Introduction
Acute kidney injury (AKI) is related to the toxic
effects of various chemical agents and reactive oxygen species
(ROS) and results in increased risk for progression to chronic
kidney disease. The incidence of AKI in hospital patients has
generally been reported to range from 2 to 7%, and AKI has been
shown to be associated with mortality (1). Ischemia/reperfusion (I/R) injury is
one of the major causes of AKI, resulting from a generalized
impairment of oxygen and nutrient delivery along with a mismatch of
local tissue oxygen supply (2).
I/R kidney injury is characterized by inflammation, peroxidation,
DNA damage, apoptosis, vascular leakage, immune activation,
endothelial cell activation, leukocyte adhesion and compromised
microvascular blood flow (3,4).
Moreover, I/R kidney injury increases ROS levels (5).
ROS, including hydrogen peroxide
(H2O2), enhance tubular stress and epithelial
cell injury, interfere with normal regenerative processes and lead
to fibrosis (6,7). Furthermore, ROS induced by oxidative
stress are implicated in the pathogenesis of many renal diseases,
such as acute glomerulo nephritis, acute interstitial nephritis and
tubular cell injury (8).
Additionally, ROS induce pro-inflammatory and chemotactic
cytokines, such as cyclooxygenase (COX)-2, inducible nitric oxide
synthase (iNOS), tumor necrosis factor-α (TNF-α), transforming
growth factor-β (TGF-β), interleukin-1β (IL-1β), IL-6, IL-8 and
activated inflammatory cells in the kidneys (9,10).
In response to oxidative stress, tubular cells also express
Toll-like receptor, complement and complement receptors, and
costimulatory molecules, which regulate T-lymphocyte activity
(11). COX-2 and iNOS are
important components in a network of inflammatory cytokines
activated by ROS in the kidney (12,13). The expression of COX-2 and iNOS is
controlled through the transcription factors, nuclear factor-κB
(NF-κB) and activator protein-1 (AP-1) (14–16). NF-κB and AP-1 have been shown to
be crucial for the induction of genes involved in inflammation
(17). Moreover, NF-κB and AP-1
are important ROS-sensitive transcription factors that regulate the
transcription of genes encoding inflammatory cytokines and
chemokines (18).
The small heterodimer partner (SHP, also known as
NR0B2) is an atypical orphan nuclear receptor that is structurally
related to nuclear hormone receptors but lacks both a known
physiological ligand and a DNA binding domain (19). The human SHP gene is
expressed in various tissues, including the heart, pancreas, lung,
spleen, smooth muscle and kidney (20–23). SHP functions as a transcriptional
co-regulator by directly interacting with other nuclear receptors
and transcription factors (24–27). Moreover, SHP plays a crucial role
in negatively regulating the transactivation of various
transcription factors involved in diverse physiological and
metabolic pathways (26). Recent
studies have demonstrated that the NF-κB p65 protein complex
requires interaction with SHP, which is an intrinsic negative
regulator of Toll-like receptor-triggered inflammation (28). These findings suggest that SHP may
exert anti-inflammatory effects.
In the present study, we aimed to ascertain whether
SHP is effective in preventing H2O2-induced
oxidative stress, which can trigger inflammation in tubular
epithelial cells, and to explore the molecular mechanisms
underlying the protective effects of SHP. We examined whether SHP
attenuates H2O2-induced COX-2 and iNOS
expression through suppression of the transcription factors NF-κB
and AP-1 in human renal proximal tubule epithelial (HK-2)
cells.
Materials and methods
Cell culture and reagents
Human renal proximal tubule epithelial HK-2 cells
(ATCC, Manassas, VA, USA), were cultured. Cells were passaged every
3–4 days in 100-mm dishes containing combined Dulbecco's modified
Eagle's medium (DMEM)-F-12 medium supplemented with 10% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin
(all from Sigma, St. Louis, MO, USA). The cells were incubated in a
humidified atmosphere of 5% CO2 and 95% air at 37°C for
24 h and sub-cultured at 70–80% confluence. For experimental use,
the HK-2 cells were plated onto 60-mm dishes in medium containing
10% FBS for 24 h and cells were then switched to DMEM-F12 with 1%
FBS for 16 h. The cells were then treated with
H2O2 (0, 100, 300, 500 and 1,000 μM).
The cells were harvested at the end of treatment for further
analysis. SP600125 (a specific JNK inhibitor) was obtained from
Calbiochem (San Diego, CA, USA). N-acetyl-L-cysteine (NAC)
was obtained from Sigma-Aldrich (Steinheim, Germany). Bay11-7082
was obtained from BioMol (Plymouth Meeting, PA, USA).
Animals
The animal experiments were approved by the Animal
Care Regulations (ACR) Committee of Chonnam National University
Medical School and our protocols conformed to the institution
guidelines for experimental animal care and use. Male 8-week-old
C57BL6 mice were purchased from Samtako (Osan, Korea). Mice were
divided into two groups. The control group (n=8) underwent a sham
operation without clamping of the renal pedicle. In the
experimental group, in order to induce I/R kidney injury, both
renal pedicles of the mice (n=8) were clamped for 30 min.
Twenty-four hours later, the mice were anesthetized with 2%
isoflurane and 100% oxygen. Blood samples were collected from the
left ventricle and analyzed for creatinine. Plasma creatinine was
measured using the Jaffe method (Olympus 5431; Olympus Optical,
Tokyo, Japan). The kidney was rapidly removed, and then processed
for semi-quantitative immunoblotting. Another series of experiment
was carried out for the assay of real-time polymerase chain
reaction (PCR). The mice were decapitated and their kidneys were
excised and maintained at −70°C until assayed for the mRNA
expression by real-time PCR.
Real-time PCR
Total RNA was isolated with TRIzol reagent
(Invitrogen, Carlsbad, CA, USA). cDNA was constructed by reverse
transcribing 1 μg of total RNA using oligo(dT) priming and
superscript reverse transcriptase II (Invitrogen). cDNA was
quantified using SmartCycler II System (Cepheid, Sunnyvale, CA,
USA) and SYBR-Green was used for detection. Each PCR reaction was
performed using 10 μM forward primer, 10 μM reverse
primer, 2X SYBR-Green Premix Ex Taq (Takara Bio, Inc., Shiga,
Japan), 0.5 μl cDNA and H2O to bring the final
volume to 20 μl. Relative levels of mRNA were determined by
real-time PCR, using a Rotor-Gene™ 3000 detector system (Corbett
Research, Mortlake, New South Wales, Australia). The specific
primers sequences were: hSHP forward, 5′-CAATGTGGGAGGCGGCT-3′ and
reverse, 5′-TGAAAGGGACCATCCTCTTCA-3′ (60 bp); hCOX-2 forward,
5′-CGAGGTGTATGTATGAGTGT-3′ and reverse, 5′-TCTAGCCAGAGTTTCACCGT-3′
(594 bp); hiNOS forward, 5′-ACGTGCGTTACTCCACCAACA-3′ and reverse,
5′-CATAGCGGATGAGCTGAGCATT-3′ (114 bp); hIL-1β forward,
5′-TGATGTTCCCATTAGACAGC-3′ and reverse, 5′-GAGGTGCTGATGTACCAGTT-3′
(378 bp); hTNF-α forward, 5′-GCATGATCCGCGACGTGGAA-3′ and reverse,
5′-AGATCCATG CCGTTGGCCAG-3′ (352 bp); hGAPDH forward,
5′-GCCAAAAGGGTCATCATCTC-3′ and reverse, 5′-GGCCATCCACAGTCTTCT-3′
(229 bp). The PCR was performed according to the following steps:
i) 95°C for 5 min; ii) 95°C for 20 sec; iii) 58 to 62°C for 20 sec
(optimized for each primer pair); iv) 72°C for 30 sec. Steps 2–4
were repeated for an additional 40 cycles, while at the end of the
last cycle, the temperature was increased from 60 sec to 95°C to
produce a melting curve. Data from the reaction were collected and
analyzed with Corbett Research Software. The comparative critical
threshold (Ct) values from quadruplicate measurements were used to
calculate the gene expression, with normalization to GAPDH as an
internal control. Melting curve analysis was performed to enhance
specificity of the amplification reaction.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Viability of the HK-2 cells was determined using the
MTT assay. HK-2 cells were subcultured in a 96-well plate at an
initial density of 5×103 cells/ml. Cells were incubated
with fresh medium containing 0, 300, 500 and 1000 μM of
H2O2 for 6 h. After incubation, 50 μl
of 5 mg/ml MTT (Sigma) was added to each well of the 96-well plates
and subsequently incubated for 4 h at 37°C. Supernatants were
removed by aspiration and then dimethysulfoxide (DMSO) was added to
solubilize the precipitated dyes. Absorbance was measured at a
wavelength of 570 nm. The viability of the cells was expressed as
the fraction of surviving cells relative to the untreated
controls.
Intracellular level of ROS
HK-2 cells were cultured in 96-well plates until
they reached confluence. Cells were incubated with fresh medium
containing 0, 300, 500 or 1,000 μM of
H2O2 for 6 h. Cells were washed twice with
Hanks' Balanced Salt Solution (HBSS) and incubated with HBSS
(without phenol red) containing 10 μM
2′,7′-dichlorofluorescein diacetate (DCF-DA; Molecular Probes,
Camarillo, CA, USA) for 30 min at 37°C. Fluorescence intensity was
analyzed by a fluorescence reader (Fluoroscan Ascent FL;
Labsystems, Helsinki, Finland) using 485 nm excitation and 538 nm
emission filter. The images were obtained.
Protein extraction and western blot
analysis
The kidney was homogenized in ice-cold isolation
solution containing 0.3 M sucrose, 25 mM imidazole, 1 mM EDTA, 8.5
μM leupeptin, and 1 mM phenylmethylsulfonyl fluoride (pH
7.2). The homogenates were centrifuged, and the total protein
concentration was measured using the Pierce BCA protein assay kit
(Thermo Fisher Scientific, Rockford, IL, USA). All samples were
adjusted with isolation solution to normalize the protein
concentrations, solubilized at 65°C for 15 min in sodium dodecyl
sulfate (SDS)-containing sample buffer, and then stored at -20°C.
The HK-2 cells were harvested, washed twice with ice-cold
phosphate-buffered saline (PBS) and re-suspended in lysis buffer
(20 mM Tris-HCl, pH 7.4, 0.01 mM EDTA, 150 mM NaCl, 1 mM PMSF, 1
μg/ml leupeptin, 1 mM Na3VO4) and
sonicated briefly. After centrifugation, the supernatant was
prepared as protein extract, and protein concentrations were
measured (Pierce BCA protein assay reagent kit; Thermo Fisher
Scientific). Equal amounts of protein were separated on 9 or 12%
sodium dodecyl sulfate polyacrylamide gels. The proteins were
electrophoretically transferred onto nitrocellulose membranes using
Bio-Rad Mini Protean II apparatus (Bio-Rad, Hercules, CA, USA). The
blots were blocked with 5% milk in PBS-T (80 mM
Na2HPO4, 20 mM NaH2PO4,
100 mM NaCl, and 0.1% Tween-20 at pH 7.5) for 2 h. The anti-SHP
antibody was provided by Professor Heung-Sik Choi (Chonnam National
University, Korea). The NF-κB p65 (8242; Cell Signaling Technology,
Beverly, MA, USA), anti-IκBα (SC-1643; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), anti-iNOS (610600; BD Transduction
Laboratories, San Jose, CA, USA), anti-COX-2 (160107; Cayman
Chemical, Ann Arbor, MI, USA), and β-actin (a5316; Sigma)
antibodies were diluted in a blocking buffer and incubated with the
blots overnight at 4°C. The bound antibodies were detected with a
1:2,500 dilution of horseradish peroxidase-conjugated secondary
antibody according to the instructions provided with the ECL kit
(Amersham, Franklin Lakes, NJ, USA).
Small interfering RNA transfection
For knockdown of SHP expression, siRNAs for SHP were
chemically synthesized (Dharmacon Inc., Lafayelle, CA, USA) and
transfected according to the manufacturer's instructions. HK-2
cells were transfected with siRNA using DhamaFECT 2 reagent
(Dharmacon Inc.). Efficiency of knockdown was performed through
western blot analysis.
Transient transfection of the plasmid
construct, and SHP, AP-1 and NF-κB reporter
pcDNA3-mSHP and the reporter construct was kindly
provided by Professor Heung-Sik Choi (Chonnam National University).
The mouse SHP was subcloned into the NcoI/XhoI site
of the pcDNA3 vector. pcDNA3-mSHP or pcDNA3 was introduced into the
HK-2 cells by FuGene HD reagent (Promega, Madison, WI, USA). Two
days after transfection, we identified the overexpression of SHP
and Flag in the HK-2 cells by western blot analysis. AP-1 and NF-κB
reporter construct were purchased from Clontech (Palo Alto, CA,
USA). Once the cells had reached 60–70% confluence, they were
washed with DMEM-F-12 medium and incubated in the medium without
serum and antibiotics for 18 h. The cells were then transfected
with SHP, AP-1 and NF-κB reporter containing the pGL3 vector using
FuGene HD reagent. Reporter transfected cells were pretreated with
NAC and Bay for 1 h and incubated with 500 μM
H2O2 for 6 h. Also, c-Jun dominant-negative
construct and pcDNA3-mSHP were co-transfected with the reporter
construct. The luciferase activity was measured using a
luminometer.
Promotor activity of SHP, AP-1 and
NF-κB
The transcriptional regulation of SHP, AP-1 and
NF-κB was examined by transient transfection of an SHP, AP-1 and
NF-κB promoter-luciferase reporter construct (pGL3-SHP, pGL3-AP-1
and pGL3-NF-κB). HK-2 cells (5×105) were seeded and
grown until they reached 60–70% confluence and pGL3-SHP, pGL3-AP-1
and pGL3-NF-κB wild-type and pGL3-empty were trans-fected into the
cells using FuGene HD reagent, according to the manufacturer's
protocol. The pRL-null plasmid encoding Renilla luciferase
was included in all the samples to monitor transfection efficiency.
At 24 h post-transfection, the levels of Firefly and Renilla
luciferase activity were measured sequentially from a single sample
using the Dual-Glo Luciferase assay system (Promega). Firefly
luciferase activity was normalized to Renilla activity and
the relative amount of luciferase activity in the untreated
cells.
Electrophoretic mobility shift assay
Nuclear extracts of HK-2 cells were prepared with
the NE-PER nuclear extraction reagent (Pierce Biotechnology).
Biotin labeled oligonucleotides were
5′-biotin-AGTTGAGGGGACTTTCCCAGGC-3′ for NF-κB and
5′-biotin-CGCTTGATGACTCAGCGGAA-3′ for AP-1 as well as nonlabeled
NF-κB oligonucleotide. The binding reactions contained 10 μg
of nuclear extract protein, buffer (10 mM Tris, pH 7.5, 50 mM KCl,
5 mM MgCl2, 1 mM dithiothreitol, 0.05% Nonidet P-40, and
2.5% glycerol), 1 μg of poly(dI-dC) and 2 nM of
biotin-labeled DNA. The reactions were incubated at 23°C for 20
min. The competition reactions were performed by adding 10-fold
excess unlabeled double-stranded NF-κB consensus oligonucleotide to
the reaction mixture. The reactions were electrophoresed on a 6%
precasted Tris-borate-EDTA gel (Invitrogen) at 100 V for 1 h 30 min
in a 100 mM Tris-borate-EDTA buffer. The reactions were transferred
to a nylon membrane. The biotin-labeled DNA was detected with
LightShift chemiluminescent electrophoretic mobility shift assay
kit (Pierce Biotechnology).
Statistical analysis
The results are expressed as mean ± SEM. Multiple
comparisons among 3 groups were performed using one-way ANOVA and
the post hoc Tukey's honestly significant difference test.
Differences with values of p<0.05 were considered
significant.
Results
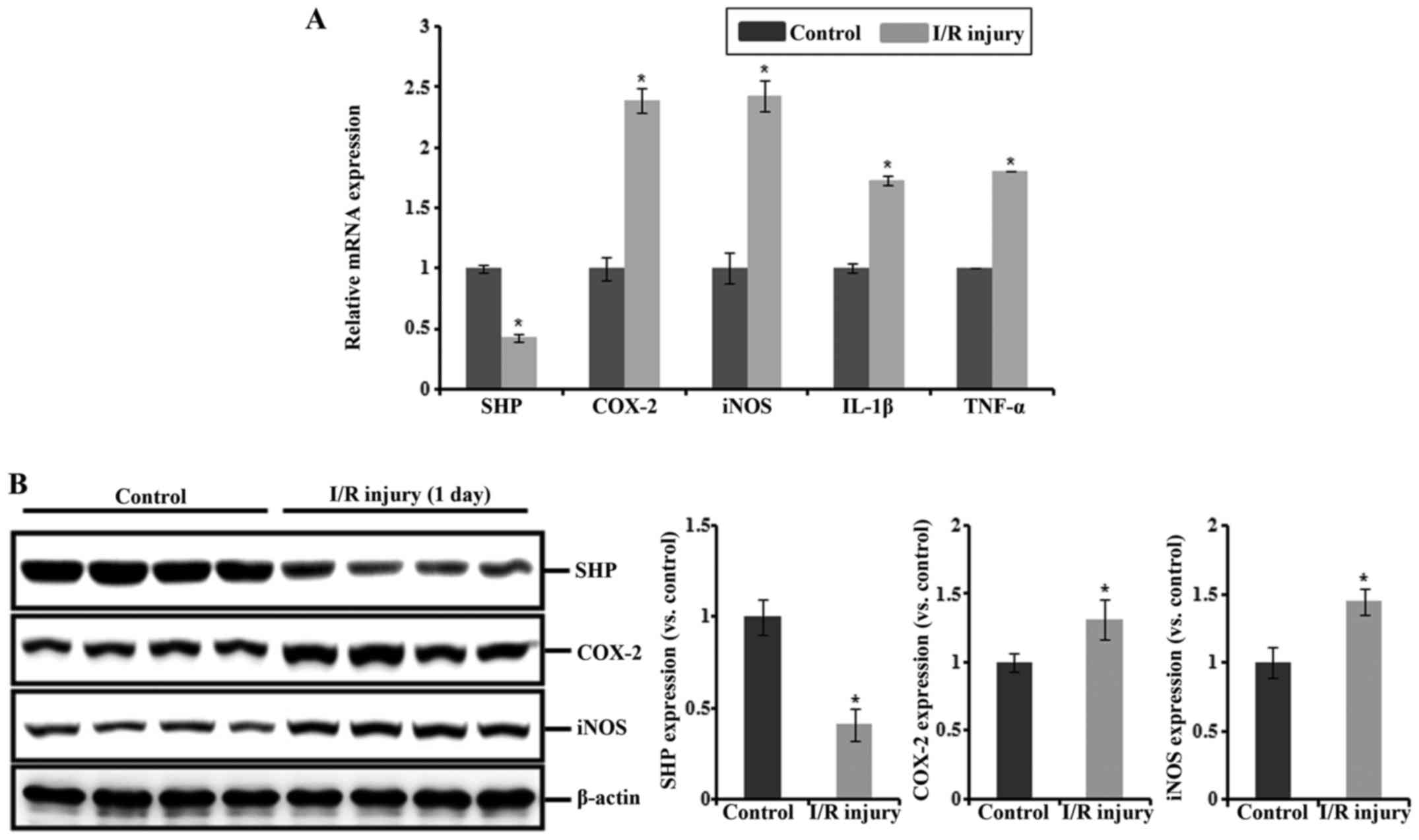
Expression of SHP and inflammatory
proteins in I/R-induced kidney injury
Serum creatinine levels were significantly increased
in the I/R injury mice compared with that in the sham-operated
controls. The mRNA expression levels of COX-2, iNOS, IL-1β and
TNF-α were increased in the I/R injury mice compared with those in
the controls, whereas that of SHP was reduced in these mice
(Fig. 1A). Consistent with this,
the protein expression of SHP was decreased after I/R injury,
whereas the expression levels of COX-2 and iNOS proteins were
increased (Fig. 1B).
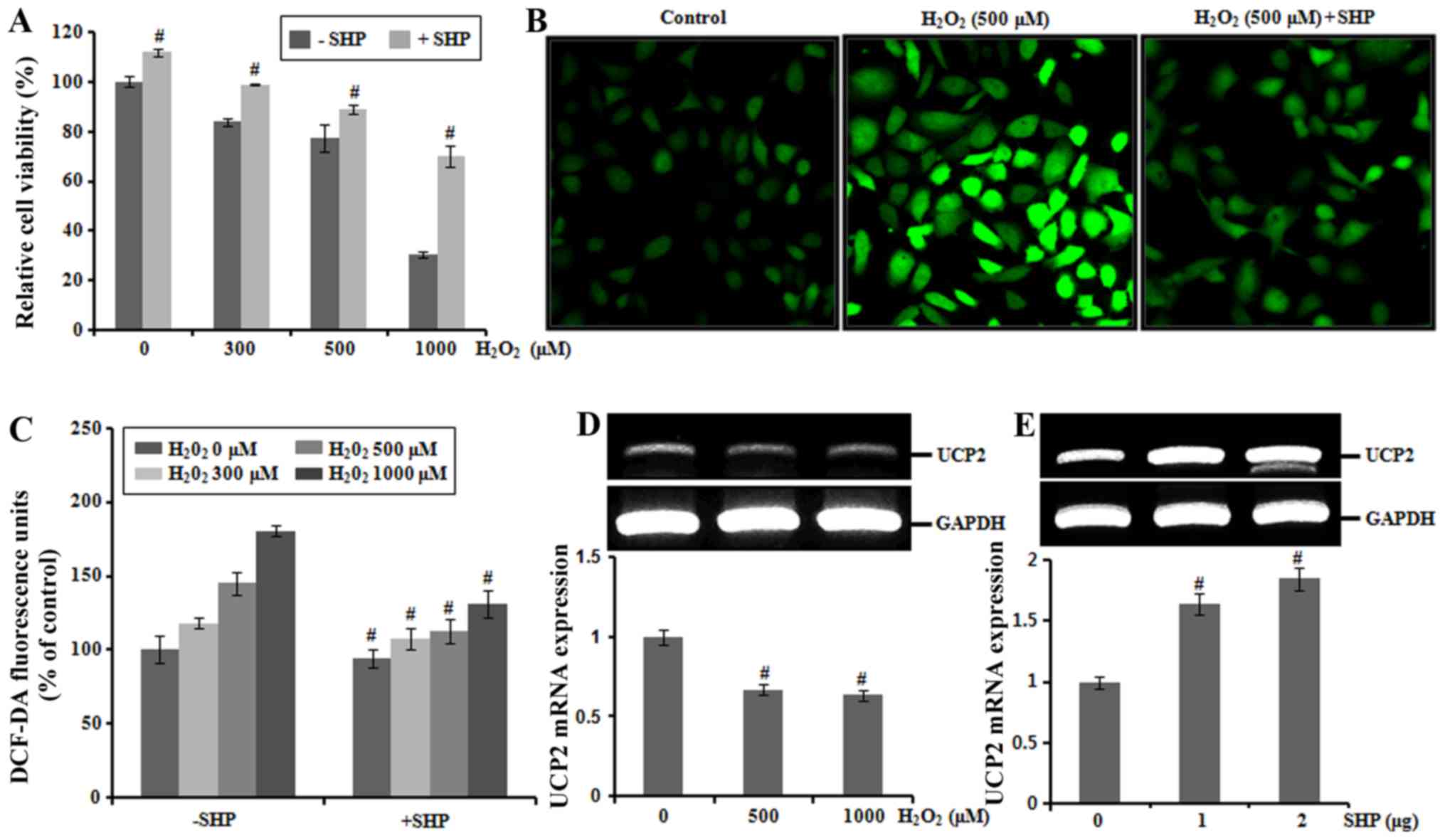
Expression of SHP, COX-2, and iNOS and
the effect of SHP on the production of ROS by
H2O2 exposure in HK-2 cells
H2O2 treatment (300, 500 and
1,000 μM) for 6 h decreased HK-2 cell viability in a
concentration-dependent manner as determined by MTT assays. To
examine the physiological effects of SHP on HK-2 cells, we
determined the viability of SHP-transfected HK-2 cells treated with
H2O2. Decreased cell viability induced by
H2O2 treatment was recovered by
overexpression of SHP (Fig. 2A).
We next assessed the formation of ROS using the ROS-sensitive
fluorescent dye DCF-DA in HK-2 cells. The level of intracellular
ROS increased progressively after incubation of cells with 500
μM H2O2, reaching a peak at 30 min,
whereas overexpression of SHP attenuated the increased production
of ROS (Fig. 2B).
H2O2 exposure increased ROS production in a
concentration-dependent manner in the HK-2 cells. In contrast,
following H2O2 exposure at 0, 300, 500 and
1,000 μM, ROS production was attenuated by 10, 11, 12 and
13% in the SHP-transfected HK-2 cells, respectively compared with
levels in the non-SHP-transfected cells (Fig. 2C). We also performed additional
experiments to examine whether SHP may play a role in the
inhibition of ROS production through mitochondrial uncoupling
protein 2 (UCP2). H2O2 exposure decreased
UCP2 mRNA levels in a concentration-dependent manner in the HK-2
cells (Fig. 2D). SHP transfection
induced gene expression of UCP2, suggesting that UCP2 is involved
in the SHP-mediated suppression of ROS production (Fig. 2E).
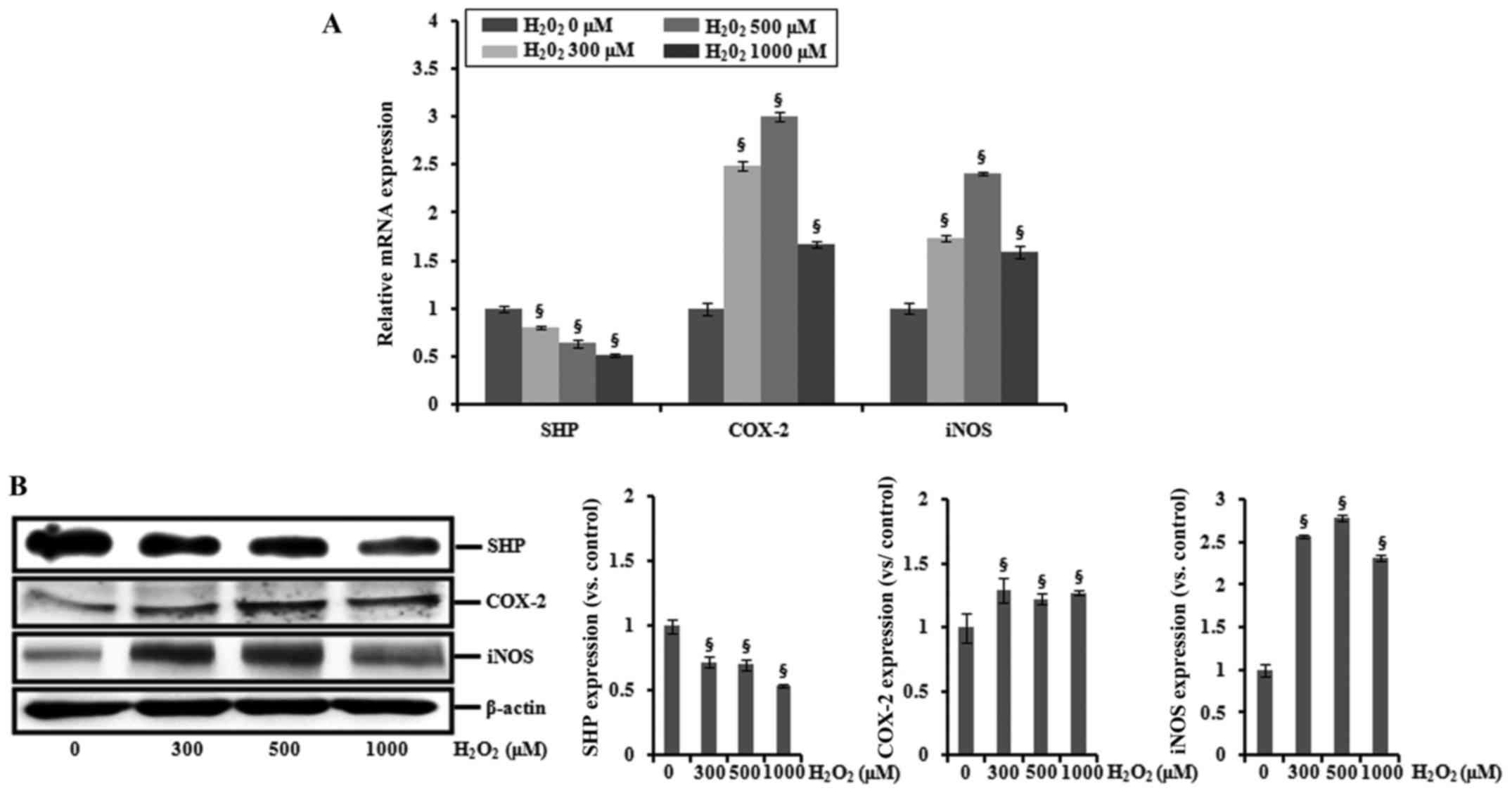
HK-2 cells were incubated with 0, 300, 500 or 1,000
μM H2O2 for 6 h, and the expression
levels of SHP, COX-2 and iNOS were determined by real-time PCR and
western blotting. As shown in Fig.
3, H2O2 treatment increased the mRNA and
protein expression of COX-2 and iNOS, whereas levels of SHP mRNA
and protein were decreased.
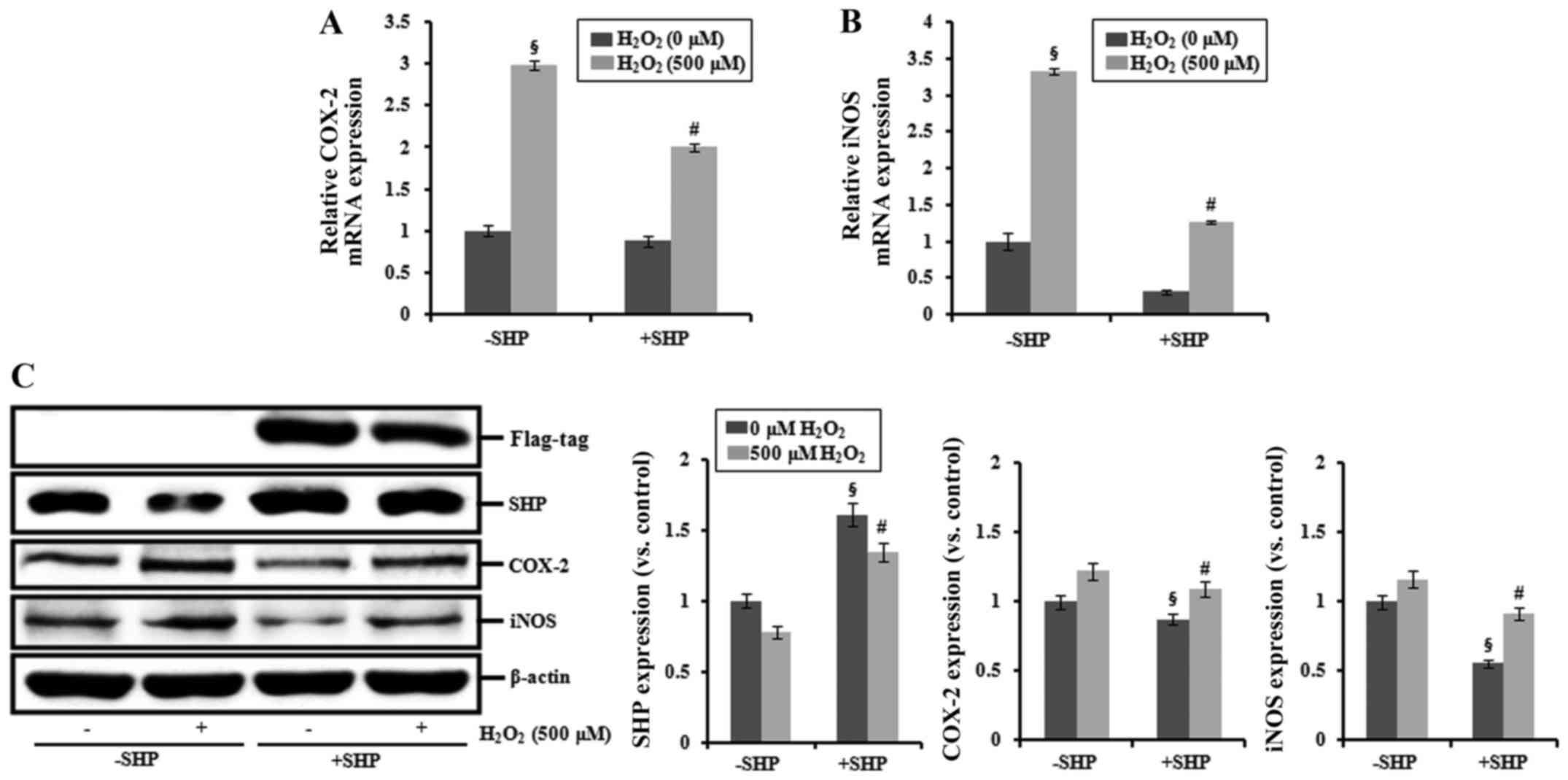
Effects of SHP on the expression of COX-2
and iNOS
COX-2 and iNOS mRNAs were increased in cells exposed
to 500 μM H2O2, and transfection with
SHP blocked this increase (Fig. 4A
and B). SHP mRNA expression was significantly increased in the
SHP-transfected cells (data not shown). In addition, SHP-Flag-tag
construct transfection induced increased protein expression of SHP
compared with that noted in the non-SHP-transfected cells.
Additionally, the expression levels of COX-2 and iNOS proteins were
increased in cells exposed to 500 μM
H2O2 compared with those in the untreated
controls, and transfection with SHP suppressed this effect
(Fig. 4C).
Effects of an NF-κB inhibitor,
dominant-negative c-Jun, and NAC on the transcriptional activation
of SHP
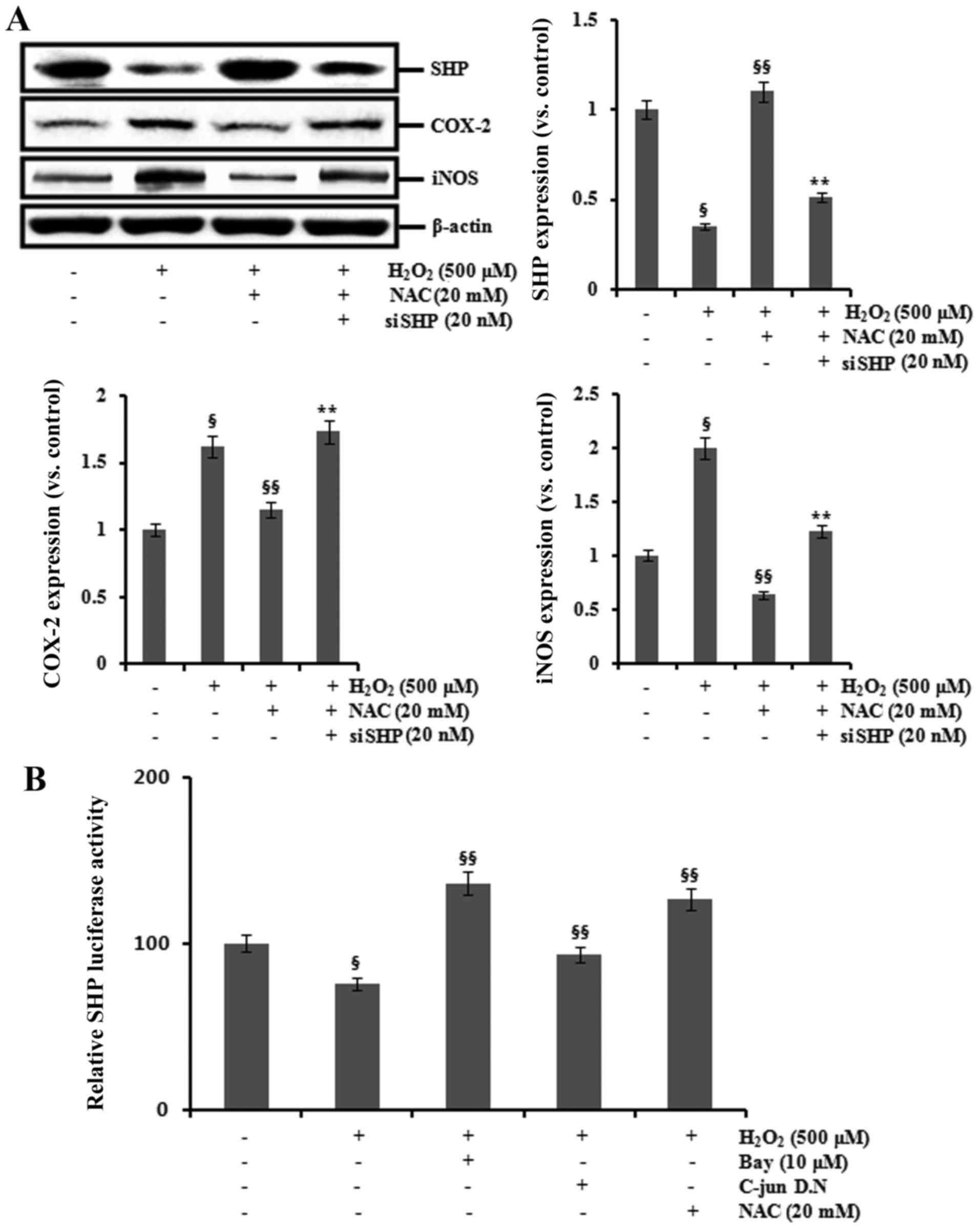
Next, we investigated whether NAC could modulate the
expression of SHP in HK-2 cells exposed to
H2O2. H2O2 exposure
increased the expression of COX-2 and iNOS, but decreased SHP
expression. These changes were counteracted by pretreatment with
NAC for 1 h. In addition, transfection with SHP siRNA attenuated
the inhibitory effects of NAC on the expression of COX-2 and iNOS
in the H2O2-treated HK-2 cells (Fig. 5A).
To further investigate the transcriptional
regulation of SHP, HK-2 cells were transiently transfected with a
mouse SHP promoter luciferase reporter construct (pGL3-SHP). HK-2
cells were pretreated with 10 μM Bay11-7082 (an NF-κB
inhibitor) and 20 mM NAC and cotransfected with dominant-negative
c-Jun before H2O2 exposure.
H2O2 exposure decreased SHP promoter
activity, and this decrease was blocked by treatment with
Bay11-7082 or NAC and by cotransfection with dominant-negative
c-Jun owing to competitive inhibition of AP-1 activation (Fig. 5B).
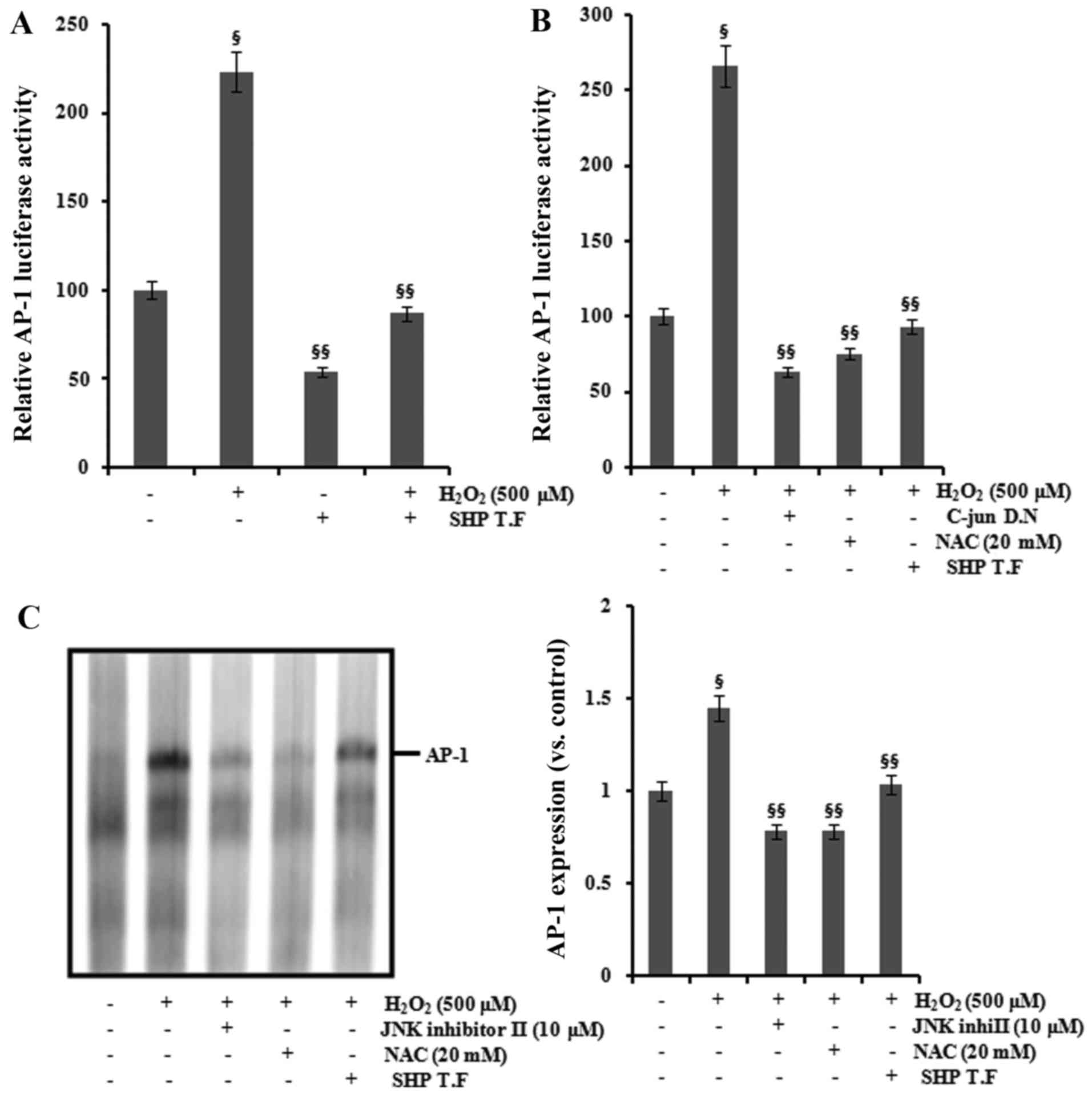
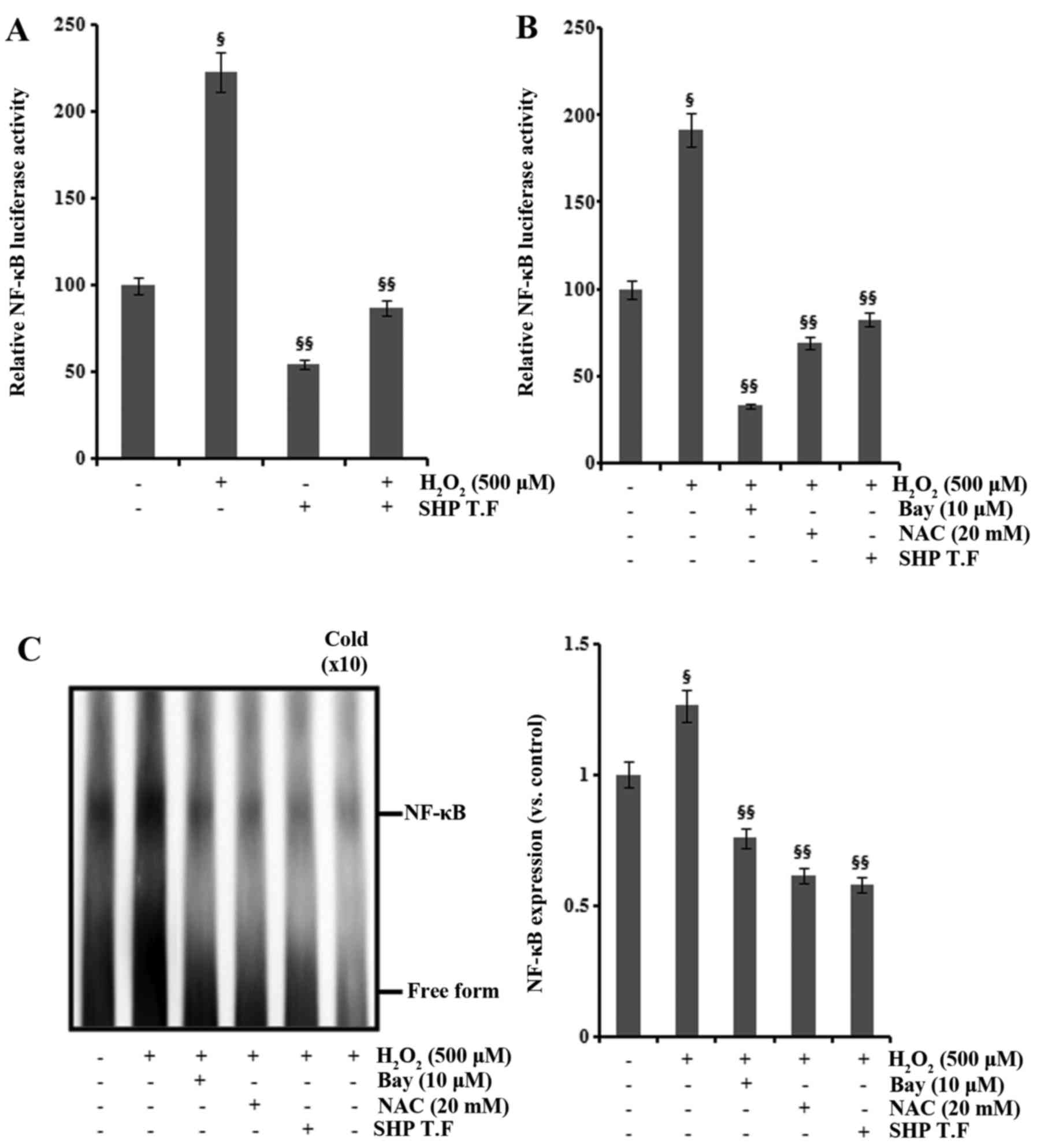
Effect of SHP on the transcriptional
activation of AP-1 and NF-κB
AP-1 and NF-κB are important transcription factors
activating the expression of COX-2 and iNOS (14–16), and AP-1 and NF-κB are activated by
ROS (17,18). We examined the role of SHP in the
H2O2-induced activation of AP-1 and NF-κB.
The promoter activity of AP-1 and NF-κB was increased following
H2O2 exposure in the HK-2 cells, and this
increase was attenuated by SHP transfection and NAC treatment
(Figs. 6A and B and 7A and B). Cotransfection with
dominant-negative c-Jun inhibited the
H2O2-induced increase in AP-1 promoter
activity (Fig. 6B). Furthermore,
pretreatment with 10 μM Bay11-7082 reduced
H2O2-induced NF-κB promoter activity
(Fig. 7B). The nuclear extracts
from cells analyzed by EMSA for activated AP-1 and NF-κB confirmed
these findings (Figs. 6C and
7C).
Discussion
I/R kidney injury is widely utilized as an
experimental model of AKI. Increased generation of ROS, endothelial
dysfunction, tubular necrosis and inflammation are major players in
the pathogenesis of I/R kidney injury (29). Post-ischemic tissues generate
inflammatory mediators that can stimulate circulating neutrophils
(30). Inflammation involves a
complex cascade of intercellular cytokine signals. Activated
monocytes and macrophages release a variety of inflammatory
mediators, such as TNF-α, IL-1β, nitric oxide and ROS. Nitric oxide
has various effects in renal physiology and pathophysiology
(31). Moreover, nitric oxide
produced by constitutive NOS (eNOS and nNOS) is essential and plays
a role in maintaining cellular function, whereas nitric oxide
produced by iNOS is an important mediator of inflammation (32). In addition, COX-2 is also an
inducible enzyme involved in the pathogenesis of inflammation. Many
studies have reported that iNOS-derived nitrogen reactive species
and COX-2-derived oxidative stress play roles in inflammatory
kidney injury (33,34). In the present study, we examined
changes in the expression levels of inflammatory mediators in
response to induction of AKI. Consistent with previous studies, the
expression of COX-2 and iNOS was increased, and associated with
upregulation of IL-1β and TNF-α (33,34). Moreover, renal dysfunction caused
by I/R-induced kidney injury resulted in marked reduction in SHP
expression. These results suggest that SHP is associated with the
pathogenesis of renal inflammation in I/R kidney injury.
Recently, we demonstrated the effect of SHP on
cisplatin-induced kidney injury using a farnesoid X receptor ligand
(35). Farnesoid X receptor
ligand prevented cisplatin-induced kidney injury by inhibiting
renal inflammation, fibrosis and apoptosis through induction of
SHP. In the present study, we examined the hypothesis that SHP is
involved in the inflammatory signaling pathway in HK-2 cells. An
imbalance between cell survival and death, a key process in many
degenerative and inflammatory diseases, may be caused by aberrant
turnover of ROS, which regulates the crosstalk between
mitogen-activated protein kinases (MAPKs) and NF-κB (36). Because H2O2
is a strong inducer of ROS production, we examined the effects of
SHP on H2O2-mediated ROS production using the
fluorescent dye H2DCF-DA. H2O2
exposure strongly induced ROS production, which was ameliorated by
the overexpression of SHP. Accordingly, cell viability was
decreased by H2O2 exposure, which was again
attenuated by SHP transfection. Thus, SHP increased the cell
viability following H2O2-mediated kidney
tubule cell injury in HK-2 cells through inhibition of ROS
production. Mitochondrial uncoupling proteins may play a role in
minimizing mitochondrial ROS production and function in the
protection against oxidative stress (37). We examined whether SHP may play a
role in the inhibition of ROS production through mitochondrial
uncoupling protein 2 (UCP2). SHP transfection induced gene
expression of UCP2, suggesting that UCP2 is involved in
SHP-mediated suppression of ROS production. In addition,
H2O2 exposure increased the expression of
COX-2 and iNOS, which was ameliorated by NAC pretreatment. These
findings suggest that the protective activity of SHP on
ROS-mediated inflammation is through suppression of ROS
production.
We then aimed to ascertain whether SHP prevents
H2O2-induced inflammation in HK-2 cells. Both
iNOS and COX-2 exhibited increased expression after
H2O2 treatment, and SHP transfection
prevented this H2O2-mediated increase in iNOS
and COX-2 expression. Therefore, SHP expression may be essential
for suppression of inflammatory markers such as COX-2 and iNOS in
H2O2-induced injury of proximal tubular
cells.
Next, we investigated whether the antioxidant, NAC
modulates the expression of SHP, COX-2 and iNOS in HK-2 cells
following exposure to H2O2.
H2O2 exposure increased the expression of
COX-2 and iNOS, but decreased SHP expression. These changes were
ameliorated by NAC pretreatment. In addition, transfection with SHP
siRNA attenuated the inhibitory effects of NAC on the expression of
COX-2 and iNOS in the H2O2-treated HK-2
cells. These findings indicated that the inhibitory effects of NAC
on the expression of COX-2 and iNOS in the
H2O2-treated HK-2 cells may be attributed in
part to SHP.
Our results also showed that
H2O2 exposure decreased SHP promoter activity
and that this effect was blocked by treatment with an NF-κB
inhibitor or cotransfection with dominant-negative c-Jun. These
findings indicate that the promoter activity of SHP is regulated by
AP-1 and NF-κB in kidney-related inflammatory signaling. In
addition, SHP promoter activation was increased by elimination of
ROS using NAC treatment. Because AP-1 and NF-κB are activated by
ROS (17,18), these data suggest that SHP
promoter activation may be inhibited by ROS through the activation
of NF-κB and AP-1. However, further studies are needed to elucidate
the exact interactive mechanisms that couple NF-κB and AP-1 to
oxidative stress and the role of these mechanisms in the regulation
of SHP in kidney injury.
In the present study, H2O2
exposure increased the promoter activity of AP-1 and NF-κB. COX-2
may induce stimulation of pro-inflammatory cytokines and growth
factors (38), and the COX-2
promoter has transcription binding sites for AP-1, GATA BOX, C/EBP,
CRE and NF-κB (39).
Additionally, the iNOS promoter has binding sites for NF-κB, AP-1,
STAT1, C/EBP and IRF-1 (40).
Importantly, in the present study, the promoter activity of AP-1
and NF-κB was increased by H2O2 exposure in
the HK-2 cells, and this effect was blocked by SHP transfection.
Taken together with the observation that SHP transfection prevented
the H2O2-mediated increases in iNOS and COX-2
expression, these findings suggest that SHP decreased the
expression of COX-2 and iNOS through inhibition of NF-κB and AP-1
promoter activities. Pretreatment with NAC and cotransfection with
dominant-negative c-Jun ameliorated the
H2O2-induced increase in AP-1 promoter
activity. Moreover, pretreatment with NAC and an NF-κB inhibitor
reduced the H2O2-induced increase in NF-κB
promoter activity. Furthermore, EMSA results indicated that
H2O2 exposure markedly increased the amount
of AP-1 and NF-κB that could form complexes with the biotin-labeled
oligonucleotide probe. In contrast, the promoter activities of AP-1
and NF-κB were decreased by pretreatment with JNK inhibitor II and
Bay11-7082, respectively. The present study demonstrated that SHP
protected HK-2 cells from H2O2-induced
tubular injury by inhibition of COX-2 and iNOS through inhibition
of AP-1 and NF-κB promoter activities. This knowledge may lead to
an important new therapeutic target for the treatment of AKI such
as I/R kidney injury.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (no. 2014R1A1A2008333),
by the Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Science, ICT
and future Planning (no. 2016R1A2B4007870), by the Pioneer Research
Center Program through the National Research Foundation of Korea
funded by the Ministry of Science, ICT and Future Planning (no.
2014M3C1A3053036), and by a grant of the Korea Health Technology
R&D Project through the Korea Health Industry Development
Institute (KHIDI), funded by the Ministry of Health and Welfare,
Republic of Korea (grant no. HI14C2084).
References
|
1
|
Lameire N, Van Biesen W and Vanholder R:
The changing epidemiology of acute renal failure. Nat Clin Pract
Nephrol. 2:364–377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Le Dorze M, Legrand M, Payen D and Ince C:
The role of the microcirculation in acute kidney injury. Curr Opin
Crit Care. 15:503–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonventre JV and Zuk A: Ischemic acute
renal failure: An inflammatory disease? Kidney Int. 66:480–485.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thurman JM: Triggers of inflammation after
renal ischemia/reperfusion. Clin Immunol. 123:7–13. 2007.
View Article : Google Scholar
|
|
5
|
Sasaki M and Joh T: Oxidative stress and
ischemia-reperfusion injury in gastrointestinal tract and
antioxidant, protective agents. J Clin Biochem Nutr. 40:1–12. 2007.
View Article : Google Scholar
|
|
6
|
Basile DP: The endothelial cell in
ischemic acute kidney injury: Implications for acute and chronic
function. Kidney Int. 72:151–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwon O, Hong SM, Sutton TA and Temm CJ:
Preservation of peritubular capillary endothelial integrity and
increasing pericytes may be critical to recovery from postischemic
acute kidney injury. Am J Physiol Renal Physiol. 295:F351–F359.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubattu S, Mennuni S, Testa M, Mennuni M,
Pierelli G, Pagliaro B, Gabriele E, Coluccia R, Autore C and Volpe
M: Pathogenesis of chronic cardiorenal syndrome: Is there a role
for oxidative stress? Int J Mol Sci. 14:23011–23032. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ruiz S, Pergola PE, Zager RA and Vaziri
ND: Targeting the transcription factor Nrf2 to ameliorate oxidative
stress and inflammation in chronic kidney disease. Kidney Int.
83:1029–1041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nath KA and Norby SM: Reactive oxygen
species and acute renal failure. Am J Med. 109:665–678. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gentle ME, Shi S, Daehn I, Zhang T, Qi H,
Yu L, D'Agati VD, Schlondorff DO and Bottinger EP: Epithelial cell
TGFβ signaling induces acute tubular injury and interstitial
inflammation. J Am Soc Nephrol. 24:787–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lessio C, de Assunção Silva F, Glória MA,
Di Tommaso AB, Gori Mouro M, Di Marco GS, Schor N and Higa EM:
Cyclosporine A and NAC on the inducible nitric oxide synthase
expression and nitric oxide synthesis in rat renal artery cultured
cells. Kidney Int. 68:2508–2516. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ostergaard M, Christensen M, Nilsson L,
Carlsen I, Frøkiær J and Nørregaard R: ROS dependence of
cyclooxygenase-2 induction in rats subjected to unilateral ureteral
obstruction. Am J Physiol Renal Physiol. 306:F259–F270. 2014.
View Article : Google Scholar
|
|
14
|
Jedinak A, Dudhgaonkar S, Wu QL, Simon J
and Sliva D: Anti-inflammatory activity of edible oyster mushroom
is mediated through the inhibition of NF-κB and AP-1 signaling.
Nutr J. 10:522011. View Article : Google Scholar
|
|
15
|
Liu DY, Li XW, Li H, Li XM and Ye WL:
Expression of cyclooxygenase-2 in a mouse macula densa cell lines
and signal transduction of NF-kappaB and AP-1. Zhongguo Yi Xue Ke
Xue Yuan Xue Bao. 29:78–82. 2007.In Chinese. PubMed/NCBI
|
|
16
|
Jung KJ, Lee EK, Kim JY, Zou Y, Sung B,
Heo HS, Kim MK, Lee J, Kim ND, Yu BP, et al: Effect of short term
calorie restriction on pro-inflammatory NF-κB and AP-1 in aged rat
kidney. Inflamm Res. 58:143–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahn KS and Aggarwal BB: Transcription
factor NF-kappaB: A sensor for smoke and stress signals. Ann NY
Acad Sci. 1056:218–233. 2005. View Article : Google Scholar
|
|
18
|
Roebuck KA: Oxidant stress regulation of
IL-8 and ICAM-1 gene expression: Differential activation and
binding of the transcription factors AP-1 and NF-κB (Review). Int J
Mol Med. 4:223–230. 1999.PubMed/NCBI
|
|
19
|
Seol W, Choi HS and Moore DD: An orphan
nuclear hormone receptor that lacks a DNA binding domain and
heterodimerizes with other receptors. Science. 272:1336–1339. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HK, Lee YK, Park SH, Kim YS, Park SH,
Lee JW, Kwon HB, Soh J, Moore DD and Choi HS: Structure and
expression of the orphan nuclear receptor SHP gene. J Biol Chem.
273:14398–14402. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sanyal S, Kim JY, Kim HJ, Takeda J, Lee
YK, Moore DD and Choi HS: Differential regulation of the orphan
nuclear receptor small heterodimer partner (SHP) gene promoter by
orphan nuclear receptor ERR isoforms. J Biol Chem. 277:1739–1748.
2002. View Article : Google Scholar
|
|
22
|
Nishizawa H, Yamagata K, Shimomura I,
Takahashi M, Kuriyama H, Kishida K, Hotta K, Nagaretani H, Maeda N,
Matsuda M, et al: Small heterodimer partner, an orphan nuclear
receptor, augments peroxisome proliferator-activated receptor gamma
transactivation. J Biol Chem. 277:1586–1592. 2002. View Article : Google Scholar
|
|
23
|
Masuda N, Yasumo H, Tamura T, Hashiguchi
N, Furusawa T, Tsukamoto T, Sadano H and Osumi T: An orphan nuclear
receptor lacking a zinc-finger DNA-binding domain: Interaction with
several nuclear receptors. Biochim Biophys Acta. 1350:27–32. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim YD, Park KG, Lee YS, Park YY, Kim DK,
Nedumaran B, Jang WG, Cho WJ, Ha J, Lee IK, et al: Metformin
inhibits hepatic gluconeogenesis through AMP-activated protein
kinase-dependent regulation of the orphan nuclear receptor SHP.
Diabetes. 57:306–314. 2008. View Article : Google Scholar
|
|
25
|
Lee YS, Chanda D, Sim J, Park YY and Choi
HS: Structure and function of the atypical orphan nuclear receptor
small heterodimer partner. Int Rev Cytol. 261:117–158. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chanda D, Park JH and Choi HS: Molecular
basis of endocrine regulation by orphan nuclear receptor Small
Heterodimer Partner. Endocr J. 55:253–268. 2008. View Article : Google Scholar
|
|
27
|
Båvner A, Sanyal S, Gustafsson JA and
Treuter E: Transcriptional corepression by SHP: Molecular
mechanisms and physiological consequences. Trends Endocrinol Metab.
16:478–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW,
Jin HS, Yang CS, Park KA, Chanda D, Kim DK, et al: The orphan
nuclear receptor SHP acts as a negative regulator in inflammatory
signaling triggered by Toll-like receptors. Nat Immunol.
12:742–751. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carden DL and Granger DN: Pathophysiology
of ischaemia-reperfusion injury. J Pathol. 190:255–266. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Molitoris BA, Sandoval R and Sutton TA:
Endothelial injury and dysfunction in ischemic acute renal failure.
Crit Care Med. 30(Suppl 5): S235–S240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mount PF and Power DA: Nitric oxide in the
kidney: Functions and regulation of synthesis. Acta Physiol (Oxf).
187:433–446. 2006. View Article : Google Scholar
|
|
32
|
Chatterjee PK, Patel NS, Sivarajah A,
Kvale EO, Dugo L, Cuzzocrea S, Brown PA, Stewart KN, Mota-Filipe H,
Britti D, et al: GW274150, a potent and highly selective inhibitor
of iNOS, reduces experimental renal ischemia/reperfusion injury.
Kidney Int. 63:853–865. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Choi YJ, Kim HS, Lee J, Chung J, Lee JS,
Choi JS, Yoon TR, Kim HK and Chung HY: Down-regulation of oxidative
stress and COX-2 and iNOS expressions by dimethyl lithospermate in
aged rat kidney. Arch Pharm Res. 37:1032–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Villanueva S, Céspedes C, González AA, Vio
CP and Velarde V: Effect of ischemic acute renal damage on the
expression of COX-2 and oxidative stress-related elements in rat
kidney. Am J Physiol Renal Physiol. 292:F1364–F1371. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bae EH, Choi HS, Joo SY, Kim IJ, Kim CS,
Choi JS, Ma SK, Lee J and Kim SW: Farnesoid X receptor ligand
prevents cisplatin-induced kidney injury by enhancing small
heterodimer partner. PLoS One. 9:e865532014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nakano H, Nakajima A, Sakon-Komazawa S,
Piao JH, Xue X and Okumura K: Reactive oxygen species mediate
crosstalk between NF-kappaB and JNK. Cell Death Differ. 13:730–737.
2006. View Article : Google Scholar
|
|
37
|
Mailloux RJ and Harper ME: Uncoupling
proteins and the control of mitochondrial reactive oxygen species
production. Free Radic Biol Med. 51:1106–1115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Murakami M and Kudo I: Prostaglandin E
synthase: A novel drug target for inflammation and cancer. Curr
Pharm Des. 12:943–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Corral RS, Iñiguez MA, Duque J,
López-Pérez R and Fresno M: Bombesin induces cyclooxygenase-2
expression through the activation of the nuclear factor of
activated T cells and enhances cell migration in Caco-2 colon
carcinoma cells. Oncogene. 26:958–969. 2007. View Article : Google Scholar
|
|
40
|
Guo Z, Shao L, Du Q, Park KS and Geller
DA: Identification of a classic cytokine-induced enhancer upstream
in the human iNOS promoter. FASEB J. 21:535–542. 2007. View Article : Google Scholar
|