Introduction
The DNA damage response (DDR), which is caused by
genotoxic stresses involves various cellular responses, such as the
activation of DNA damage signaling, cell cycle checkpoints, DNA
damage repair and cell death (1).
The DDR is regulated by concerted activities of the DNA damage
responses factors (DDRFs). The DDRFs function as DNA damage
sensors, transducers of signaling and effectors for the DDR
(2). For example, ataxia
telangiectasia mutated (ATM) and ATM-Rad3-related (ATR) initiate
DNA damage signaling upon the induction of DNA damage, and trigger
a cascade of downstream signaling pathways, which results in the
activation of transducing kinases, such as checkpoint kinase 1
(Chk1) and Chk2 (3). The
transducing kinases in turn relay the signals to effector
molecules, such as CDC25A to regulate the cell cycle checkpoint
(4,5).
Chk1 is an essential kinase in the DDR and serves as
an important checkpoint regulator in the S- and G2 checkpoints, as
well as in the mitotic spindle checkpoint (6). The domain structure of Chk1 shows a
highly conserved N-terminal kinase domain and a less conserved
C-terminal regulatory domain (7,8).
Activity and stability of Chk1 is regulated by its phosphorylation
following genotoxic stresses. ATR phosphorylates Chk1 at the highly
conserved serine (S)317 and S345 residues (8). The phosphorylation of S345 is
commonly used as a biomarker of Chk1 activation. Since the
Chk1-mediated checkpoints are crucial for the DDR, protein
stability is tightly regulated. The phosphorylation and subsequent
activation of Chk1 by ATR-mediated phosphorylation on S317 and S345
lead to conformational changes and the degradation of Chk1
(9). Chk1 is degraded via the
ubiquitin-mediated degradation pathway by E3 ubiquitin ligases that
employ CUL1 and CUL4A (9–11). As Chk1 activation and modulation
of its stability are tightly coupled, the understanding of the
processes may be crucial to target Chk1 for cancer therapy. Indeed,
considering the essential nature of Chk1 for cell survival, Chk1
has been studied as a promising target for cancer therapy (12,13). Since many cancer cells defective
in p53 require the Chk1-induced G2 checkpoint for their survival
following genotoxic treatment, the combination of genotoxic
stresses and Chk1 inhibitors enhances the efficacy of DNA damaging
agents (14,15). A similar effect has also been
observed by the combination of Chk1 knockdown and ionizing
radiation in p53-defective cancer cells (16). As the inhibition of Chk1 enhances
tumor cell killing by genotoxic stresses, a number of Chk1
inhibitors, including UCN-01 and AZD7762 have been investigated
(12).
In addition to chemical modulators of Chk1, we
reasoned that specific Chk1-binding peptides could be delivered
into the cells and modulate the activity of Chk1 and Chk1-regulated
responses. To achieve this goal, in this study, we screened a phage
display library and identified a specific Chk1-binding 12-mer
peptide. This peptide specifically binds the N-terminal kinase
catalytic domain of Chk1. We report the characterization of the
Chk1-binding peptide in terms of intracellular delivery,
Chk1-binding and cellular cytotoxicity, as well as in terms of its
effect on Chk1 activity and radiosensitivity. Our data may
facilitate the development of Chk1-based methods to enhance tumor
cell killing by DNA-damaging agents.
Materials and methods
Expression and purification of the
recombinant N-terminal fragment of Chk1 protein
PCR was used to amplify DNA fragments encoding the
N-terminal kinase domain of Chk1 (amino acid residues 2-270) using
forward and reverse primers (forward, 5′-CGCGGATCCGCAGTGCCCTTTGTGGAA
GACT-3′ and reverse, 5′-CGCCAAGCTTACTTGAGGGGTTT
GTTGTACCATC-3′). The underlined letters indicate restriction sites
for subcloning after PCR steps. The amplification product
incorporated a hexahistidine-tag at the beginning of the coding
region. An amplified fragment was digested with the BamHI
and HindIII restriction enzymes and cloned into a similarly
digested pQE30 vector (Qiagen, Venlo, The Netherlands), resulting
in a plasmid, which was designated as pQE30-Chk1N-His. The His-Chk1
fusion protein was expressed by subjecting the bacteria to 0.1 mM
isopropyl-1-thio-β-D-galactopyranoside (IPTG) induction for 6 h.
The bacterial extracts containing the expressed Chk1 proteins were
subjected to Ni-Affinity chromatography (Qiagen) to the purify
N-terminal Chk1 fragment (amino acids 2-270) and subsequently
washed with 3 column volumes of 300 mM NaCl and 20 mM sodium
phosphate, pH 8.0. The protein was eluted with the same buffer
containing 300 mM imidazole. The purified Chk1 protein fragment was
concentrated, and the protein concentration was determined using
the BCA protein assay kit (Thermo Fisher Scientific Inc., Rockford,
IL, USA).
Screening of phage display library
The PhD-12 Phage display library kit (#E8111L; New
England Biolabs, Beverly, MD, USA) was used for all panning
experiments. A randomized 12-mer peptide was expressed and
incorporated into the N-terminus of the pIII coat protein of the
M13 bacteriophage separated by a 4 amino acid linker.
A solution of 100 µg/ml of recombinant Chk1
protein in 0.1 M NaHCO3 (pH 8.6) was added to each well
of a 12-well plate and swirled repeatedly until the surface was
completely wet followed by incubation overnight at 4°C with gentle
agitation in a humidified container. Phages (2×1011 pfu)
were added to the recombinant Chk1-coated plate after blocking with
1 ml 5% skimmed milk for 1 h at 25°C. The mixture was incubated for
2 h at 25°C with gentle shaking. Unbound phages were removed by
approximately 6 washes with TBST buffer (50 mM Tris-HCl pH 7.5, 150
mM NaCl with 0.1% Tween-20). The bound phage particles were eluted
with 0.2 M glycine-HCl (pH 2.2), neutralized with 1 M Tris-HCl (pH
9.1), and used for the titer assay and amplification in E.
coli ER2738. The phage titer was evaluated by the blue
plaque-forming assay on an agar plate containing IPTG and X-gal. To
amplify the selected phage clones, the phages were mixed with 20 ml
E. coli ER2738 culture and incubated at 37°C with vigorous
shaking for 4.5 h. The phage particles were then harvested,
resuspended in 50 µl TBS (50 mM Tris-HCl pH 7.5, 150 mM
NaCl) with 0.02% sodium azide and titred as per the manufacturer's
instructions. Round 2 and 3 of the selections with
2×1010 and 2×109 phages, respectively, which
were harvested from the first selection, were performed with
recombinant protein-coated plates. The above-mentioned procedures
were repeated for the specific Chk1-binding peptides. Following 3
rounds of positive panning, 50 single plaques were selected for DNA
sequencing.
Peptide pull-down assay
Biotin-labeled peptides were purchased from Peptron
(Daejeon, Korea). The R9 and N-terminal Chk1-binding peptide
(Chk1-NP) peptide have the following amino acid sequences, R9,
Biotin-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg; and Chk1-NP,
Biotin-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Ala-Pro-Asn-Lys-Thr-Leu-Ser-Val-Asn-Lys-Met-Val.
The peptides were dissolved in distilled water. HeLa
(ATCC® CCL-2TM; ATCC, Manassas, VA, USA) were treated
with 5 µM of each peptide followed by incubation at 37°C for
6 h. To prepare cell extracts, the cells were lysed in an IP buffer
(1% Triton X-100, 50 mM Tris-Cl, pH 7.4, 300 mM NaCl, 5 mM EDTA)
containing protease inhibitors. A high-speed centrifugation step
removed the cell debris from the cell lysates. An equal amount (1
mg) of cell extracts was used in the pull-down assay. Pull-down
assay for the biotinylated peptide and its interacting Chk1 was
carried out by following manufacturer's instructions (Pierce™
Biotinylated protein interaction pull-down kit #21115; Thermo
Scientific, Inc.). Specific peptide-bound Chk1 was detected by
western blot analysis using anti-Chk1 antibody (mouse monoclonal
antibody against human Chk1, sc-8408; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). Western blot analysis was performed as
previously described (17).
Western blot analysis
Specific peptide-bound Chk1 was detected by western
blot analysis using anti-Chk1 antibody (mouse monoclonal antibody
against human Chk1, sc-8408; Santa Cruz Biotechnology, Inc.).
Western blot analysis was performed as previously described
(17). Briefly, protein extracts
(35 mg) were separated on 10% SDS-polyacrylamide gels and then
transferred onto nitrocellulose membranes. The membranes were
incubated with 5% skim milk solution in Tris-Tween Buffered Saline
(TTBS) solution overnight at 4°C with the indicated antibodies.
After washing 3 times in TTBS, horseradish peroxidase-conjugated
secondary antibodies were applied. The proteins were visualized
using enhanced chemiluminescence (GE Healthcare Life Sciences,
Pittsburgh, PA, USA). β-actin is a control for protein loading and
detected in western blot analysis using a specific antibody
(sc-81178; Santa Cruz Biotechnology, Inc.).
Fluorescence microscopy
The HeLa cells were cultured on sterile coverslips
in a 12-well plate. The cells were incubated with the peptides (5
µM of R9 or Chk1-NP) for 2 h at 37°C in 5% CO2
humidified air and washed 3 times with phosphate-buffered saline
(PBS; Thermo Fisher Scientific Inc.). The cells were then fixed
with methanol, stained with anti-Chk1 antibody (mouse monoclonal
antibody against human Chk1, sc-8408; Santa Cruz Biotechnology,
Inc.) and Alexa Fluor 594 goat anti-mouse secondary antibody
(A-11005; Invitrogen™, Grand Island, NY, USA), washed with PBS, and
re-stained with FITC-conjugated streptavidin (SA100-02) (both from
Invitrogen) before staining with 4′,6-diamidino-2-phenylin-dole
(DAPI). The nuclei were fluorescently labeled with DAPI (D9542;
Sigma-Aldrich, Inc., St. Louis, MO, USA). The cells were then
imaged using a Leica DMIRB microscope (Leica Microsystems Inc.,
Wetzlar, Germany) with a X20 objective. The fluorescent images were
merged. HJURP was also detected by fluorescence microscopy as a
nuclear resident protein and compared with localization of
Chk1.
Cell viability assay
The HeLa or NCI-H460 (ATCC) were treated with the R9
or Chk1-NP peptide, or no peptide as a control; 0, 5, 10 or 20
µM of the peptides were used for cell treatment. To measure
cell survival, the peptide-treated cells were incubated up to 48 h.
Cell viability was measured by MTT assay as previously described
(17). For combination treatment
of the cells with peptide and genotoxic stresses, the HeLa or
NCI-H460 cells were used. The cells were treated with 5 µM
of the R9 or Chk1-NP peptide for 1 h pior to treatment with
genotoxic agents, such as 500 nM camptothecin (CPT; #C9911) 5 mM
hydroxyurea (HU; #H8627) (both from Sigma-Aldrich) or 10 Gy
ionizing radiation (IR). Cell survival was measured by MTT assay
after 24 h. To measure and compare effects of the peptides on the
radiation sensitivity of the different cell lines, the HeLa or
NCI-H460 cells were treated with 5 µM of the R9 or Chk1-NP
peptide for 1 h prior to treatment with 5 Gy IR. MTT assay was used
to measure cell viability after 24 h. Cell cultures were irradiated
using a 137Cs gamma-ray source (Atomic Energy of Canada, Ltd.,
Chalk River, ON, Canada) at a dose rate of 3.81 Gy/min.
3D modeling to predict the Chk1-NP
peptide-binding surface of Chk1
The PEP-Site Finder program was used to identify
candidate sites of peptide interactions on the Chk1 protein
surface, as previously described (18). To use the program, data of the 3D
structure of Chk1 (PDB ID: 2E9N) and peptide sequence information
(APNKTLSVNKMW) were used as inputs. The optimal peptide-binding
Chk1 surface was generated. Information about the 2D and 3D
structure of the Chk1 kinase domain was obtained from the RCSB
Protein Data Bank (www.rcsb.org).
Statistical analysis
All analyses were carried out using the two-tailed
Student's t-test. The means of 3 independent experiments were
graphed and data are presented as the means ± standard deviation. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
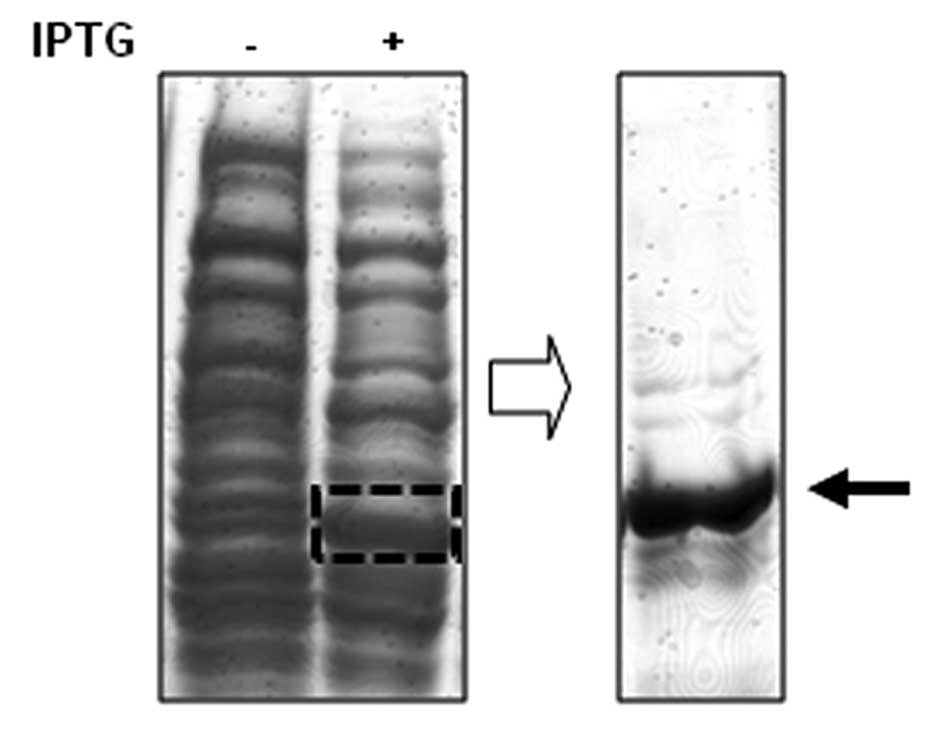
In order to screen specific Chk1-binding peptides,
we first isolated the recombinant Chk1 protein fragment to use as a
target for potential binding peptides (Fig. 1). The recombinant fragment of Chk1
protein is the N-terminal portion of Chk1 protein which is composed
of amino acid residues from 2 to 270 of Chk1. A 6-mer
histidine-tagged Chk1 fragment was isolated from the bacterial
extracts subjected to IPTG induction (Fig. 1).
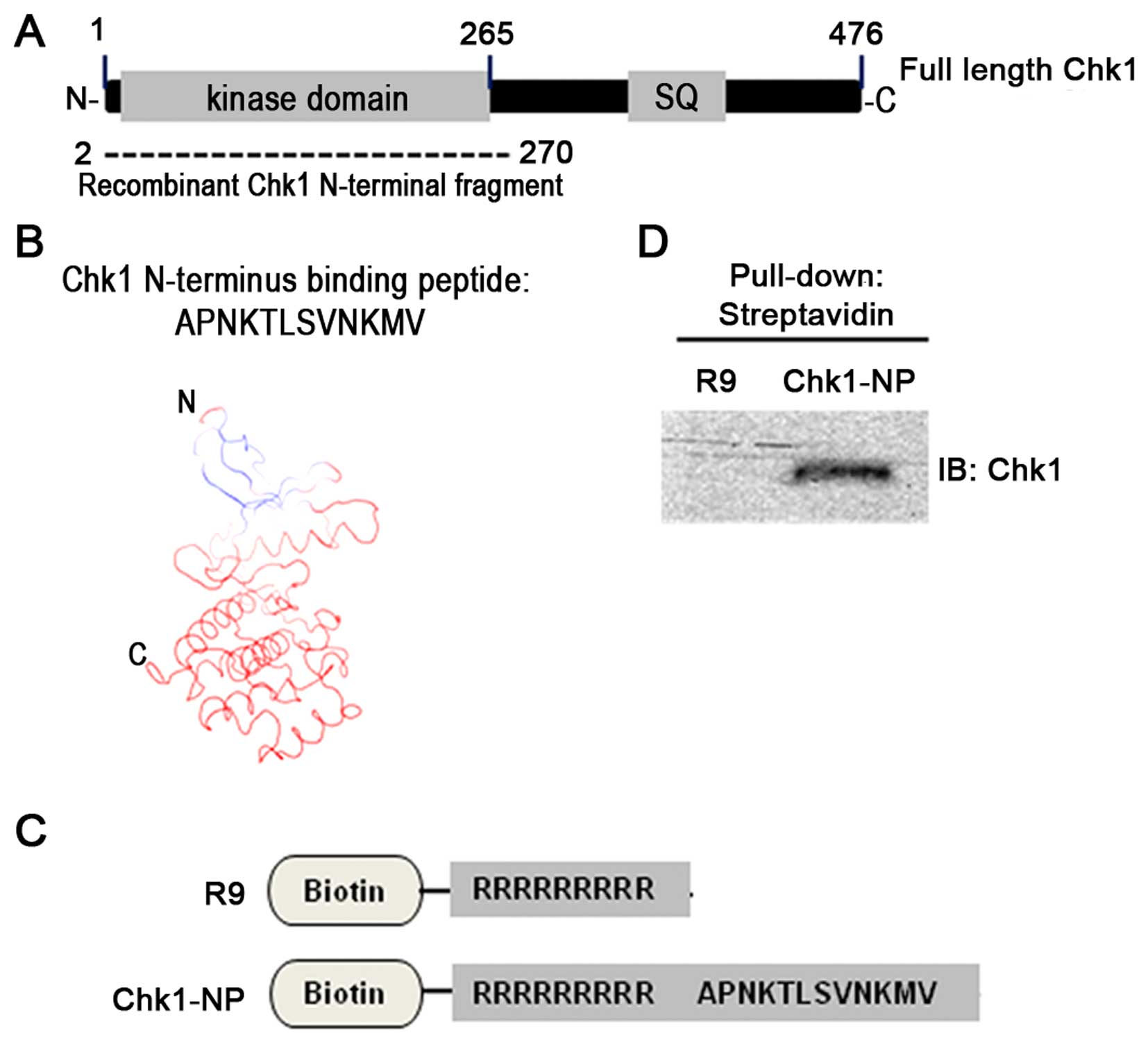
The N-terminal Chk1 fragment encompasses the Chk1
kinase domain as shown in Fig.
2A. Moreover, the secondary structure of the N-terminal Chk1
fragment is mainly composed of α-helices and β-sheets (19,20). A Chk1 fragment-bound 12-mer
peptide was screened from the phage display library, which has an
amino acid sequence of APNKTLSVNKMV (Fig. 2B).
Simulation of the 12-mer peptide-binding to the
N-terminal Chk1 fragment revealed potential sites of Chk1 for the
peptide binding (Fig. 2B). Models
of the peptide Chk1 binding predict that the very N-terminus of
Chk1, which is composed of mostly a β-sheet secondary structure is
the peptide-binding site of Chk1. In order to test the activity of
the peptide in the cell, we used a 9-mer poly-arginine sequence for
an internalization domain. A poly-arginine peptide (R9 peptide)
(Fig. 2C) has been shown to
efficiently transport peptides and proteins into mammalian cells
(21). We used two different
peptides, such as an R9 internalization domain alone peptide as a
control and a specific Chk1-binding peptide, named Chk1-NP, which
is composed of R9 plus the identified Chk1-binding 12-mer peptide
sequence (Fig. 2C). These two
peptides were tagged with a biotin at their N termini for easy
detection in in vitro experiments. In order to confirm the
Chk1-binding activity of the identified 12-mer peptide, we carried
out a pull-down experiment with cell extracts and the two peptides.
Immunoblot detection of the precipitated Chk1 demonstrated that
only the Chk1-NP peptide was able to bind Chk1, but not the R9
control peptide (Fig. 2D).
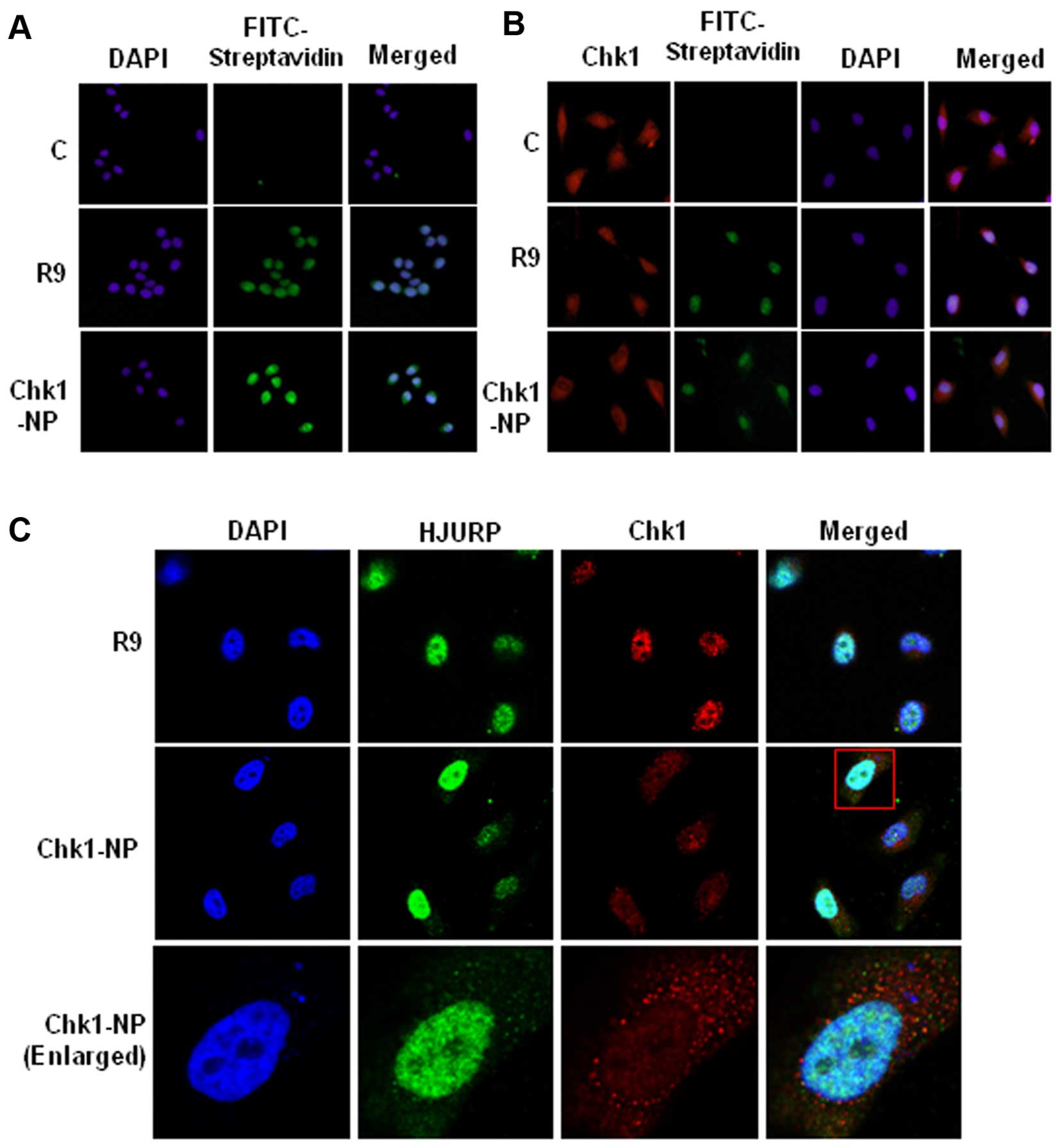
To explore the activities of the peptides in the
cells, we examined the delivery of the fusion peptides into the
cell. Treatment of the HeLa cells with R9 or Chk1-NP at a
concentration of 5 µM showed a significant cellular uptake
of the peptides (Fig. 3A).
Detection of the cellular localization of the biotin-labeled
peptides by FITC-conjugated streptavidin revealed cytoplasmic and
nuclear localization of the peptides, indicating that the peptide
could be efficiently delivered into the cellular compartments. Of
the cellular compartments, the nuclear staining was more intense
than that of the cytoplasm, which may reflect more densely
populated peptides in the nucleus. The localization of Chk1 was
also examined in the presence or absence of the peptides (Fig. 3B). As shown in Fig. 3A, the peptides, R9 and Chk1-NP,
were shown mainly in the nucleus, whereas the no-peptide control
(C) cells showed no fluorescence, as expected (Fig. 3B). Chk1 was visible both in the
cytoplasm and nucleus, which is consistent with a previous study
(22). The nuclear staining of
Chk1 is dense compared to the diffuse cytoplasmic staining. The
no-peptide control (C) or R9-treated control (R9) exhibited clear
nuclear staining of Chk1. However, Chk1 staining in the Chk1-NP
treated cells showed a sparse nuclear staining compared to the
cytoplasmic staining (Fig. 3B).
The sparse nuclear staining of Chk1 may imply that Chk1-NP
redistributes nuclear Chk1 to other cellular compartments.
To examine the Chk1 localization further, we
compared the localization of Chk1 and HJURP in the R9 or Chk1-NP
peptide-treated cells. HJURP, a deposition factor of CENP-A at
centromeres, is a nuclear protein (23). The cellular localization of HJURP
was unaffected by the R9 and the Chk1-NP peptide, as it showed
consistent nuclear staining (Fig.
3C). By contrast, as shown in Fig. 3B, the nuclear staining of Chk1 in
the Chk1-NP peptide-treated cells was prominently weak when
compared to that of the R9 control peptide-treated cells (Fig. 3C). This finding indicates that
Chk1-NP redistributes nuclear Chk1 in a Chk1-specific manner.
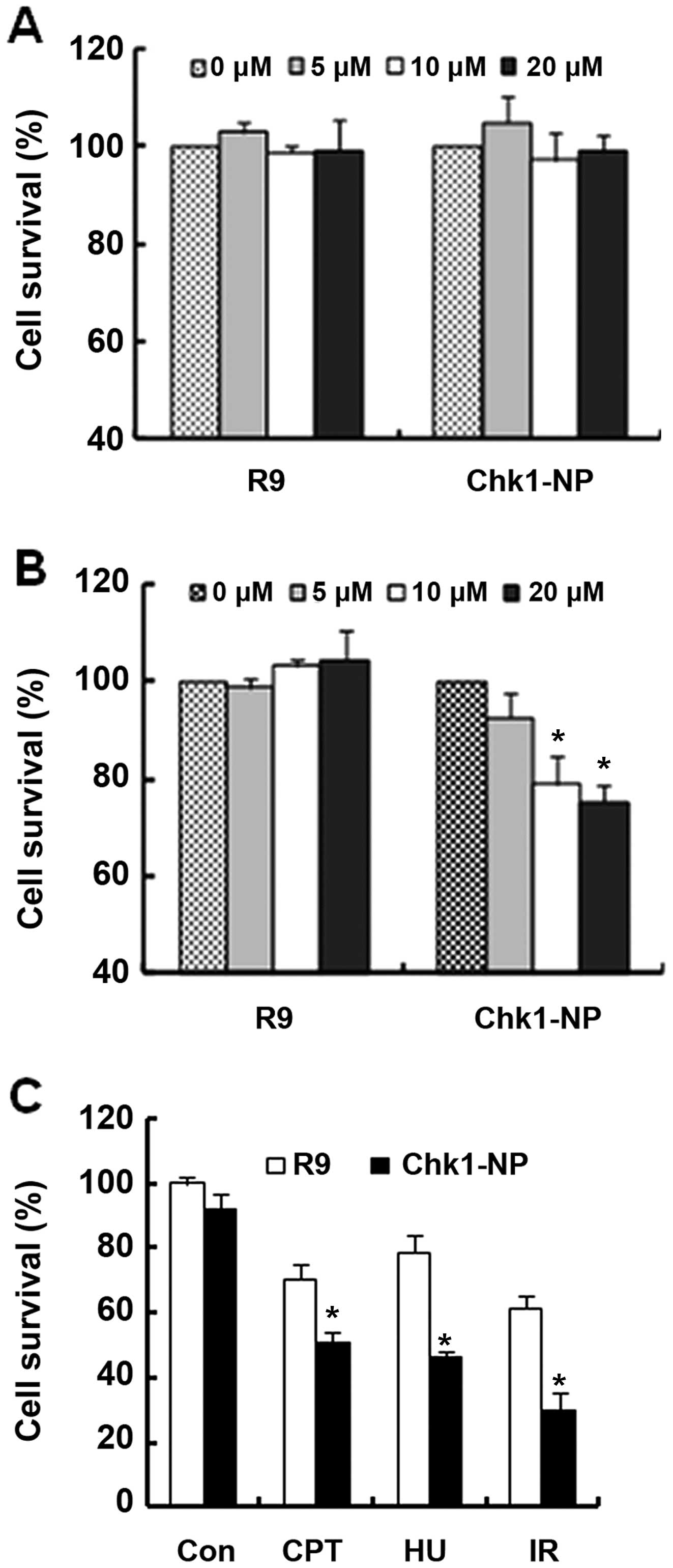
Since Chk1 is a critical component in cellular DNA
damage responses, we wished to determine whether the Chk1-binding
peptide has an effect on the function of Chk1 following treatment
with genotoxic stresses. We first evaluated the cytotoxicity of the
R9 and Chk1-NP peptides in NCI-H460 or in HeLa cells (Fig. 4A and B, respectively). The two
cell lines have a different genetic background, in that NCI-H460
cells are functional in p53, whereas HeLa cells are not.
NCI-H460 cell survival was unaffected by treatment
with either peptide at a concentration of up to 20 µM
(Fig. 4A). By contrast, the
survival of the HeLa cells exhibited a striking difference between
the peptides used (Fig. 4B).
Whereas the R9 control peptide had no effect on HeLa cell survival,
the Chk1-binding Chk1-NP peptide decreased cell survival by
approximately 30% at a concentration of 20 µM of the peptide
(Fig. 4B). This finding may imply
that the essential function of Chk1 for HeLa cell survival was
affected by the Chk1-binding peptide, Chk1-NP. Moreover, the
results suggest that the effect of the Chk1-binding peptide on cell
survival may be cell type-specific or more specifically,
genotype-specific.
HeLa cells are non-functional in p53, and have been
reported to be dependent on Chk1 function for cell survival
following exposure to genotoxic stress. Therefore without Chk1
function, HeLa cells are vulnerable to genotoxic stresses and
undergo increased cell death by caspase-2 activation (24). In this study, we examined whether
Chk1-NP has an effect on HeLa cell survival following treatment
with various genotoxic stresses (Fig.
4C). Treatment with CPT, HU, or with IR decreased the survival
of the HeLa cells. Whereas decrease in HeLa cell survival was
observed in the cells treated with either the R9 or Chk1-NP
peptides following exposure to genotoxic stresses, the decrease was
more prominent in the cells treated with Chk1-NP (Fig. 4C). The survival of the HeLa cells
treated with Chk1-NP was about half that of the R9-treated cells
following treatment with IR (Fig.
4C). These findings suggest that Chk1-NP interferes with the
functions of Chk1 and signifi-cantly increases the death of
vulnerable cells, such as HeLa cells following DNA damage.
We further carried out a comparison between the HeLa
and NCI-H460 cells as regards radiation sensitivity following
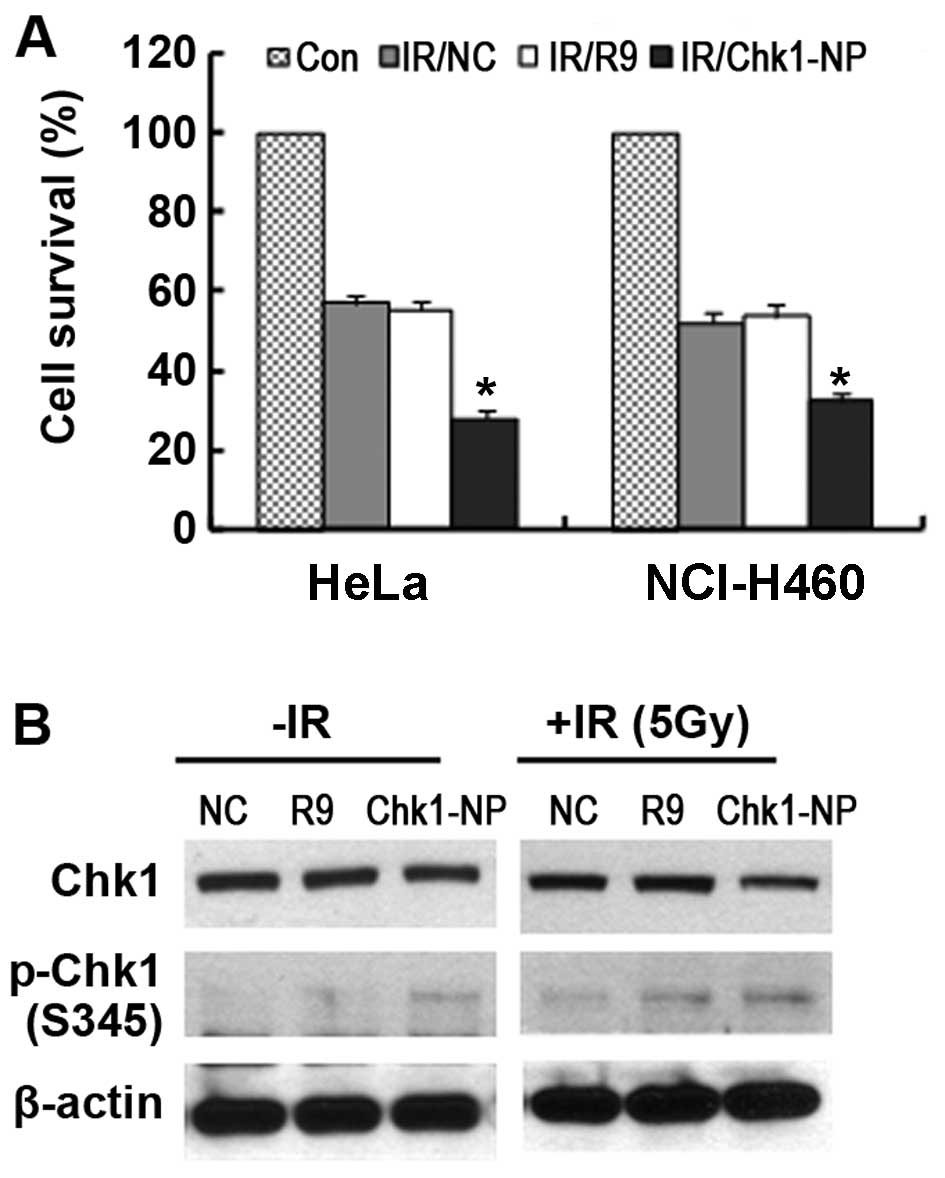
treatment with the peptides (Fig.
5A). Both the HeLa and NCI-H460 cells showed a significantly
increased radiation sensitivity following treatment with the
Chk1-NP peptide. The survival of the Chk1-NP-treated HeLa and
NCI-H460 cells following exposure to IR was approximately 35 and
50%, respectively when compared to that of no peptide control (NC)
or the R9 control-treated cells. Thus, these findings demonstrate
that Chk1-NP is able to enhance the radiation sensitivity of the
cells with a greater enhancement observed in p53-defective cancer
cells, such as HeLa cells.
Examination of Chk1 phosphorylation in the
Chk1-NP-treated HeLa cells following expsoure to IR showed
increased the phosphorylation of Chk1 S345 residue (pS345 Chk1)
when compared to that of the NC or the R9 control-treated cells
(Fig. 5B). The results are
reminiscent of the findings that the Chk1 inhibitor, AZD7762,
enhanced chemosensitization with a concomitant induction of pS345
Chk1 (25). We also noted that
the total levels of Chk1 in the Chk1-NP-treated HeLa cells were
less than those in the NC or the R9 control-treated cells following
exposure to IR (Fig. 5B). Since
Chk1 is degraded in a Chk1 phosphorylation-dependent manner
following exposure to IR, these findings suggest that the Chk1-NP
peptide is able to facilitate the activation-induced degradation of
Chk1 following exposure to IR. Furthermore, our results suggest the
potential use of Chk1-NP in the artificial modulation of Chk1
stability.
Discussion
Since Chk1 is essential for cell survival following
DNA damage, a mechanistic understanding and potential modulation of
Chk1 is of importance to cancer therapy. To facilitate such
efforts, in this study, we screened and identified a specific
Chk1-binding peptide. This peptide, named Chk1-NP, bound the
N-terminal kinase domain of Chk1. Simulation of the peptide binding
revealed that the very N-terminus of the Chk1 kinase domain was the
potential peptide binding site. The effect of the Chk1-NP peptide
on endogenous Chk1 was examined following internalization of the
peptide. Cell viability was significantly decreased by combined
treatment with the peptide and genotoxic agents. Our findings
suggest that the decreased nuclear localization of endogenous Chk1
following the internalization of the Chk1-NP peptide is a likely
cause of the decreased cell survival following genotoxic
treatments.
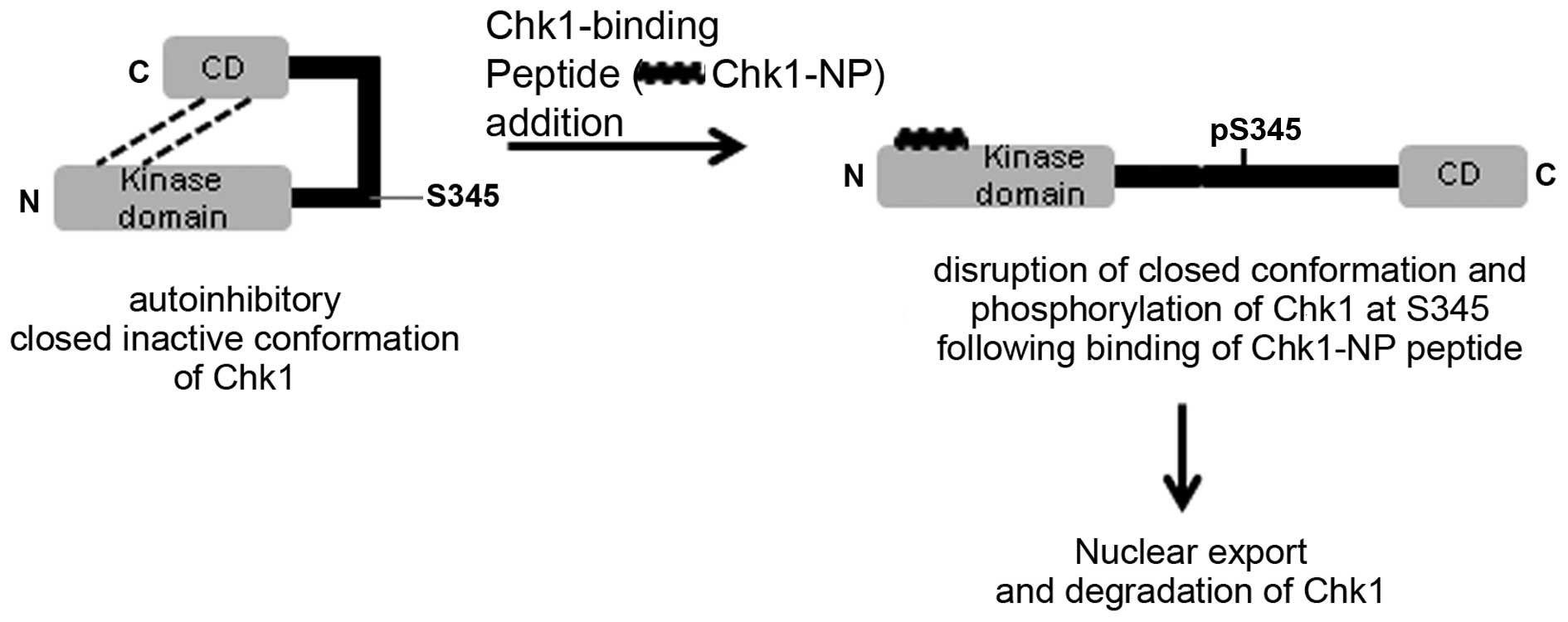
In the absence of genotoxic stresses, Chk1 seems to
undergo an intramolecular interaction between the N-terminus and
the C-terminus (Fig. 6) (26). By this interaction, Chk1 blocks
the active site of the kinase domain of Chk1, which results in
autoinhibition (11,26). Upon DNA damage, Chk1 is
phosphorylated by ATR on chromatin. Chk1 phosphorylation seems to
release the protein from the autoinhibitory conformation and leads
to activation of the protein. Moreover, upon Chk1 phosphorylation,
the protein is released from the chromatin and moves into the
soluble nucleoplasm and later into the cytoplasm (9,27,28). Considering these facts, we
hypothesized the mechanisms through which Chk1-NP may affect
multiple aspects of Chk1 activities. These may involve peptide
binding-induced conformational changes and the cellular
redistribution of Chk1 (Fig. 6).
The Chk1-binding peptide, Chk1-NP may affect the conformation of
endogenous Chk1, and this in turn may lead to the decreased nuclear
localization of Chk1. Since our simulation of the peptide binding
to Chk1 revealed that the very N-terminus of the Chk1 kinase domain
was the potential peptide binding site (Fig. 2B), the closed inactive
conformation of Chk1 may be released by the disruption of the
intramolecular interaction upon binding of Chk1-NP peptide to the
N-terminus of Chk1 (Fig. 6). This
conformational change may expose the C-terminus of Chk1 and lower
the threshold for Chk1 activation to Chk1-activating kinases upon
DNA damage. Of note, treatment of the cells with the Chk1-binding
peptide alone without DNA damage slightly increased Chk1
phosphorylation at the Ser345 residue (Fig. 5B). This finding is reminiscent of
that of a previous study, where the disruption of the closed
conformation of Chk1 resulted in Chk1 activation even in the
absence of DNA damage (29). In
that study, the authors showed that the N-terminal kinase domain of
Chk1 prevents Chk1 phosphorylation at the C-terminus by ATR in the
absence of DNA damage. Disruption of the closed inactive
conformation of Chk1 by mutation of C-terminal conserved residues
and loss of the inhibitory effect by the N-terminus result in
phosphorylation of Chk1 by ATR even in the absence of DNA damage
(29). We speculate that the
Chk1-binding peptide causes similar effect as the Chk1 mutation in
disrupting closed conformation of Chk1. A specific binding of
Chk1-NP to the Chk1 N-terminus may disrupt the closed conformation
of Chk1 and release the C-terminus from the inhibitory N-terminus
which may lead to Chk1 activation by phosphorylation. Irradiation
of the Chk1-NP peptide-treated cells increased further Chk1
phosphorylation at the S345 residue, which is an indication of Chk1
activation (Fig. 5B). Open
conformation of Chk1 upon Chk1-NP binding may have facilitated the
phosphorylation of Chk1 following IR (Fig. 6).
As conformational changes of Chk1 are closely tied
to its spatial redistribution (9,27,28,30) we may expect changes in Chk1
localization following Chk1-NP peptide binding. Chk1-NP
significantly decreased the nuclear localization of endogenous Chk1
(Fig. 3C). Chk1 phosphorylation
triggers a release of Chk1 from chromatin upon DNA damage into the
nucleoplasm and the cytoplasm (9,27,28). In our study, Chk1 was present both
in the cytoplasm and the nucleus under normal conditions (Fig. 3B). Upon treatment with the
Chk1-binding peptide, however, nuclear Chk1 was significantly
decreased even in the absence of genotoxic treatment (Fig. 3C). Since Chk1 phosphorylation is
slightly increased by treatment with the Chk1-binding peptide
(Fig. 5B) under normal and IR
conditions when compared to the controls, we hypothesized that the
increased Chk1 phosphorylation was partly responsible for the
decreased nuclear localization of Chk1. More importantly,
considering the fact that the nuclear pool of Chk1 supports cell
viability (22), the decreased
cell viability of the Chk1-NP peptide-treated cells upon DNA damage
may have been caused by the decreased nuclear pool of Chk1
(Figs. 4C and 5A).
The phosphorylation and subsequent activation of
Chk1 lead to conformational changes and degradation of the protein
(9). Our findings demonstrated
that the expression levels of Chk1 were decreased in the Chk1-NP
peptide treated cells upon DNA damage (Fig. 5B). The slightly increased
phosphorylation of Chk1 in the Chk1-NP peptide treated cells may
have triggered Chk1 degradation. Therefore, the decreased levels of
Chk1, as well as the decreased nuclear pool of Chk1 may have caused
decreased cell viability following genotoxic treatments in the
Chk1-NP peptide-treated cells.
The enhanced activation of Chk1 results in the
resistance of cancer cells to chemo- or radiotherapy (31–34). Therefore, the artificial
inhibition of Chk1 activity may reverse such therapy resistance.
The combination of Chk1 inhibition with chemo- or radiotherapy
results in synthetic lethality in cancer therapy. As many tumors
with defective p53 function depend on Chk1-dependent S and G2/M
checkpoints for survival, such combination treatment should be more
effective against p53-defective cancer cells (35,36). In our study, the combination of
Chk1-NP and genotoxic agents significantly decreased the survival
of both HeLa and NCI-H460 cells (Figs. 4C and 5A). The combined treatment with IR and
the peptide was slightly more effective in killing HeLa cells when
compared to that of NCI-H460 cells (Fig. 5A). HeLa cells are p53-defective,
whereas NCI-H460 cells are p53-functional. p53-functional NCI-H460
cells treated with Chk1-NP may better deal with the DNA damage
response by mobilizing p53-dependent cell cycle checkpoints which
may contribute to enhanced cell survival.
Of note, our findings demonstrated that treatment
with the Chk1-NP peptide alone significantly decreased the
viability of HeLa cells, but not that of NCI-H460 cells (Fig. 4A and B). These results are
reminiscent of the study by Wang et al (29). In that study, the authors
demonstrated that the expression of the constitutively active
mutant form of Chk1 in the absence of DNA damage inhibited HeLa
cell proliferation. In our study, the Chk1-binding peptide also
showed decreased cell survival with increased Chk1 activation in
HeLa cells (Figs. 4B and 5B). These findings suggest that the
artificial activation of Chk1 in the absence of DNA damage by a
specific Chk1-binding peptide like Chk1-NP may be utilized to
develop a novel method in cancer therapy.
In conclusion, as demonstrated in the present study,
although the use of a peptide as a therapeutic agent still presents
many challenges, such as toxicity and stability of the peptide, the
approach using a specific Chk1-binding peptide seems promising in
enhancing the mechanistic understanding of Chk1 activities. The
single or combined use of the Chk1-binding Chk1-NP peptide with
chemo- or radiotherapy may provide a novel rationale for
development of specific Chk1-targeting agents.
Abbreviations:
|
IR
|
ionizing radiation
|
|
DDR
|
DNA damage response
|
|
DDRFs
|
DNA damage response factors
|
|
Chk1
|
checkpoint kinase 1
|
|
Chk1-NP
|
N-terminal Chk1-binding peptide
|
Acknowledgments
This study was supported by the nuclear research and
development program through the National Research Foundation of
Korea (NRF) funded by the Ministry of Science, ICT and Future
Planning of Korea (grant no. NRF-2012M2A2A7012377).
References
|
1
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sørensen CS, Syljuåsen RG, Falck J,
Schroeder T, Rönnstrand L, Khanna KK, Zhou BB, Bartek J and Lukas
J: Chk1 regulates the S phase checkpoint by coupling the
physiological turnover and ionizing radiation-induced accelerated
proteolysis of Cdc25A. Cancer Cell. 3:247–258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Falck J, Mailand N, Syljuåsen RG, Bartek J
and Lukas J: The ATM-Chk2-Cdc25A checkpoint pathway guards against
radio-resistant DNA synthesis. Nature. 410:842–847. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patil M, Pabla N and Dong Z: Checkpoint
kinase 1 in DNA damage response and cell cycle regulation. Cell Mol
Life Sci. 70:4009–4021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tapia-Alveal C, Calonge TM and O'Connell
MJ: Regulation of chk1. Cell Div. 4:82009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y and Hunter T: Roles of Chk1 in
cell biology and cancer therapy. Int J Cancer. 134:1013–1023. 2014.
View Article : Google Scholar
|
|
9
|
Zhang YW, Otterness DM, Chiang GG, Xie W,
Liu YC, Mercurio F and Abraham RT: Genotoxic stress targets human
Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell.
19:607–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leung-Pineda V, Huh J and Piwnica-Worms H:
DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling
cells and in cells experiencing replication stress. Cancer Res.
69:2630–2637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang YW, Brognard J, Coughlin C, You Z,
Dolled-Filhart M, Aslanian A, Manning G, Abraham RT and Hunter T:
The F box protein Fbx6 regulates Chk1 stability and cellular
sensitivity to replication stress. Mol Cell. 35:442–453. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB
and Grant S: CHK1 inhibitors in combination chemotherapy: Thinking
beyond the cell cycle. Mol Interv. 11:133–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Merry C, Fu K, Wang J, Yeh IJ and Zhang Y:
Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle.
9:279–283. 2010. View Article : Google Scholar
|
|
14
|
Suganuma M, Kawabe T, Hori H, Funabiki T
and Okamoto T: Sensitization of cancer cells to DNA damage-induced
cell death by specific cell cycle G2 checkpoint abrogation. Cancer
Res. 59:5887–5891. 1999.PubMed/NCBI
|
|
15
|
Kawabe T: G2 checkpoint abrogators as
anticancer drugs. Mol Cancer Ther. 3:513–519. 2004.PubMed/NCBI
|
|
16
|
Zhao H, Watkins JL and Piwnica-Worms H:
Disruption of the checkpoint kinase 1/cell division cycle 25A
pathway abrogates ionizing radiation-induced S and G2 checkpoints.
Proc Natl Acad Sci USA. 99:14795–14800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim KS, Heo JI, Choi KJ and Bae S:
Enhancement of cellular radiation sensitivity through degradation
of Chk1 by the XIAP-XAF1 complex. Cancer Biol Ther. 15:1622–1634.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saladin A, Rey J, Thévenet P, Zacharias M,
Moroy G and Tufféry P: PEP-SiteFinder: a tool for the blind
identification of peptide binding sites on protein surfaces.
Nucleic Acids Res. 42:W221–W226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen P, Luo C, Deng Y, Ryan K, Register J,
Margosiak S, Tempczyk-Russell A, Nguyen B, Myers P, Lundgren K, et
al: The 1.7 A crystal structure of human cell cycle checkpoint
kinase Chk1: Implications for Chk1 regulation. Cell. 100:681–692.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao B, Bower MJ, McDevitt PJ, Zhao H,
Davis ST, Johanson KO, Green SM, Concha NO and Zhou BB: Structural
basis for Chk1 inhibition by UCN-01. J Biol Chem. 277:46609–46615.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fuchs SM and Raines RT: Pathway for
polyarginine entry into mammalian cells. Biochemistry.
43:2438–2444. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Han X, Feng X, Wang Z and Zhang Y:
Coupling cellular localization and function of checkpoint kinase 1
(Chk1) in checkpoints and cell viability. J Biol Chem.
287:25501–25509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dunleavy EM, Roche D, Tagami H, Lacoste N,
Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y and
Almouzni-Pettinotti G: HJURP is a cell-cycle-dependent maintenance
and deposition factor of CENP-A at centromeres. Cell. 137:485–497.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sidi S, Sanda T, Kennedy RD, Hagen AT,
Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF,
et al: Chk1 suppresses a caspase-2 apoptotic response to DNA damage
that bypasses p53, Bcl-2, and caspase-3. Cell. 133:864–877. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Parsels LA, Qian Y, Tanska DM, Gross M,
Zhao L, Hassan MC, Arumugarajah S, Parsels JD, Hylander-Gans L,
Simeone DM, et al: Assessment of chk1 phosphorylation as a
pharmaco-dynamic biomarker of chk1 inhibition. Clin Cancer Res.
17:3706–3715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Katsuragi Y and Sagata N: Regulation of
Chk1 kinase by auto-inhibition and ATR-mediated phosphorylation.
Mol Biol Cell. 15:1680–1689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scorah J, Dong MQ, Yates JR III, Scott M,
Gillespie D and McGowan CH: A conserved proliferating cell nuclear
antigen-interacting protein sequence in Chk1 is required for
checkpoint function. J Biol Chem. 283:17250–17259. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smits VA, Reaper PM and Jackson SP: Rapid
PIKK-dependent release of Chk1 from chromatin promotes the
DNA-damage checkpoint response. Curr Biol. 16:150–159. 2006.
View Article : Google Scholar
|
|
29
|
Wang J, Han X and Zhang Y: Autoregulatory
mechanisms of phosphorylation of checkpoint kinase 1. Cancer Res.
72:3786–3794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shimada M, Niida H, Zineldeen DH, Tagami
H, Tanaka M, Saito H and Nakanishi M: Chk1 is a histone H3
threonine 11 kinase that regulates DNA damage-induced
transcriptional repression. Cell. 132:221–232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Perego P, Gatti L, Righetti SC, Beretta
GL, Carenini N, Corna E, Dal Bo L, Tinelli S, Colangelo D, Leone R,
et al: Development of resistance to a trinuclear platinum complex
in ovarian carcinoma cells. Int J Cancer. 105:617–624. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bartucci M, Svensson S, Romania P, Dattilo
R, Patrizii M, Signore M, Navarra S, Lotti F, Biffoni M, Pilozzi E,
et al: Therapeutic targeting of Chk1 in NSCLC stem cells during
chemotherapy. Cell Death Differ. 19:768–778. 2012. View Article : Google Scholar :
|
|
34
|
Wang X, Ma Z, Xiao Z, Liu H, Dou Z, Feng X
and Shi H: Chk1 knockdown confers radiosensitization in prostate
cancer stem cells. Oncol Rep. 28:2247–2254. 2012.PubMed/NCBI
|
|
35
|
Koniaras K, Cuddihy AR, Christopoulos H,
Hogg A and O'Connell MJ: Inhibition of Chk1-dependent G2 DNA damage
checkpoint radiosensitizes p53 mutant human cells. Oncogene.
20:7453–7463. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma CX, Cai S, Li S, Ryan CE, Guo Z,
Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, et al: Targeting
Chk1 in p53-deficient triple-negative breast cancer is
therapeutically beneficial in human-in-mouse tumor models. J Clin
Invest. 122:1541–1552. 2012. View Article : Google Scholar : PubMed/NCBI
|