH1 gene may have prevented the synthesis
of a functional protein and, consequently, determine the mutator
phenotype (MSI-H). In the present study, it was not possible to
assess the segregation of this variant with the disease in the
family of the DS-05 patient. However, in light of the results of
the present study and as this silent mutation was not identified in
the 100 healthy control subjects, it is indicated that this variant
is likely to be pathogenetic.
In the present study, the missense mutation
c.1844T>C in exon 16 of the MLH1 gene was identified in a
patient with rectal cancer. This mutation was identified also in
the mother of this proband, who developed stomach cancer at age 75.
The mutation c.1844T>C was in the highly conserved region of the
MLH1 protein and caused an amino acid change from leucine to
proline. In silico analysis by PolyPhen, SIFT and
PredictProtein software showed that this mutation caused severe
damage to the protein functionality. For this mutation, familial
segregation with the disease was also observed. Therefore, this
mutation was considered as pathogenetic.
The relatives of the two patients with the novel
gene mutations (DS-05 and 08-01) are recommended to undergo
pre-symptomatic genetic testing.
Finally, it is of note that in the present study,
all germline mutations identified in the MLH1 and
MSH2 genes were missense or splicing mutations, and no
truncating mutation was identified. Due to their nature, these
mutations may lead to variations in the phenotypic expression of
the disease alleles; indeed, the patients of the present study had
a familial background of atypical LS.
In conclusion, the findings of the present study
broadened the spectrum of known mutations of the MLH1 gene
and reaffirmed that the combination of MSI testing and V600E
genotyping for the BRAF gene associated with clinical
features, including familial clustering of LS-associated tumors and
early age of onset, are relevant predictors to identify LS
patients.
Identifying pathogenic mutations in these families
will greatly facilitate pre-symptomatic diagnosis and genetic
counseling, making better therapeutic decisions for carriers prior
to disease manifestation.
Acknowledgments
The present study was supported by the agreement
2010–2012 between CEINGE and Campania Regional Authority; POR
Campania fSe2007–2013.
References
|
1
|
De Rosa M, Dourisboure RJ, Morelli G,
Graziano A, Gutiérrez A, Thibodeau S, Halling K, Avila KC, Duraturo
F, Podesta EJ, et al: First genotype characterization of
Argentinean FAP patients: Identification of 14 novel APC mutations.
Hum Mutat. 23:523–524. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De Rosa M, Galatola M, Borriello S,
Duraturo F, Masone S and Izzo P: Implication of adenomatous
polyposis coli and MUTYH mutations in familial colorectal
polyposis. Dis Colon Rectum. 52:268–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch PM: The hMSH2 and hMLH1 genes in
hereditary nonpolyposis colorectal cancer. Surg Oncol Clin N Am.
18:611–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with Lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar
|
|
5
|
Barrow E, Hill J and Evans DG: Cancer risk
in Lynch Syndrome. Fam Cancer. 12:229–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasen HF, Watson P, Mecklin JP and Lynch
HT: New clinical criteria for hereditary nonpolyposis colorectal
cancer (HNPCC, Lynch syndrome) proposed by the International
Collaborative group on HNPCC. Gastroenterology. 116:1453–1456.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wolf B, Gruber S, Henglmueller S, Kappel
S, Bergmann M, Wrba F and Karner-Hanusch J: Efficiency of the
revised Bethesda guidelines (2003) for the detection of mutations
in mismatch repair genes in Austrian HNPCC patients. Int J Cancer.
118:1465–1470. 2006. View Article : Google Scholar
|
|
8
|
Wang L, Cunningham JM, Winters JL,
Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK,
Schaid DJ and Thibodeau SN: BRAF mutations in colon cancer are not
likely attributable to defective DNA mismatch repair. Cancer Res.
63:5209–5212. 2003.PubMed/NCBI
|
|
9
|
Domingo E, Laiho P, Ollikainen M, Pinto M,
Wang L, French AJ, Westra J, Frebourg T, Espín E, Armengol M, et
al: BRAF screening as a low-cost effective strategy for simplifying
HNPCC genetic testing. J Med Genet. 41:664–668. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rost B, Yachdav G and Liu J: The
PredictProtein server. Nucleic Acids Res. 32:W321–W326. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human Splicing Finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Costabile V, Duraturo F, Delrio P, Rega D,
Pace U, Liccardo R, Rossi GB, Genesio R, Nitsch L, Izzo P, et al:
Lithium chloride induces mesenchymal to epithelial reverting
transition in primary colon cancer cell cultures. Int J Oncol.
46:1913–1923. 2015.PubMed/NCBI
|
|
15
|
Woods MO, Williams P, Careen A, Edwards L,
Bartlett S, McLaughlin JR and Younghusband HB: A new variant
database for mismatch repair genes associated with Lynch syndrome.
Hum Mutat. 28:669–673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Genuardi M, Viel A, Bonora D, Capozzi E,
Bellacosa A, Leonardi F, Valle R, Ventura A, Pedroni M, Boiocchi M,
et al: Characterization of MLH1 and MSH2 alternative splicing and
its relevance to molecular testing of colorectal cancer
susceptibility. Hum Genet. 102:15–20. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mester JL, Moore RA and Eng C: PTEN
germline mutations in patients initially tested for other
hereditary cancer syndromes: Would use of risk assessment tools
reduce genetic testing? Oncologist. 18:1083–1090. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galatola M, Paparo L, Duraturo F, Turano
M, Rossi GB, Izzo P and De Rosa M: Beta catenin and cytokine
pathway dysregulation in patients with manifestations of the ‘PTEN
hamartoma tumor syndrome’. BMC MedGenet. 13:282012.
|
|
19
|
Paparo L, Rossi GB, Delrio P, Rega D,
Duraturo F, Liccardo R, Debellis M, Izzo P and De Rosa M:
Differential expression of PTEN gene correlates with phenotypic
heterogeneity in three cases of patients showing clinical
manifestations of PTEN hamartoma tumour syndrome. Hered Cancer Clin
Pract. 11:82013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lynch HT and Lynch J: Lynch syndrome:
Genetics, natural history, genetic counseling, and prevention. J
Clin Oncol. 18(Suppl): 19S–31S. 2000.PubMed/NCBI
|
|
21
|
Galatola M, Miele E, Strisciuglio C,
Paparo L, Rega D, Delrio P, Duraturo F, Martinelli M, Rossi GB,
Staiano A, et al: Synergistic effect of interleukin-10-receptor
variants in a case of early-onset ulcerative colitis. World J
Gastroenterol. 19:8659–8670. 2013. View Article : Google Scholar :
|
|
22
|
Duraturo F, Cavallo A, Liccardo R, Cudia
B, De Rosa M, Diana G and Izzo P: Contribution of large genomic
rearrangements in Italian Lynch syndrome patients: characterization
of a novel alu-mediated deletion. Biomed Res Int. 2013:2198972013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rahner N, Friedrichs N, Wehner M, Steinke
V, Aretz S, Friedl W, Buettner R, Mangold E, Propping P and
Walldorf C: Nine novel pathogenic germline mutations in MLH1, MSH2,
MSH6 and PMS2 in families with Lynch syndrome. Acta Oncol.
46:763–769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lipkin SM, Wang V, Stoler DL, Anderson GR,
Kirsch I, Hadley D, Lynch HT and Collins FS: Germline and somatic
mutation analyses in the DNA mismatch repair gene MLH3: evidence
for somatic mutation in colorectal cancers. Hum Mutat. 17:389–396.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Doss CG and Sethumadhavan R: Investigation
on the role of nsSNPs in HNPCC genes - a bioinformatics approach. J
Biomed Sci. 16:422009. View Article : Google Scholar
|
|
26
|
Cartegni L, Chew SL and Krainer AR:
Listening to silence and understanding nonsense: exonic mutations
that affect splicing. Nat Rev Genet. 3:285–298. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
De Rosa M, Morelli G, Cesaro E, Duraturo
F, Turano M, Rossi GB, Delrio P and Izzo P: Alternative splicing
and nonsense-mediated mRNA decay in the regulation of a new
adenomatous polyposis coli transcript. Gene. 395:8–14. 2007.
View Article : Google Scholar : PubMed/NCBI
|














