Introduction
Acute respiratory distress syndrome (ARDS) and acute
lung injury mark the final stages of several respiratory diseases;
the traditional supportive treatment for ARDS is mechanical
ventilation. Although such ventilation can be lifesaving, it comes
with several potential disadvantages and complications (1). High tidal volume (HTV) mechanical
ventilation can cause lung edema and activate inflammatory
pathways, a process known as ventilator-induced lung injury
(2). Mechanical ventilation and
inflammation induce the activation of alveolar macrophages at
sensitive sites, and these cells in turn help trigger and sustain
an acute inflammatory response (3). The macrophages release inflammatory
cytokines, including interleukin (IL)-1β and IL-6 (4–6).
The combination of mechanical tissue stretching and inflammation
lead to lung injury (7).
Toll-like receptors (TLRs) play a critical role in
ventilator-induced lung injury (8). In our previous study, the expression
of TLR4 and TLR9 were found to be upregulated in animal models of
ventilator-induced lung injury, and the most obvious increase in
the expression of TLRs was observed following ventilation for 4 h
(9). In particular, the
activation of the signaling pathway mediated by TLRs, myeloid
differentiation factor 88 (MyD88)/nuclear factor (NF)-κB, has been
proposed to cause the release of inflammatory cytokines [IL-1β,
IL-6 and tumor necrosis factor (TNF)-α], to strengthen the immune
response, and increase endothelial permeability, all of which
contribute to injury (10,11).
This pathway may be activated in alveolar macrophages during
mechanical ventilation, but this has never been directly examined
in vivo, at least to the best of our knowledge. Consistent
with this possibility, studies using mice have indicated that TNF-α
intensifies the inflammatory response during endotoxic shock
(12) and that signaling via the
TNF-α receptor helps drive ventilator-induced lung injury (13).
In this study, we used a combination of in
vivo and in vitro approaches in an aim to determine the
mechanisms through which TLR4-mediated signaling drives
ventilator-induced lung injury. We also examined whether
neutralizing receptor activity using an anti-TLR4 monoclonal
antibody may alleviate this type of injury.
Materials and methods
Animals
Forty-eight adult male Sprague-Dawley rats weighing
250–300 g were purchased from the Medical Laboratory Animal Center
of Guangxi Medical University, China (certificate no.
SCXK-Gui-2010-0001). The rats were fed a normal diet with water
ad libitum and were housed in accordance with the Chinese
Regulations for the Administration of Affairs Concerning
Experimental Animals. The study protocol was approved by the Animal
Care and Use Committee of Guangxi Medical University.
Rat model of ventilator-induced lung
injury
Rats were anesthetized intraperitoneally with
ketamine (100 mg/kg body weight) and xylazine (10 mg/kg) (no.
091127; Fujian Gutian Pharmaceutical Co. Ltd, Ningde, China).
Anesthesia was maintained by administering one-third of the initial
dose of anesthetic agents approximately every 45 min during
experiments. Rats were placed in a supine position on an adjustable
warming pad (Alcott Biotech, Shanghai, China) that was maintained
at 37±1°C; animal temperature was monitored continuously using a
rectal probe.
According to the relevant cell count in the
bronchoalveolar lavage fluid (BALF) extracted from each of the rats
(3–6×105 cells) (14),
and the protocol of immunohistochemistry and western blot analysis
of anti-TLR4 monoclonal antibody, the rats were subjected to
tracheal intubation and administered either 200 μl of normal
saline (n=8) or 200 μl of anti-TLR4 monoclonal antibody
(ab8376; Abcam, Cambridge, USA) at a dose of 8 μg/kg body
weight (n=8). After 1 h, both groups of animals were connected to a
small animal ventilator (Alcott Biotech) and ventilated for 4 h at
40 ml/kg with 80 breaths/min and 0 positive end-expiratory
pressure. The respiratory parameter were based on a previous study
(9) and the preliminary results
of our research groups. A third group of rats (n=8) was subjected
to tracheotomy and the intratracheal administration of saline, and
then allowed to breathe spontaneously. All 3 groups of animals
breathed ambient air. Oxygen saturation and heart rate were
continuously monitored in the anesthetized rats using the MouseOx
system (NatureGene Corp., Beijing, China). At the end of the 4-h
ventilation period, the animals were administered a lethal dose of
anesthetic agents and lung tissues, blood and BALF were
harvested.
Collection of plasma
Plasma samples were collected from the atrium
dextrum of the rats, followed by centrifugation for 15 min at 500 ×
g at 4°C to remove red cells, and the plasma was frozen at
-20°C.
Collection and culture of alveolar
macrophages
Alveolar macrophages were harvested from the BALF of
each group of rats into phosphate-buffered saline (PBS) as
previously described (15). The
cells were resuspended in RPMI-1640 medium containing 10% fetal
bovine serum (FBS) (HyClone, Logan, UT, USA) and cultured for 2 h
in 25-cm2 flasks. The cultures were then washed with
fresh RPMI-1640 to remove non-adherent cells. The viability of the
remaining macrophages was assayed using trypan blue before
conducting the TNF-α stimulation experiments described below.
In vitro model of the stimulation of
alveolar macrophages with TNF-α
The alveolar macrophages were harvested in BALF of
24 ventilated rats. The macrophage cultures were divided into 3
treatment groups, with each group containing cultures from 24
ventilated rats pre-treated with saline. One set (n=8) of
macrophage cultures was incubated for 2 h with anti-TLR4 antibody
(group TNF + Ab), and the other 2 sets (n=16 per set) with PBS. The
medium for all 3 sets of cultures was then replaced with fresh
RMPI-1640 containing 10% FBS, and the cultures pre-treated with
anti-TLR4 antibody (TNF + Ab) or PBS (TNF group) were stimulated
for a further 16 h with TNF-α (20 ng/ml), and a third set with PBS
only (PBS group).
Pulmonary edema based on the lung wet/dry
weight ratio
At the end of the 4-h ventilation period, the rats
were sacrificed and the right lung was flushed with PBS. Following
the ligation of left lungs of the rats, the lungs were weighed
immediately after removal (wet weight) and again after drying in an
oven at 65°C for 48 h (dry weight). The ratio was calculated to
serve as an index of pulmonary edema.
Lung histopathology
Tissue from the right lower lobe was removed and
processed for transmission electron microscopy as previously
described (16). Specimens were
examined on a JEOL 8000 transmission electron microscope (Hitachi
High-Technologies Corp., Tokyo, Japan) microscope. The animals were
then subjected to intratracheal instillation with 10% formalin to
fix the lungs. The lungs were then removed and embedded in
paraffin. Sections (4-μm-thick) were made using a rotary
microtome (HM 355S; Thermo Fisher Scientific, Waltham, MA, USA) and
stained with hematoxylin and eosin, and examined under an IX71
light microscope (Olympus, Tokyo, Japan).
Analysis of BALF
Lung inflammation was assessed by counting the
numbers of cells and assaying the total protein amount in BALF.
Each rat was instilled with 1.0 ml of PBS 5 times, and the
recovered volumes were kept on ice. The total recovered volume was
approximately 80% of the original 5 ml. The recovered BALF was
centrifuged for 5 min at 500 × g at 4°C. The pellet was resuspended
in RPMI-1640 containing 10% FBS, and analyzed on a hemocytometer
(YA0810; Solarbio, Beijing, China) to determine the total cell
number. Supernatants were frozen immediately on dry ice and stored
at -80°C for cytokine assays.
Cytokine levels in BALF, plasma and
culture medium
Commercial kits based on the enzyme-linked
immunosorbent assay (ELISA) (R&D Systems, Inc., Minneapolis,
MN, USA) were used to assay TNF-α, IL-1β and IL-6 in BALF and
plasma from mechanically ventilated or spontaneously breathing
rats, as well as in the medium of alveolar macrophage cultures.
Each sample was assayed in duplicate following the manufacturer's
instructions (rat TNF-α Quantikine ELISA RTA00; rat IL-1β/IL-1F2
Quantikine ELISA RLB00; Rat TNF-α Quantikine ELISA R6000B; R&D
Systems).
Western blot analysis of specific
proteins in alveolar macrophage cultures
Alveolar macrophages (2×106 cells) were
suspended in 50 μl ice-cold RIPA lysis buffer with protease
inhibitors (Beyotime Institute of Biotechnology, Haimen, China),
homogenized and centrifuged at 12,000 × g for 5 min at 4°C to
remove insoluble material. The total protein concentration in the
supernatant was determined using a BCA Protein Assay kit (Beyotime
Institute of Biotechnology). The supernatant was mixed with 4X
loading buffer (Takara Bio, Dalian, China), and equal amounts of
protein were fractionated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene fluoride membranes (both from Bio-Rad Laboratories,
Inc., Hercules, USA). The blots were blocked for 2 h at room
temperature with Tris-buffered saline containing 0.1% Tween-20 and
5% non-fat milk, then incubated overnight with primary antibody,
followed by horseradish peroxidase-conjugated secondary antibody
for chemiluminescent visualization (Beyotime Institute of
Biotechnology). Processed membranes were scanned using the ChemiDoc
MP system (Bio-Rad Laboratories, Inc.), and densitometry was
performed using in-house software developed at the Affiliated Tumor
Hospital of Guangxi Medical University (Nanning, China). Antibodies
against TLR4 (ab8376) and TLR9 (ab12121) were purchased from Abcam
(Cambridge, MA, USA), while antibodies against MyD88 (Cat. no.
4283) and NF-κB (Cat. no. 3033) were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Horseradish
peroxidase-conjugated secondary anti-rabbit (SA00001-2) and
anti-mouse (SA00001-1) antibodies were purchased from the
ProteinTech Group, Inc. (Bioconnect, Wuhan, China).
Analysis of gene expression in alveolar
macrophage cultures by reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the alveolar macrophage
cultures using the GeneJET RNA Purification kit (Thermo Fisher
Scientific). Reverse transcription was used to generate cDNA
encoding TLR4, TLR9, MyD88 and NF-κB, which was then amplified by
PCR. Specific primers to target regions of the corresponding genes
were designed based on sequences in GenBank (Table I). In parallel, the gene
expressing glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was
amplified as an internal control and used to normalize gene
expression levels.
 | Table IPrimer sequences used to detect mRNAs
encoding TLR4, TLR9, MyD88, NF-κB and GAPDH. |
Table I
Primer sequences used to detect mRNAs
encoding TLR4, TLR9, MyD88, NF-κB and GAPDH.
| Gene | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| GAPDH | F:
GGCACAGTCAAGGCTGAGAATG | 143 |
| R: ATGGTGGTGAGA
CGCCAGTA | |
| TLR4 | F:
CATCCAAAGGAATACTGCAACA | 398 |
| R:
GTTTCTCACCCAGTCCTCATTC | |
| TLR9 | F: CAGCTAAAGGCCCTG
ACCAA | 160 |
| R:
CCACCGTCTTGAGAATGTTGTG | |
| MyD88 | F:
TATACCAACCCTTGCACCAAGTC | 525 |
| R:
TCAGGCTCCAAGTCAGCTCATC | |
| NF-κB | F: GAG
GACTTGCTGAGGGTTGG | 148 |
| R:
TGGGGTGGTTGATAAGGAGTG | |
Statistical analysis
Data are reported as the means ± SD from 8 animals
for each experimental condition, unless otherwise indicated in the
figures or tables. Inter-group differences were assessed for
statistical significance using the Student's t-test and analysis of
variance. All statistical tests were performed using SPSS 13.0
software (IBM, Chicago, IL, USA). All P-values were two-tailed, and
a value of P<0.05 was defined as the threshold of statistical
significance.
Results
Anti-TLR4 monoclonal antibody reduces
ventilation-induced lung edema and injury
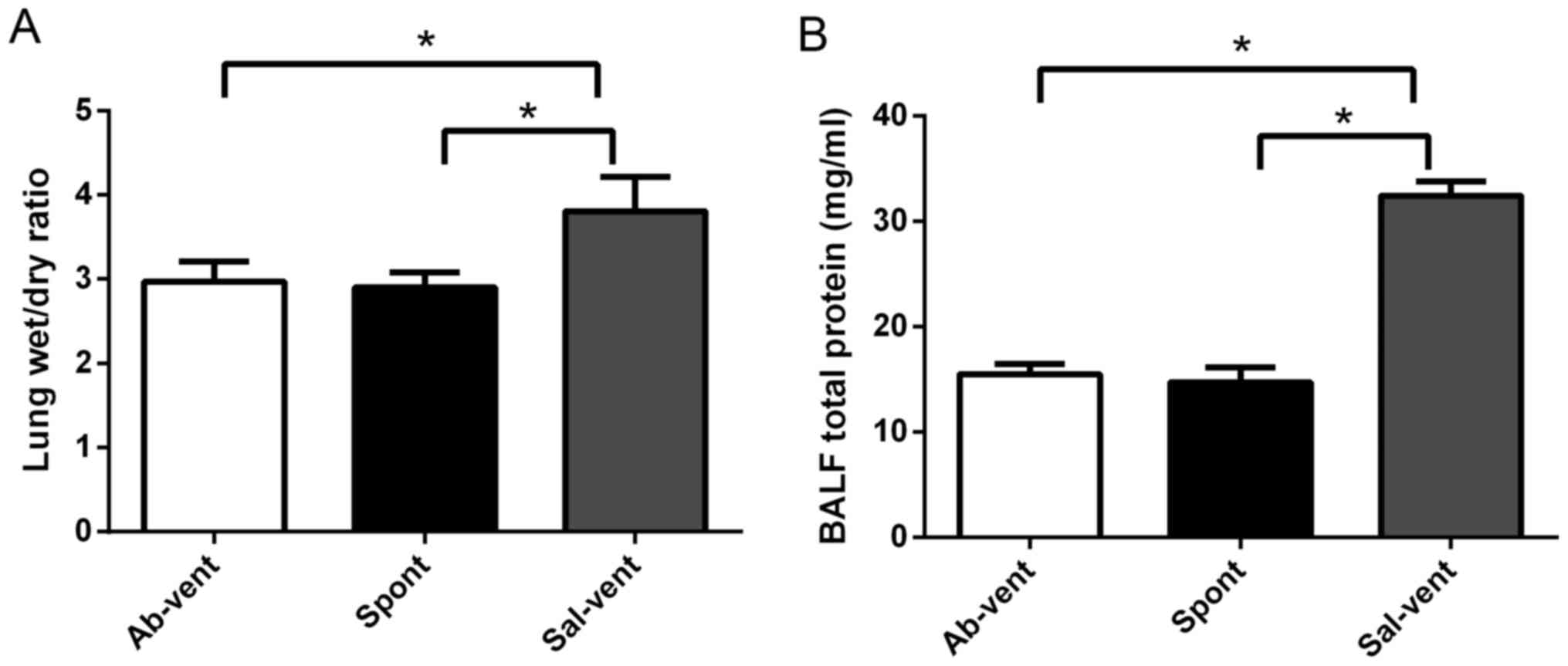
The rats treated intratracheally with anti-TLR4
antibody prior to mechanical ventilation exhibited significantly
less pulmonary edema and BALF total protein than the rats treated
with saline prior to ventilation by determining the lung wet/dry
ratio (Fig. 1). In fact, edema in
the rats pre-treated with antibody was similar to that observed in
the rats treated with saline that were then allowed to breathe
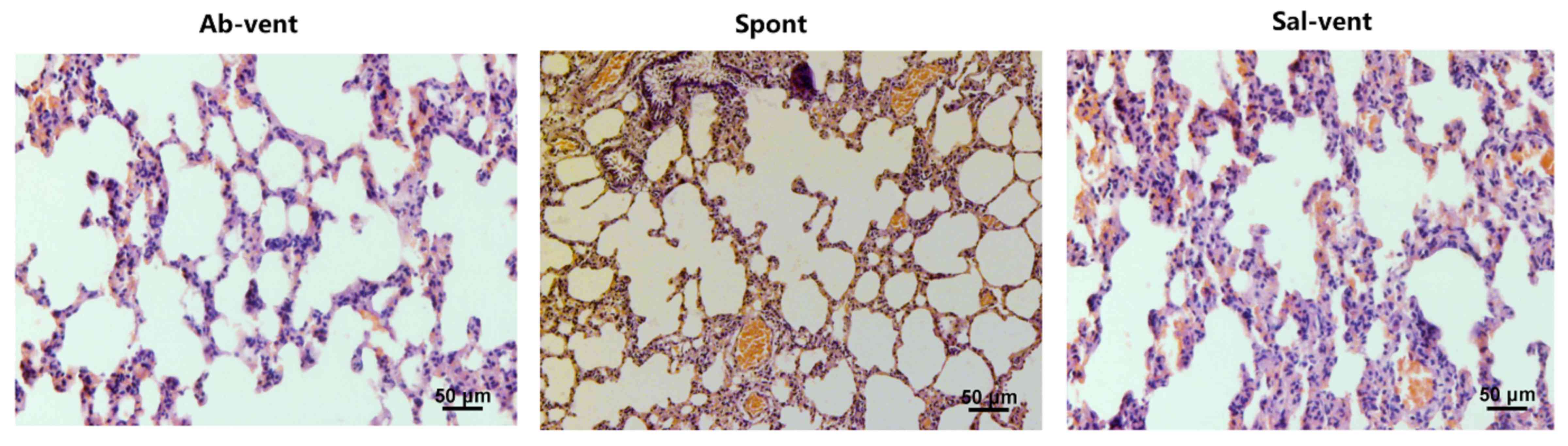
spontaneously. Similarly, the ventilated rats pre-treated with
saline exhibited significantly more alveolar septal thickening than
the ventilated rats pre-treated with antibody and the spontaneously
breathing rats (Fig. 2).
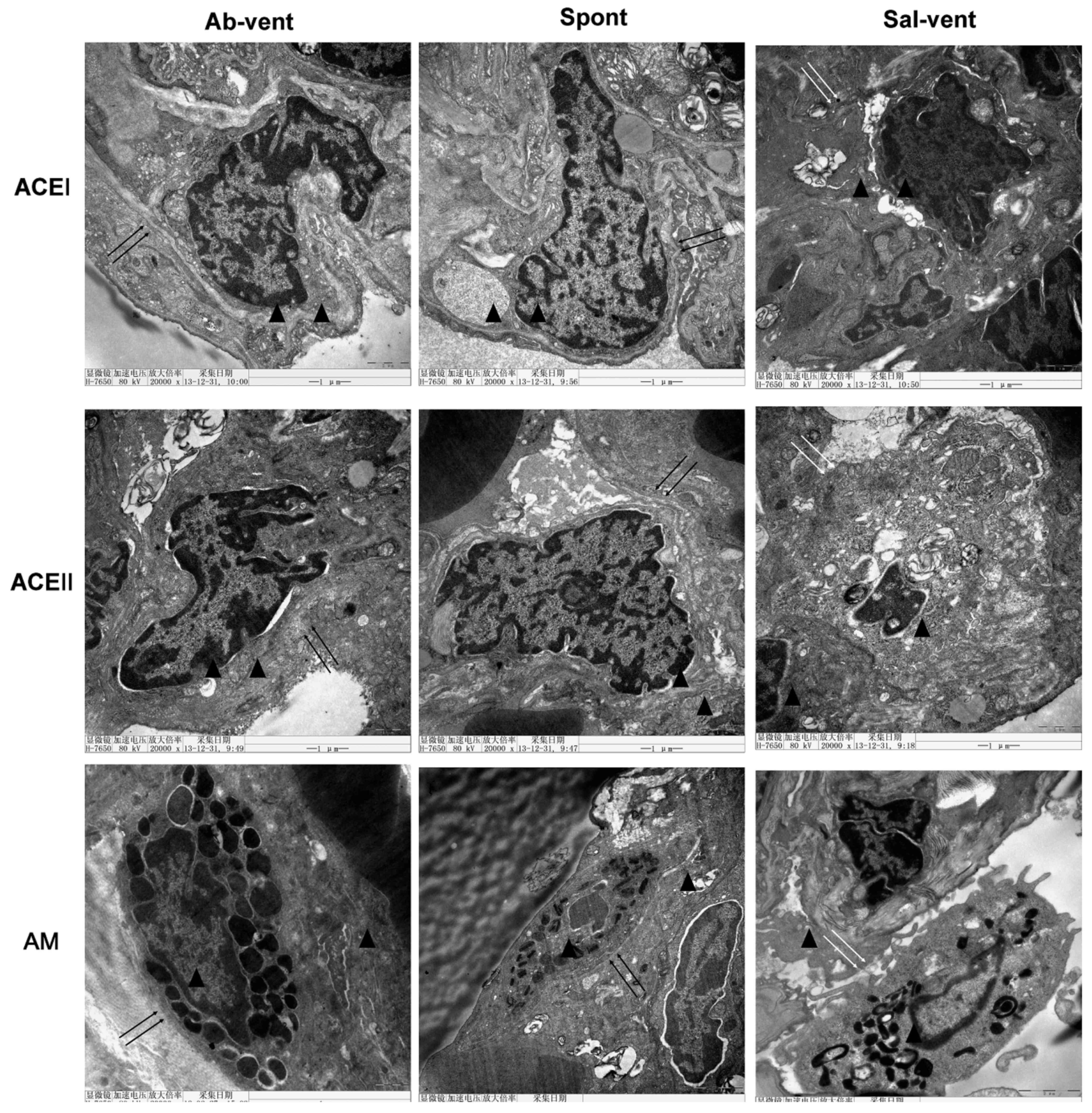
Using electron microscopy to examine alveolar
histopathology in greater detail, we found that, as expected,
alveolar cells in the tissue of the ventilated rats pre-treated
with saline exhibited a disrupted cytoplasmic and nuclear
structure, as well as cell membrane discontinuities (Fig. 3). By contrast, tissue from the
ventilated rats pre-treated with antibody exhibited a normal
cytoplasmic and nuclear structure and continuous cell membrane for
types I and II alveolar epithelial cells and for alveolar
macrophages.
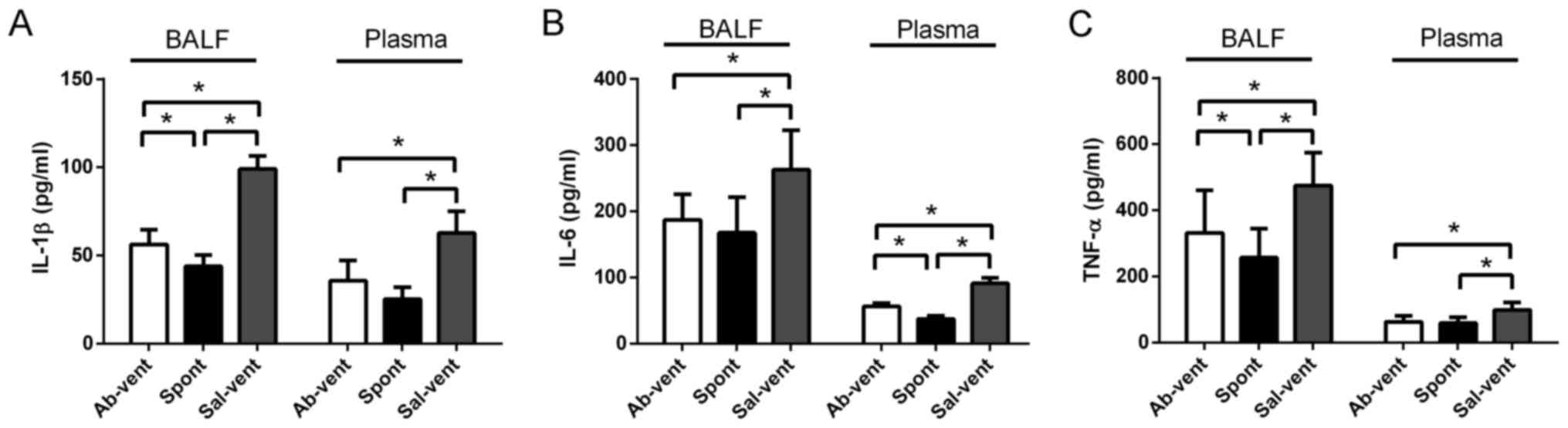
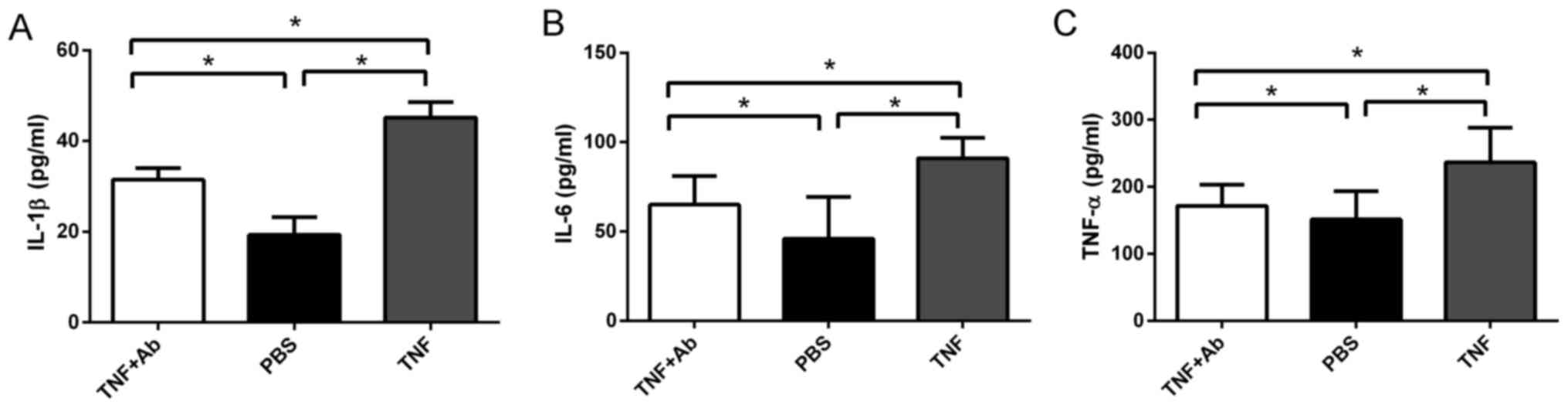
Anti-TLR4 monoclonal antibody reduces the
ventilation-induced secretion of pro-inflammatory cytokines
The levels of IL-1β, IL-6 and TNF-α in BALF and
plasma were significantly higher in the ventilated rats pre-treated
with saline than in the ventilated rats pre-treated with antibody
(Fig. 4). In fact, the levels of
these cytokines were similar between the antibody- pre-treated rats
and the control rats that were not ventilated.
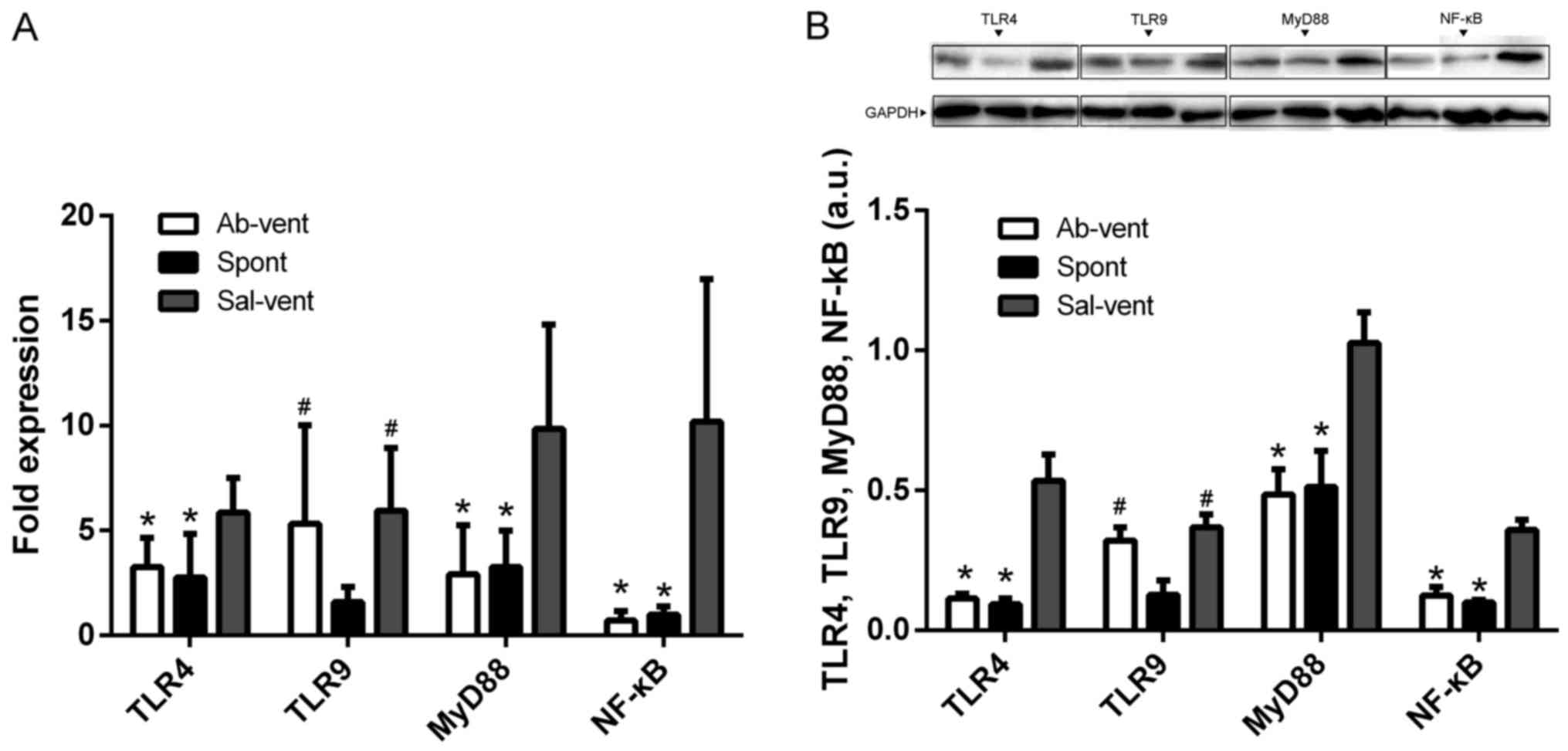
Anti-TLR4 monoclonal antibody reduces the
ventilation-induced activation of NF-κB
Since most TLR signaling pathways stimulated by
mechanical ventilation culminate in the activation of the
transcription factor, NF-κB (8,9,17),
we examined whether pre-treatment with anti-TLR4 monoclonal
antibody attenuates NF-κB activation in alveolar macrophages
cultured from BALF. Indeed, we found that the protein and mRNA
levels of TLR4, NF-κB and MyD88 were significantly higher in the
macrophages obtained from the ventilated rats pre-treated with
saline than in the macrophages obtained from the ventilated rats
pre-treated with anti-TLR4 antibody or in the macrophages from the
rats allowed to breathe spontaneously (Fig. 5).
Since TLR9 is upregulated in lung diseases (8,18),
we wished to determine whether ventilation-induced injury is
associated with changes in the expression of TLR9. The protein and
mRNA levels of TLR9 were similar in the ventilated rats, regardless
of whether they had been pre-treated with saline or anti-TLR4
antibody, and these levels were higher than in the rats allowed to
breathe spontaneously (Fig.
5).
Anti-TLR4 monoclonal antibody reduces
TNF-α-induced cytokine secretion by alveolar macrophages
Since separate studies have demonstrated that TNF-α
is upregulated in ventilation-induced lung injury (8,9,13,19), and that TNF-α can induce the
secretion of some inflammatory cytokines (13), we wished to observe directly
whether TNF-α stimulates the secretion of IL-1β and IL-6 by
alveolar macrophages in our rat model of lung injury. We found that
the levels of IL-1β, IL-6 and TNF-α were significantly higher in
the macrophages from the high tidal ventilated rats stimulated with
TNF-α than in those stimulated with PBS (Fig. 6). Pre-treatment with anti- TLR4
antibody reduced the levels of IL-1β, IL-6 and TNF-α to similar
values as in the macrophages stimulated with PBS.
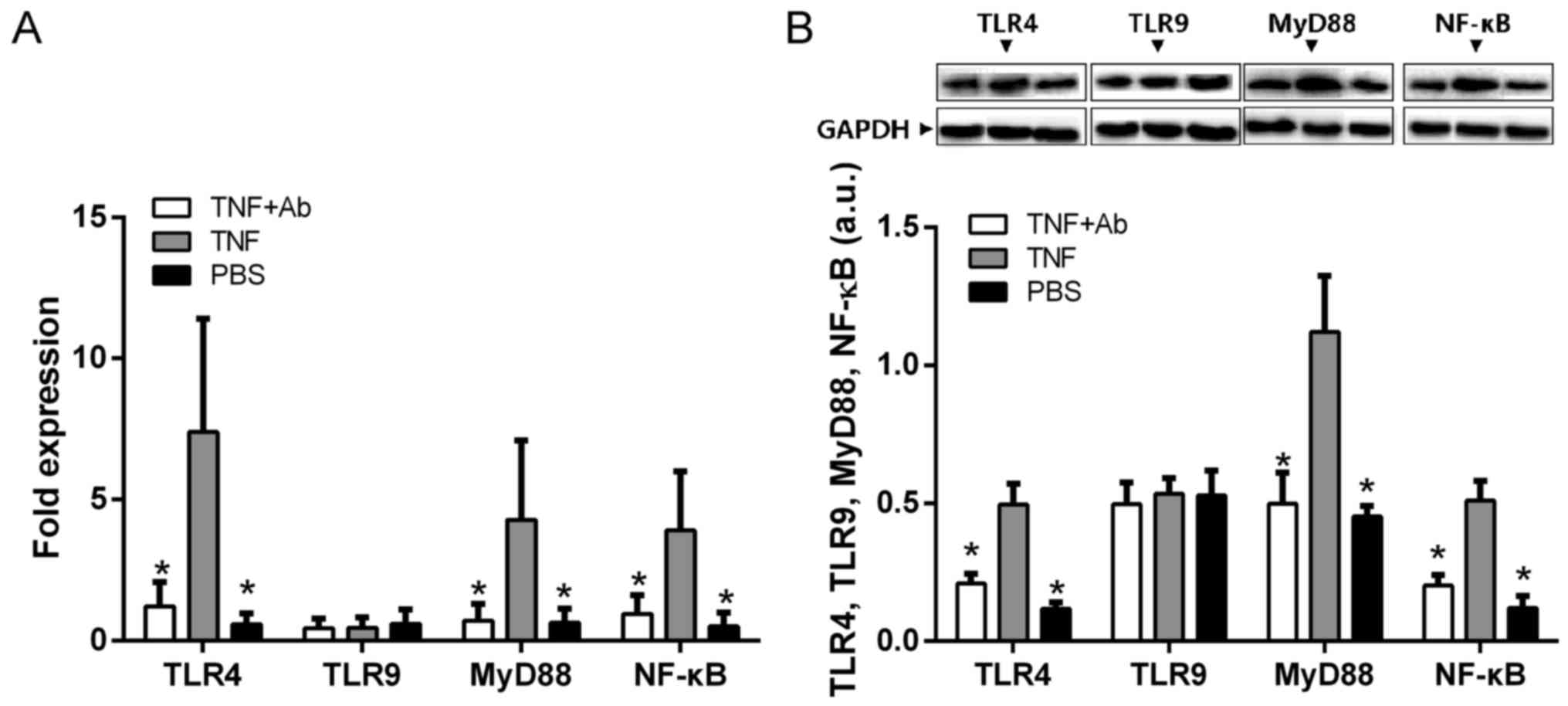
Anti-TLR4 monoclonal antibody attenuates
the TNF-α-induced activation of NF-κB and MyD88 in alveolar
macrophages
Since previous studies have suggested, but not shown
directly, that TNF-α may promote inflammation through the
TLR/NF-κB/MyD88 pathway (20,21), we examined the protein and mRNA
levels of TLR4, NF-κB and MyD88 in alveolar macrophages in the
presence and absence of TNF-α stimulation. All levels were
significantly higher in the stimulated macrophages than in the mock
(PBS)-stimulated macrophages (Fig.
7). The levels in the macrophages stimulated in the presence of
anti-TLR4 antibody were similar to those in the PBS-stimulated
controls.
Discussion
In this study, we combined in vivo and in
vitro approaches to clarify several of the molecular steps in
ventilation-induced lung injury in rats. We provide evidence to
indicate that in response to the stress of mechanical ventilation,
TLR4 activates the NF-κB/MyD88 pathway, thereby stimulating
alveolar macrophages to secrete the pro-inflammatory cytokines,
IL-1β and IL-6. We demonstrated that these events can be triggered
by TNF-α, and that the stimulation of alveolar macrophages with
TNF-α triggered the upregulation of TNF-α, constituting a positive
feedback loop that likely prolongs lung inflammation and
exacerbates lung injury. We also demonstrated that pre-treating
rats with anti-TLR4 monoclonal antibody prior to mechanical
ventilation almost completely eliminated ventilation-induced
changes in the production and secretion of cytokines and TNF-α.
Stimulating cultures of alveolar macrophages with TNF-α in the
presence of anti-TLR4 antibody also eliminated the upregulation and
secretion of cytokines and of TNF-α itself, that was observed when
macrophages are stimulated with TNF-α alone. Thus, the present
study provides several mechanistic insights into
ventilation-induced lung injury that can guide future research in
this area. Our findings also identify monoclonal antibody targeting
of TLR4 as a potential therapy to treat or prevent such lung
injury.
A hallmark of acute lung injury is the structural
impairment of the alveolar-capillary membrane barrier, leading to
increased pulmonary vascular permeability and inflammation
(22,23). Our rat model reproduced the
clinically important manifestations of ventilation-induced lung
injury, including increased alveolar permeability (Fig. 1), overall protein in the alveolar
space (Fig. 1), greater
inflammatory cell infiltration and cytokine production in BALF and
plasma (Fig. 4), and higher
pro-inflammatory signaling via NF-κB pathways (Fig. 3) in alveolar macrophages.
Mechanical ventilation for 4 h at 40 ml/kg caused the levels of
IL-1β, IL-6 and TNF-α to increase almost 2-fold in BALF and plasma,
consistent with other studies using rats (8,24).
Using this system, we demonstrated that treating rats with
anti-TLR4 antibody partially reversed all these ventilation-induced
changes. Not only do these findings provide strong evidence for the
key role of TLR4 in ventilation-induced lung injury, but they also
provide the basis for a molecular targeted therapy.
Non-infectious lung inflammation induced by alveolar
over-distention during mechanical ventilation contributes to
ventilation-induced lung injury (25,26). TLRs have long been recognized to
play a crucial role in both innate and adaptive immune responses to
pathogens and to non-infectious tissue injury (27,28). Indeed, both TLR4 and TLR9 have
been shown to play critical roles in acute lung injury caused by
HTV ventilation, lipopolysaccharide, acid aspiration, hemorrhage,
and ischemia and reperfusion (8,29,30). TLRs span the cell membrane and
simultaneously activate MyD88-dependent signaling pathways
(10,31) and TRIF-dependent pathways. TLR4,
for example, dimerizes upon ligand binding, then it recruits the
downstream adaptor molecule, MyD88, and ultimately activates NF-κB,
inducing the transcription of pro-inflammatory genes, including
ones encoding cytokines. Our in vitro and in vivo
findings in the present study are consistent with the hypothesis
that TLR4 signaling in response to mechanical ventilation
stimulates NF-κB-mediated transcription of pro-inflammatory
cytokines. We found that inhibiting TLR4 signaling by treating rats
with anti-TLR4 monoclonal antibody reduced ventilation-induced lung
injury, reminiscent of how Hayes et al were able to reduce
the severity of lung injury in their rat model by overexpressing
IκBα and thereby inhibiting pulmonary NF-κB (17,32). Indeed, the same pathway may be at
work in the present study and in the work of Hayes et al,
with the only difference being that we inhibited the initial step
of TLR4 activation, while Hayes et al inhibited the final
step of NF-κB activation (32).
The mechanisms through which ventilation-induced
lung injury initially activates the TLR4-MyD88 signaling pathway
are unclear, since the canonical ligand for TLR4 is
lipopolysaccharide due to Gram-negative bacterial infection
(11,33). In the absence of infection, it is
possible that TLR4 is activated by any of several endogenous
ligands, which have been proposed to include high-mobility group
box 1 protein released from necrotic cells, oxidized phospholipids
arising due to locally generated reactive oxygen species,
low-molecular-weight hyaluran and fibrinogen generated during
degradation of extracellular matrix, heat shock proteins released
from necrotic cells, and surfactant protein A (11,34,35). The cyclic stretching of lung
tissue by mechanical ventilation may trigger cell apoptosis and the
release of protein contents as well as harmful oxygen species,
giving rise to endogenous ligands that may activate TLR4.
Regardless of how lung injury initially triggers the
TLR4/MyD88 signaling pathway that activates NF-κB, alveolar
macrophages are likely to be the major cells that receive the
initial pro-inflammatory signal and transduce it into cytokine
signals that prolong and exacerbate injury. Stretching and
inflammation are sufficient to cause these macrophages to release
inflammatory cytokines (36,37), and it was shown that the
ventilation-induced activation of NF-κB in alveolar macrophages
induces the secretion of IL-6 and subsequent IL-1β (38). Therefore, we complemented our
in vivo experiments in rats with in vitro experiments
using alveolar macrophage cultures.
Although our study focused on TLR4, evidence
suggests that TLR9/MyD88 signaling in alveolar macrophages also
plays a role in ventilation-induced lung injury (8). We found that mechanically
ventilating rats with or without pre-treatment with anti-TLR4
antibody led to mRNA and protein levels of TLR9 in alveolar
macrophages that were similar to those in macrophages from
spontaneously breathing animals. We also found that TNF-α
stimulation in the presence or absence of anti- TLR4 antibody did
not significantly alter the levels of TLR9 mRNA or protein in
alveolar macrophage cultures. These findings suggest that TLR4 may
play a larger role than TLR9 in ventilation-induced lung injury.
The fact that we still observed some differences between
antibody-pre-treated rats and spontaneously breathing rats, as well
as between antibody-treated macrophage cultures and PBS-treated
cultures suggests that TLR9 does contribute to lung injury;
however, the precise extent and the signaling pathways involved
require further investigation. Some studies have suggested that
TLR9 in alveolar macrophages plays a role in pathogenesis of VILI
(9,38); thus, the role of this receptor
should be explored carefully.
A limitation to the present study, common to many
animal studies on acute lung injury, is that other factors can
affect injury severity, including the type and dose of anesthetic
use (e.g., sevoflurane, ketamine or protofol) (39,40), variations in pressure support
(41) and positive end-expiratory
pressure. Nevertheless, the insights from our study may have
important clinical implications (42). For example, our results suggest
that strategies to modulate the activation of the TLR4/MyD88
signaling pathway may help treat or even prevent
ventilation-induced lung injury. Our findings also highlight the
need for more extensive research into the potential involvement of
TLR9 in this type of injury.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81060008).
References
|
1
|
The Acute Respiratory Distress Syndrome
Network: Ventilation with lower tidal volumes as compared with
traditional tidal volumes for acute lung injury and the acute
respiratory distress syndrome. N Engl J Med. 342:1301–1308. 2000.
View Article : Google Scholar
|
|
2
|
Parker JC, Hernandez LA and Peevy KJ:
Mechanisms of ventilator-induced lung injury. Crit Care Med.
21:131–143. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frank JA, Wray CM, McAuley DF, Schwendener
R and Matthay MA: Alveolar macrophages contribute to alveolar
barrier dysfunction in ventilator-induced lung injury. Am J Physiol
Lung Cell Mol Physiol. 291:L1191–L1198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kawauchi Y, Takagi H, Hanafusa K, Kono M,
Yamatani M and Kojima N: SIGNR1-mediated phagocytosis, but not
SIGNR1-mediated endocytosis or cell adhesion, suppresses
LPS-induced secretion of IL-6 from murine macrophages. Cytokine.
71:45–53. 2015. View Article : Google Scholar
|
|
5
|
Folco EJ, Sukhova GK, Quillard T and Libby
P: Moderate hypoxia potentiates interleukin-1β production in
activated human macrophages. Circ Res. 115:875–883. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gordon S: Pattern recognition receptors:
doubling up for the innate immune response. Cell. 111:927–930.
2002. View Article : Google Scholar
|
|
7
|
Slutsky AS and Ranieri VM:
Ventilator-induced lung injury. N Engl J Med. 369:2126–2136. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu S, Pan L, Lin F, Zhao Q and Wei Y:
Expression of Toll-like receptor 3 and 4 in the lung tissue of rats
with ventilator-induced lung injury. J Clin Anesthesiol.
29:594–597. 2013.
|
|
9
|
Dai H, Pan L, Lin F, Ge W, Li W and He S:
Role and mechanism of signal pathway mediated by Toll-like receptor
9-myeloid differentiation factor 88 in alveolar macrophages in
ventilator-induced lung injury in rats. Zhonghua Wei Zhong Bing Ji
Jiu Yi Xue. 26:289–293. 2014.In Chinese. PubMed/NCBI
|
|
10
|
Liu Y, Yin H, Zhao M and Lu Q: TLR2 and
TLR4 in autoimmune diseases: a comprehensive review. Clin Rev
Allergy Immunol. 47:136–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Starkhammar M, Larsson O, Kumlien Georén
S, Leino M, Dahlén SE, Adner M and Cardell LO: Toll-like receptor
ligands LPS and poly (I:C) exacerbate airway hyperresponsiveness in
a model of airway allergy in mice, independently of inflammation.
PLoS One. 9:e1041142014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pfeffer K, Matsuyama T, Kündig TM, Wakeham
A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M and
Mak TW: Mice deficient for the 55 kd tumor necrosis factor receptor
are resistant to endotoxic shock, yet succumb to L. monocytogenes
infection. Cell. 73:457–467. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertok S, Wilson MR, Morley PJ, de Wildt
R, Bayliffe A and Takata M: Selective inhibition of intra-alveolar
p55 TNF receptor attenuates ventilator-induced lung injury. Thorax.
67:244–251. 2012. View Article : Google Scholar :
|
|
14
|
Li C, Li B, Dong Z, Gao L, He X, Liao L,
Hu C, Wang Q and Jin Y: Lipopolysaccharide differentially affects
the osteogenic differentiation of periodontal ligament stem cells
and bone marrow mesenchymal stem cells through Toll-like receptor 4
mediated nuclear factor κB pathway. Stem Cell Res Ther. 5:672014.
View Article : Google Scholar
|
|
15
|
Geissmann F, Manz MG, Jung S, Sieweke MH,
Merad M and Ley K: Development of monocytes, macrophages, and
dendritic cells. Science. 327:656–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müller HC, Hellwig K, Rosseau S, Tschernig
T, Schmiedl A, Gutbier B, Schmeck B, Hippenstiel S, Peters H,
Morawietz L, et al: Simvastatin attenuates ventilator-induced lung
injury in mice. Crit Care. 14:R1432010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ko YA, Yang MC, Huang HT, Hsu CM and Chen
LW: NF-κB activation in myeloid cells mediates ventilator-induced
lung injury. Respir Res. 14:692013. View Article : Google Scholar
|
|
18
|
Mortaz E, Adcock IM, Ito K, Kraneveld AD,
Nijkamp FP and Folkerts G: Cigarette smoke induces CXCL8 production
by human neutrophils via activation of TLR9 receptor. Eur Respir J.
36:1143–1154. 2010. View Article : Google Scholar
|
|
19
|
Haitsma JJ, Uhlig S, Göggel R, Verbrugge
SJ, Lachmann U and Lachmann B: Ventilator-induced lung injury leads
to loss of alveolar and systemic compartmentalization of tumor
necrosis factor-alpha. Intensive Care Med. 26:1515–1522. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mukherjee S and Biswas T: Activation of
TOLLIP by porin prevents TLR2-associated IFN-γ and TNF-α-induced
apoptosis of intestinal epithelial cells. Cell Signal.
26:2674–2682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anthwal A, Thakur BK, Rawat MS, Rawat DS,
Tyagi AK and Aggarwal BB: Synthesis, characterization and in vitro
anticancer activity of C-5 curcumin analogues with potential to
inhibit TNF-α-induced NF-κB activation. Biomed Res Int.
2014:5241612014. View Article : Google Scholar
|
|
22
|
Fioretto JR and Carvalho WB: Temporal
evolution of acute respiratory distress syndrome definitions. J
Pediatr (Rio J). 89:523–530. 2013. View Article : Google Scholar
|
|
23
|
Monahan LJ: Acute respiratory distress
syndrome. Curr Probl Pediatr Adolesc Health Care. 43:278–284. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pan WZ, Shi CX, Tian M and Yu JG:
Anti-CD11c antibody, Efalizumab attenuate ventilator-induced lung
injury. Eur Rev Med Pharmacol Sci. 18:2182–2190. 2014.PubMed/NCBI
|
|
25
|
Akella A, Sharma P, Pandey R and Deshpande
SB: Characterization of oleic acid-induced acute respiratory
distress syndrome model in rat. Indian J Exp Biol. 52:712–719.
2014.PubMed/NCBI
|
|
26
|
Belperio JA, Keane MP, Lynch JP III and
Strieter RM: The role of cytokines during the pathogenesis of
ventilator-associated and ventilator-induced lung injury. Semin
Respir Crit Care Med. 27:350–364. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meylan E, Tschopp J and Karin M:
Intracellular pattern recognition receptors in the host response.
Nature. 442:39–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O'Neill LA and Bowie AG: The family of
five: TIR-domain-containing adaptors in Toll-like receptor
signalling. Nat Rev Immunol. 7:353–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He Z, Gao Y, Deng Y, Li W, Chen Y, Xing S,
Zhao X, Ding J and Wang X: Lipopolysaccharide induces lung
fibroblast proliferation through Toll-like receptor 4 signaling and
the phosphoinositide3-kinase-Akt pathway. PLoS One. 7:e359262012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prakash A, Mesa KR, Wilhelmsen K, Xu F,
Dodd-o JM and Hellman J: Alveolar macrophages and Toll-like
receptor 4 mediate ventilated lung ischemia reperfusion injury in
mice. Anesthesiology. 117:822–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Perkins DJ and Vogel SN: Inflammation:
Species-specific TLR signalling - insight into human disease. Nat
Rev Rheumatol. 12:198–200. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayes M, Curley GF, Masterson C, Contreras
M, Ansari B, Devaney J, O'Toole D and Laffey JG: Pulmonary
overexpression of inhibitor κBα decreases the severity of
ventilator-induced lung injury in a rat model. Br J Anaesth.
113:1046–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bryant CE, Symmons M and Gay NJ: Toll-like
receptor signalling through macromolecular protein complexes. Mol
Immunol. 63:162–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hazen SL: Oxidized phospholipids as
endogenous pattern recognition ligands in innate immunity. J Biol
Chem. 283:15527–15531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo
Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al:
Regulation of lung injury and repair by Toll-like receptors and
hyaluronan. Nat Med. 11:1173–1179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oya K, Sakamoto N and Sato M: Hypoxia
suppresses stretch-induced elongation and orientation of
macrophages. Biomed Mater Eng. 23:463–471. 2013.PubMed/NCBI
|
|
37
|
Wu J, Yan Z, Schwartz DE, Yu J, Malik AB
and Hu G: Activation of NLRP3 inflammasome in alveolar macrophages
contributes to mechanical stretch-induced lung inflammation and
injury. J Immunol. 190:3590–3599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dai H, Pan L, Lin F, Ge W, Li W and He S:
Mechanical ventilation modulates Toll-like receptors 2, 4, and 9 on
alveolar macrophages in a ventilator-induced lung injury model. J
Thorac Dis. 7:616–624. 2015.PubMed/NCBI
|
|
39
|
Wu GJ, Chen TL, Ueng YF and Chen RM:
Ketamine inhibits tumor necrosis factor-alpha and interleukin-6
gene expressions in lipopolysaccharide-stimulated macrophages
through suppression of toll-like receptor 4-mediated c-Jun
N-terminal kinase phosphorylation and activator protein-1
activation. Toxicol Appl Pharmacol. 228:105–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang P, Yang N, Zhang X and Xu X: The
significance and mechanism of propofol on treatment of ischemia
reperfusion induced lung injury in rats. Cell Biochem Biophys.
70:1527–1532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Spieth PM, Carvalho AR, Güldner A, Pelosi
P, Kirichuk O, Koch T and de Abreu MG: Effects of different levels
of pressure support variability in experimental lung injury.
Anesthesiology. 110:342–350. 2009.PubMed/NCBI
|
|
42
|
Biehl M, Kashiouris MG and Gajic O:
Ventilator-induced lung injury: Minimizing its impact in patients
with or at risk for ARDS. Respir Care. 58:927–937. 2013. View Article : Google Scholar : PubMed/NCBI
|