Introduction
S100A8 and S100A9 (also termed MRP8 and MRP14),
which are called damage-associated molecular pattern (DAMP)
molecules, play critical roles in the inflammatory process. The
preferential forms of the S100A8/A9 heterodimers are associated
with the pathogenesis of various diseases, and coupled with an
inflammatory component, which is primarily released from activated
or necrotic neutrophils and monocytes/macrophages (1,2).
In the central nervous system (CNS), S100A8/A9 is implicated in the
pathology of numerous inflammatory diseases including Alzheimer's
disease, traumatic brain injury and stroke (3–5).
Previous studies have indentified both the Toll-like
receptor 4 (TLR4) and receptor for advanced glycation end products
(RAGE) as activated receptors of S100A8/A9 (6–8).
Previous studies have shown that activated microglia express high
levels of TLR4 and RAGE in response to neuroinflammation (9–12).
Microglial activation plays a pivotal role during the development
and progression of neurodegenerative diseases based on its great
capacity for secreting proinflammatory cytokines, such as tumor
necrosis factor-α (TNF-α) and interleukin-6 (IL-6), resulting in
acute inflammation (13). As an
endogenous ligand of TLR4 and RAGE (14), S100A8/A9 also amplifies the
production of roinflammatory cytokines and contributes to CNS
injury (2,15). However, whether S100A8/A9 could
activate BV-2 microglial cells by binding to TLR4 and/or RAGE on
the membrane and subsequently mediate the inflammatory response
through inflammatory cytokines or chemokines remains unclear
(12,17–20).
It is well known that NF-κB as a pleiotropic
regulator is involved in the production of many proinflammatory
cytokines and enzymes (21).
NF-κB is also a central regulator of microglial responses to
stimuli (21). In general,
activation of NF-κB in microglia leads to neuronal injury and
promotes the development of neurodegenerative disorders such as
stroke, severe epileptic seizures, and also chronic
neurodegenerative conditions, including Alzheimer's disease,
Parkinson's disease and Huntington's disease (22). Increasing evidence has shown that
the activated NF-κB-modulated proinflammatory effects of microglia
are regulated by mitogen-activated protein kinase (MAPK) signaling
pathways, including the c-Jun N-terminal protein kinase (JNK), the
p38 mitogen-activated protein kinase (MAPK) and the extracellular
signal related kinase (ERK) (22–25).
The purpose of this in vitro study was to
investigate whether S100A8/A9 could activate BV-2 microglial cells
by binding to TLR4 and/or RAGE on the membrane and then
subsequently amplify the secretion of proinflammatory cytokines
through the MAPK/NF-κB signaling pathways in BV-2 microglial
cells.
Materials and methods
Cell culture
The immortalized murine microglial cell line BV-2
was purchased from Cell Resource Centre of Peking Union Medical
College (Beijing, China) and maintained in Dulbecco's modified
Eagle's medium with F12 (DMEM/F12) and supplemented with 10% fetal
bovine serum (FBS) (both from Gibco, Grand Island, NY, USA), 100
U/ml penicillin and 100 µg/ml streptomycin at 37°C in a
humidified atmosphere of 95% air 5% CO2. Confluent
cultures were passaged by trypsinization. BV-2 cells were seeded
onto 24-well culture plates (105 cells/well for ELISA,
104 cells/well for immunofluorescence), 6-well plates
(2.5×105 cells/well for PCR) or 100 mm culture dishes
(1.2×106 cells/dish for western blotting and EMSA). BV-2
cells were incubated in the initial experiments with different
concentrations (0.01, 0.1 or 1.0 µM) of S100A8/A9 (USCN,
Wuhan, China). A concentration of 0.1 µM S100A8/A9 was used
in the subsequent experiments or vehicle (0.035% ethanol).
RNA isolation and real-time PCR
Total RNA was extracted from BV-2 microglial cells
with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to
the manufacturers' protocol. Total RNA (1.0 µg) was
subjected to oligo(dT)-primed RT with ReverTra Ace kit (Toyobo,
Osaka, Japan). Real-time PCR was performed for quantitative
analysis of IL-6 and TNF-α mRNA expression using SYBR-Green
Real-Time PCR Master Mix (Toyobo, Osaka, Japan) on an MX3000P
real-time PCR system (Stratagene, La Jolla, CA, USA). The following
primers were used: 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ and
5′-TGGGAGTAGACAAGGTACAACCC-3′, which amplify the 175-bp product for
TNF-α; and 5′-TGTCCACCTTCCAGCAGATGT-3′ and
5′-AGCTCAGTAACAGTCCGCCTAGA-3′, which amplify the 101-bp product for
β-actin; and 5′-ACAACCACGGCCTTCCCTACTT-3′ and
5′-CACGATTTCCCAGAGAACATGTG-3′, which amplify the 129-bp product for
IL-6. Relative gene expression was calculated using the
2−ΔΔCT method.
ELISA for IL-6 and TNF-α
BV-2 microglial cells were stimulated for 12 h with
166 µg/ml RAGE-blocking antibody (Abcam, Cambridge, UK), 1.0
µg/ml C225 (inhibitor of TLR4), 20 µM PD98059
[inhibitor of extracellular signaling kinase (ERK)], 7 µM
SB203580 (inhibitor of p38 MAP kinase), 10 µM SP600125
(inhibitor of JNK) or 50 µM PDTC (inhibitor of p65 NF-κB)
(all from R&D Systems, ON, Canada) for 1 h before addition of
0.1 µM recombinant S100A8/A9 proteins or 1 µg/ml LPS
(Escherichia coli O26:B6; Sigma-Aldrich, St. Louis, MO, USA)
in the presence of 25 µg/ml polymyxin B (R&D Systems),
and the culture supernatants were harvested. Levels of IL-6 and
TNF-α in 100 µl medium were measured by commercial ELISA
kits (Boster Biological Technology, Wuhan, China) according to the
manufacturer's instructions.
Biochemistry
BV-2 microglial cells were cultured on sterile glass
coverslips and treated according to the experimental design.
Afterward, the cells were fixed with 4% paraformaldehyde in
phosphate-buffered saline (PBS) and permeabilized with 0.1% Triton
X-100 in PBS. After rinsing, the cells were blocked with 3% bovine
serum albumin (BSA) for 1 h and incubated with primary antibodies
overnight at 4°C. The primary antibody used was rabbit anti-NF-κB
(1:1,000; ab31481; Abcam). After washing, the cells were incubated
with FITC-conjugated goat anti-rabbit IgG (1:400; Jackson
ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h and
counterstained with 4,6-diamidino-2-phenylindole (DAPI; Roche,
Shanghai, China) for the identification of the nuclei. After
washing with PBS, the coverslips were mounted with anti-fade
mounting medium (Beyotime, Shanghai, China) on slides, and the
cells were observed with an Olympus immunofluorescence microscope
(Olympus, Tokyo, Japan).
Protein extraction
For making whole cell lysates, the cells were lysed
in radioimmune precipitation assay (RIPA) buffer supplemented with
protease inhibitor cocktail (Roche). Nuclear and cytoplasmic
fractionations were performed with NE-PER nuclear and cytoplasmic
extraction reagents (Thermo Scientific, Rockford, IL, USA)
according to the manufacturer's instructions.
Western blot analysis
Equal amounts of nuclear or whole cell extracts were
electrophoresed on sodium dodecyl sulfate-poly-acrylamide gels, and
then transferred onto a polyvinylidene difluoride membrane
(Millipore, Schwalbach, Germany). The transformed membrane was
blocked with 5% non-fat dry milk in Tris-buffered saline containing
0.05% Tween-20 (TBST) for 1 h and incubated with primary antibodies
overnight at 4°C. The primary antibodies used were as follows:
rabbit anti-NF-κB (1:1,000; ab31481; Abcam), β-actin (1:1,000;
sc-1616; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and
lamin B (1:1,000; 12987-1-AP; Proteintech Group, Chicago, IL, USA).
The membrane was washed 3 times with TBST for 10 min and incubated
with anti-rabbit IgG-horseradish peroxidase (1:5,000; Jackson
ImmunoResearch Laboratories) at room temperature for 1 h. The
Supersignal West Pico chemiluminescent substrate system (Millipore)
was used to detect immunoreactive bands. The intensity of the
protein bands after western blot analysis was quantifed using
Quantity One software version 4.6.3 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and normalized against proper loading
controls.
Electrophoretic mobility shift assay
(EMSA)
Nuclear protein was harvested, and 10 µg of
nuclear protein was assayed for NF-κB binding activity using
radioactive-labeled oligonucleotides for the defined NF-κB
consensus sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′) at 50,000 cpm
(Cerenkov). Binding separation of the protein DNA complexes from
unbound DNA by electrophoresis was performed as previously
described in detail (26).
Nuclear protein after 1 h of S100A8/A9 treatment and a 200-fold
molar excess of unlabeled consensus sequence were used as the
specific competitor in the control lane.
Statistical analysis
Data are expressed as means ± SEM of the indicated
number of independent experiments. Statistical significance between
multiple groups was analyzed by one-way ANOVA. Least significant
difference (LSD) post hoc test was used for multiple comparisons.
Statistical analysis was performed using the SPSS software version
13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
statistically significant.
Results
Proinflammatory cytokine production by
BV-2 microglial cells after S100A8/A9 stimulation
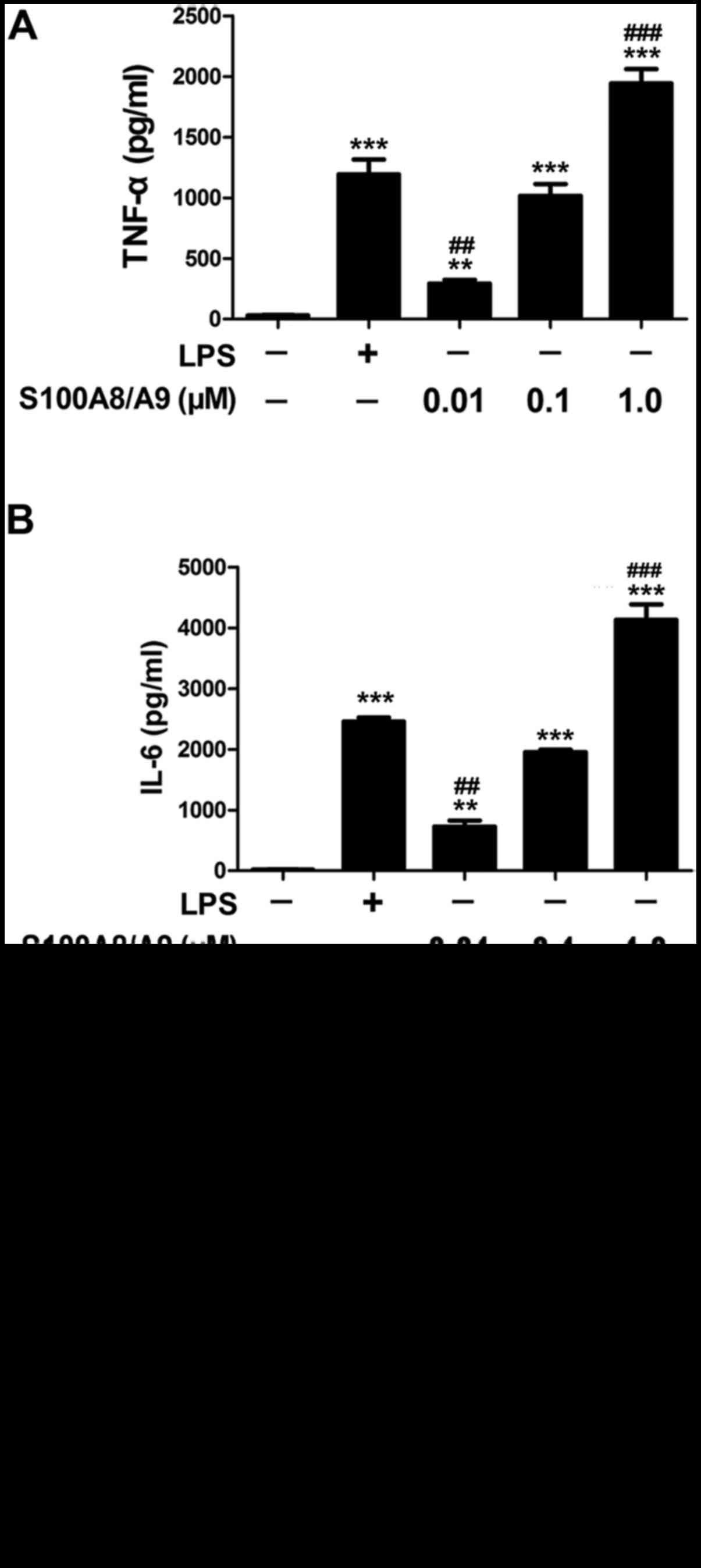
In this study, the response of BV-2 microglial cells
in culture to S100A8/A9 was evaluated by determining the expression
of inflammatory cytokine proteins. As a positive control, LPS at 1
µg/ml significantly increased the production of TNF-α and
IL-6 compared with the control group. S100A8/A9 at 0.01, 0.1 or 1.0
µM also significantly increased the production of TNF-α and
IL-6 (Fig. 1A and B). There was
no difference in the levels of TNF-α and IL-6 between the 1
µg/ml LPS-treated group and the 0.1 µM
S100A8/A9-treated group (Fig. 1A and
B). Thus, the dose of 0.1 µM S100A8/A9 was chosen for
further study.
Polymyxin B effectively blocked the TNF-α production
induced by 1 µg/ml LPS. However, polymyxin B had no effect
on the expression of TNF-α induced by S100A8/A9 in the cultured
BV-2 microglial cells, suggesting that the effect of S100A8/A9 on
the secretion of proinflammatory cytokines was not blocked by the
addition of the LPS inhibitor polymyxin B (Fig. 1C).
Effects of RAGE and TLR4 blockade on
S100A8/A9 stimulation
To examine whether S100A8/A9 uses RAGE and TLR4 as
signal transducing receptors on BV-2 microglial cells, BV-2 cells
were stimulated for 12 h using 0.1 µM S100A8/A9 with or
without the RAGE-blocking antibody and TLR4 inhibitor C225. We
found that the S100A8/A9-stimulated release of TNF-α and IL-6 was
significantly reduced by blockade of RAGE or TLR4. Therefore, our
data suggested that both RAGE and TLR-4 may be relevant to
S100A8/A9 stimulation in BV-2 microglial cells (Fig. 2).
Involvement of MAPK signaling pathways
and NF-κB activation in S100A8/A9-stimulated secretion of TNF-α and
IL-6
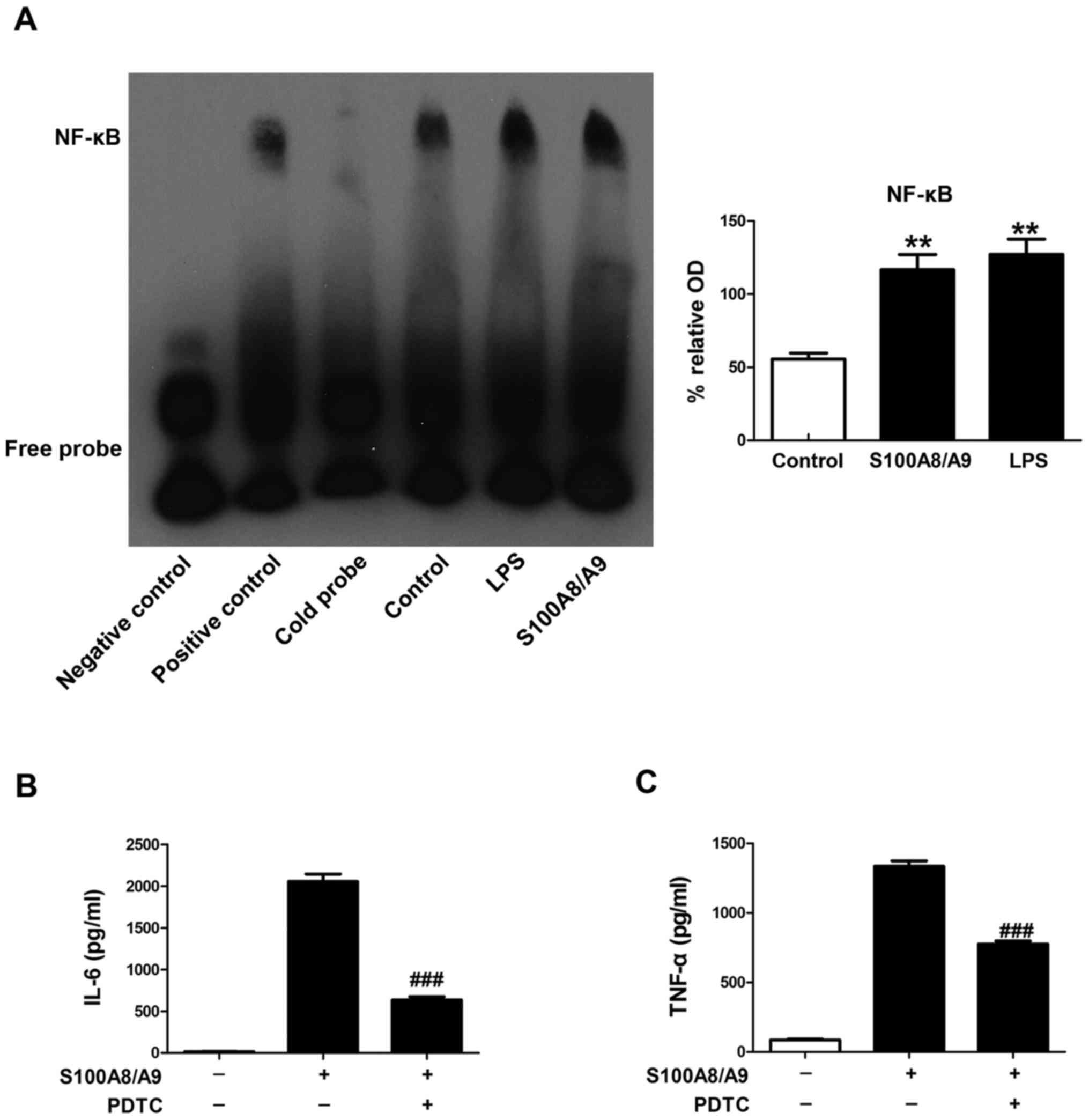
EMSA was performed to determine the effect of
S100A8/A9 on the activity of NF-κB in this study. BV-2 microglial
cells were pretreated with vehicle, 0.1 µM S100A8/A9 or 1
µg/ml LPS for 1 h. We found that the binding activities of
NF-κB were induced by S100A8/A9 treatment, which had an effect
similiar to that of LPS treatment (Fig. 3A).
To determine whether S100A8/A9-stimulated secretion
of TNF-α and IL-6 involves NF-κB activation, PDTC, a specific
inhibitor of NF-κB, was applied in our study. PDTC treatment
significantly reduced the secretion of TNF-α and IL-6 induced by
S100A8/A9 (Fig. 3B and C).
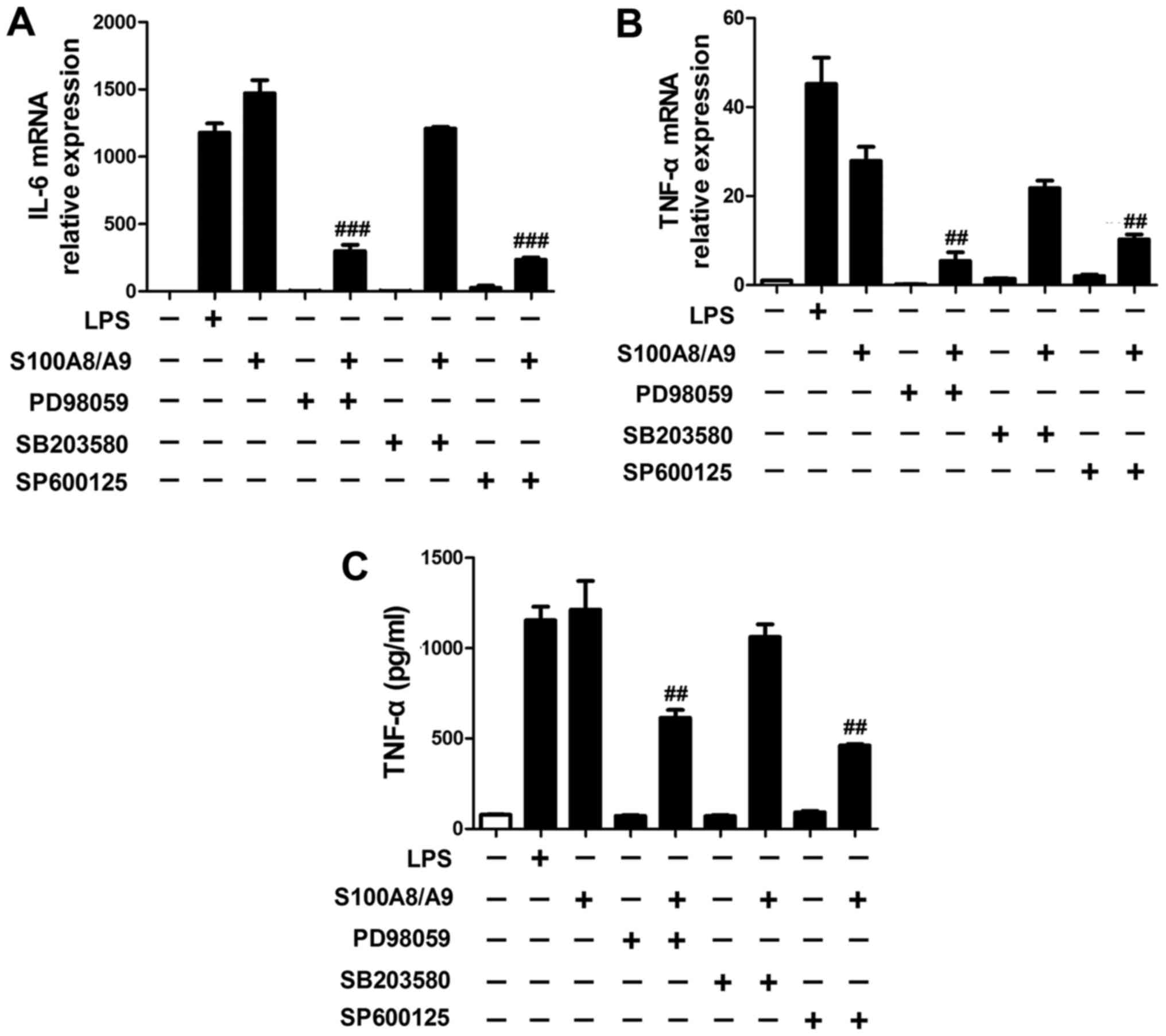
Our data also showed that the expression of IL-6
(Fig. 4A) and TNF-α (Fig. 4B and C) induced by S100A8/A9 were
significantly reduced by the addition of PD98059 and SP600125,
respectively.
MAPK signaling pathway acts upstream of
NF-κB in S100A8/A9 stimulation
To explore whether S100A8/A9-induced secretion of
proinflammatory cytokines including TNF-α and IL-6 was through
MAPK-mediated activation of NF-κB in BV-2 microglial cells, a
specific ERK inhibitor PD98059, p38 MAP kinase inhibitor SB203580
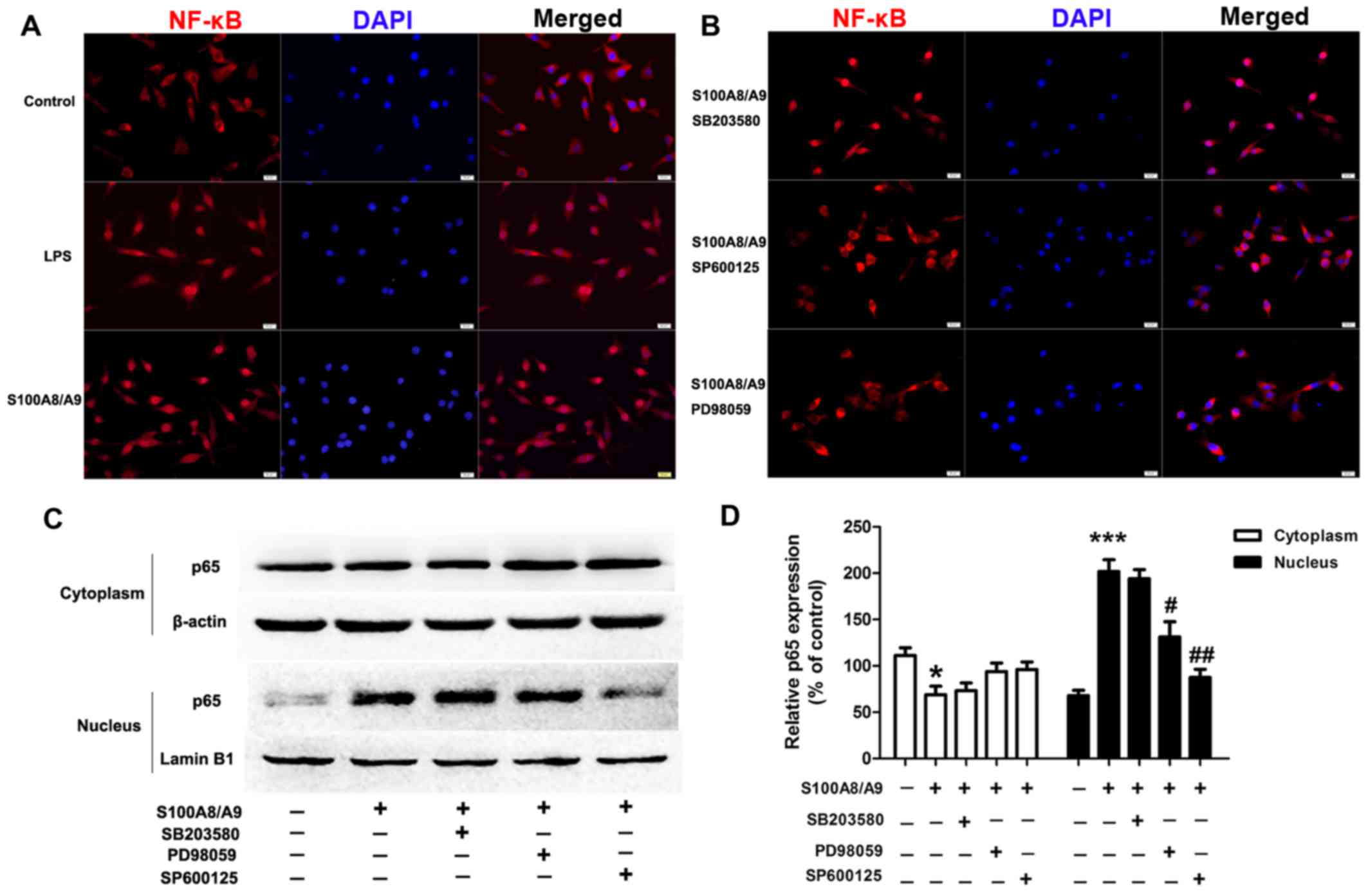
and JNK inhibitor SP600125 were used. S100A8/A9 treatment caused
obvious translocation of NF-κB p65 from the cytoplasm into the
nucleus compared with the vehicle treatment, which had an effect
similar to that of LPS treatment (Fig. 5A); whereas the presence of PD98059
and SP600125 reduced S100A8/A9-induced translocation of NF-κB p65
from the cytoplasm into the nucleus (Fig. 5B). To further verify the p65
nuclear translocation data, we analyzed the cells by western
blotting and found that pretreatment of cells with PD98059 and
SP600125 prevented p65 nuclear localization induced by S100A8/A9
(Fig. 5C and D).
Discussion
This study has the following major findings. i) The
proinflammatory effects of S100A8/A9 are partially dependent on the
TLR4 and RAGE signaling pathway in microglial BV-2 cells; and ii)
S100A8/A9 promotes inflammatory cytokine production via ERK and
JNK-mediated NF-κB activity in microglia.
Activated macrophages and microglia release an array
of proinflammatory cytokines, including TNF-α and IL-6, which play
important roles in neuroinflammation (27,28). Previous studies have demonstrated
that TNF-α and IL-6 play important roles in S100A8/A9-induced
inflammation in neutrophils, monocytes, macrophages, human
umbilical vein endothelial cells (HUVECs) and keratinocytes
(29–33). We demonstrated that S100A8/A9
acted on microglial BV-2 cells and subsequently amplified the
secretion of TNF-α and IL-6, as demonstrated by our findings that
S100A8/A9 treatment significantly increased the gene and protein
expression of TNF-α and IL-6 in the BV-2 microglial cells.
TLR4, which is mainly expressed in microglia,
mediates microglial activation and the expression of
proinflammatory mediators in response to a variety of ligands
(34). TLR4 signaling has been
confirmed to result in the activation of NF-κB, and subsequently
drives the transcriptional abundance of proinflammatory signals,
which then activates the innate immune system and produces tissue
destruction (35–38). Recently, S100A8/A9, as the
endogenous activator of TLR4 signaling, promotes lethal
endotoxin-induced shock (6).
Thus, our data, which demonstrated that S100A8/A9 contributed to
inflammation via TLR4, further revealed that neuroinflammation and
ischemic brain injury is modulated by TLR4 (39–41). However, S100A8/A9 not only
activates TLR-4, but also RAGE (42). It cannot be excluded that further
receptors besides TLR4 may be involved in S100A8/A9-mediated
inflammation. Our data showed that the S100A8/A9-stimulated
secretion of TNF-α and IL-6 was also significantly reduced by the
blockade of RAGE. Therefore, both RAGE and TLR4 may be relevant to
S100A8/A9-induced inflammation in BV-2 microglial cells.
Our subsequent results demonstrated that S100A8/A9
had a strong enhancement effect on inflammatory signaling pathways
including NF-κB and MAPK. Likewise, activation of NF-κB in
microglia contributes to neuronal injury and plays a crucial role
in the development of neurodegenerative disorders (22). NF-κB is also a central regulator
of microglial responses to activating stimuli, including cytokines
(21). Evidence has shown that
the MAPK signaling pathway, such as JNK, p38 MAPK and ERK, play
important roles in the activation of NF-κB in microglial-induced
inflammation (23,25,26). However, whether the MAPK signaling
pathway is involved in the activation of NF-κB in activated
microglia stimulated by S100A8/A9 is unclear. In the present study,
our data showed that S100A8/A9 treatment markedly enhanced the
nuclear translocation of NF-κB p65 and the DNA-binding activities
of NF-κB in BV-2 microglial cells. S100A8/A9-induced activation of
NF-κB and secretion of proinflammatory cytokines TNF-α and IL-6
were attenuated by the suppression of ERK and JNK signaling
pathways by PD98059 and SP600125, respectively. The data indicate
that the ERK and JNK signaling pathways are essential to the
activation of NF-κB in S100A8/A9-induced inflammation. In addition,
our data also showed that inhibition of activation of NF-κB reduced
S100A8/A9-induced secretion of TNF-α and IL-6 from cultured BV-2
microglial cells. Taken together, our results indicate that the
ERK/NF-κB and JNK/NF-κB signaling pathways are involved in
S100A8/A9-induced inflammation in BV-2 microglial cells.
Notably, both TLR4 and RAGE inhibitors caused
complete inhibition of S100A8/A9-mediated proinflammatory cytokine
expression, respectively. Boyd et al also demonstrated that
activated TLR4 gives rise to an upregulation of S100A8/A9
expression earlier in cardiomyocytes (19). When S100A8/A9 was overexpressed in
prostate cancer cells, NF-κB and MAPK also remained activated
(43). It provides one
possibility that a positive-feedback loop may exist both upstream
and down-stream of S100A8/A9. In regards to S100A8/A9 or other
ligands, activated RAGE also leads to further enhancement of
S100A8/A9 production, and creates a putative positive feedback loop
in acute inflammation (29,44). Therefore, blocking TLR4 or RAGE
may weaken the positive feedback, and decrease the production of
S100A8/A9 obviously, and completely suppress the S100A8/A9-mediated
elevation of the cytokines IL-6 and TNF-α.
More studies also demonstrated that major risk
factors for sepsis, systemic inflammatory response syndrome and
septic shock are related to TNF-α-independent mechanisms (45,46). However, it was confirmed that a
monoclonal antibody to TNF-α given early in the course of severe
sepsis has a harmful rather than a beneficial consequence in
clinical trials of human sepsis (46). Our present data demonstrated that
S100A8/A9 represents a molecular system involved in the
pathogenesis of inflammatory response syndrome upstream of TNF-α
induction. In consideration of the high abundance of S100A8/A9 in
inflammatory diseases, a potential strategy for blocking
uncontrolled inflammatory processes may be by targeting these
proteins with immune intervention. This strategy may be very
specific, as both proteins are secreted via a so-called
'alternative' pathway (47). The
inhibition of this alternative transport mechanism should not
affect the classical secretion of other proteins through the
endoplasmic reticulum and Golgi complex and may thus avoid major
side effects.
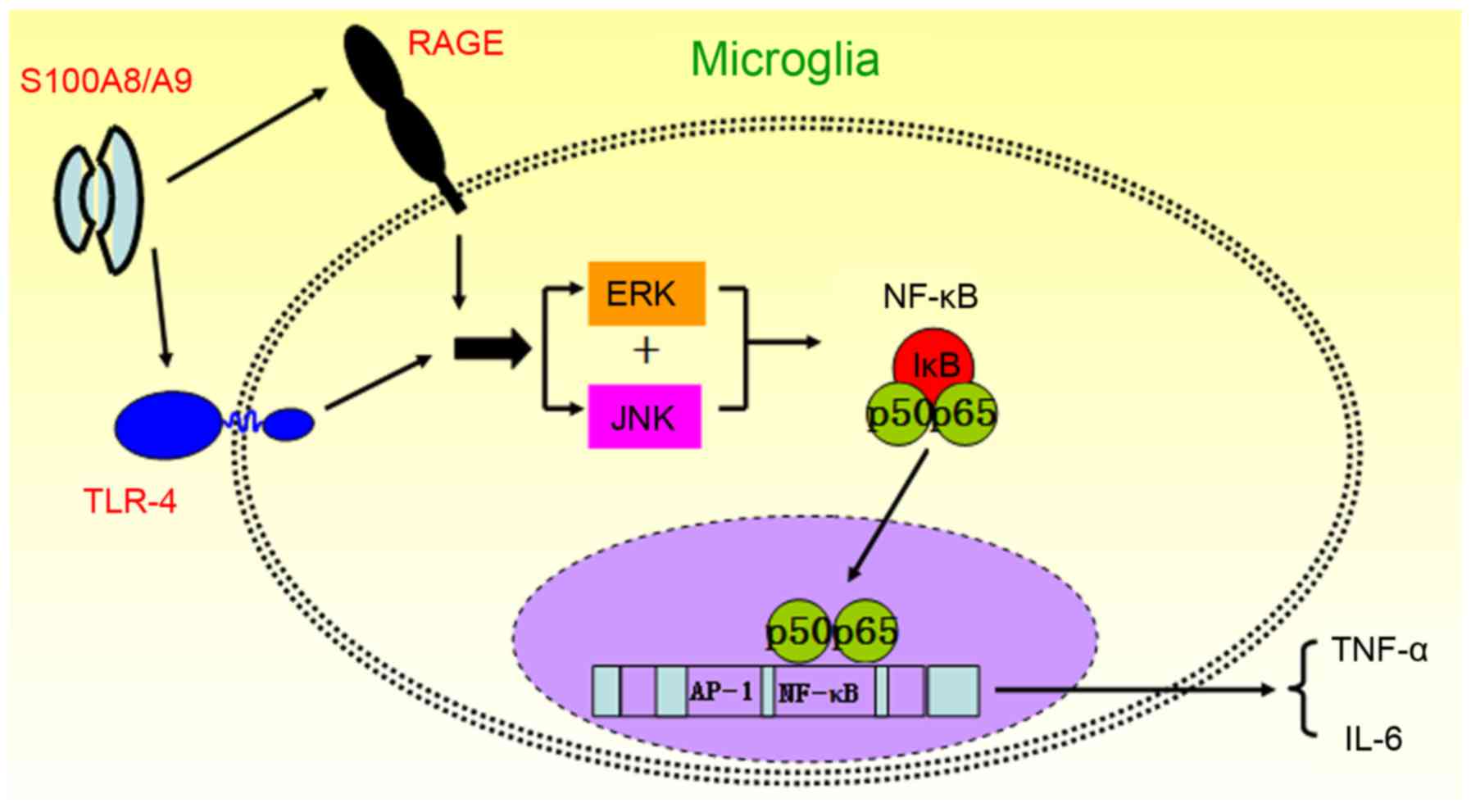
In conclusion, we found that S100A8/A9 activated
microglia via binding RAGE and TLR4 on the membrane and promoted
the production of proinflammatory cytokines in microglia through
the activation of the ERK/NF-κB and JNK/NF-κB signaling pathways
(Fig. 6). Thus, inhibition of
S100A8/A9 release may be another promising therapeutic approach,
and S100A8/A9 may represent a useful biomarker and therapeutic
target in microglial-mediated neuroinflammatory diseases.
Abbreviations:
|
CNS
|
central nervous system
|
|
DAMP
|
damage-associated molecular
pattern
|
|
DAPI
|
6-diamidino-2-phenylindole
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
IL-6
|
interleukin-6
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LPS
|
lipopolysaccharide
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NF-κB
|
nuclear factor-κB
|
|
RAGE
|
receptor for advanced glycation
endproducts
|
|
TLR4
|
Toll-like receptor 4
|
|
TNF-α
|
tumor necrosis factor-α
|
Acknowledgments
This study was supported by grants from the National
Nature Science Foundation of China (nos. 81201444 and
81101401).
References
|
1
|
Nacken W, Roth J, Sorg C and Kerkhoff C:
S100A9/S100A8: Myeloid representatives of the S100 protein family
as prominent players in innate immunity. Microsc Res Tech.
60:569–580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ehrchen JM, Sunderkötter C, Foell D, Vogl
T and Roth J: The endogenous Toll-like receptor 4 agonist
S100A8/S100A9 (calprotectin) as innate amplifier of infection,
autoimmunity, and cancer. J Leukoc Biol. 86:557–566. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Engel S, Schluesener H, Mittelbronn M,
Seid K, Adjodah D, Wehner HD and Meyermann R: Dynamics of
microglial activation after human traumatic brain injury are
revealed by delayed expression of macrophage-related proteins MRP8
and MRP14. Acta Neuropathol. 100:313–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shepherd CE, Goyette J, Utter V, Rahimi F,
Yang Z, Geczy CL and Halliday GM: Inflammatory S100A9 and S100A12
proteins in Alzheimer's disease. Neurobiol Aging. 27:1554–1563.
2006. View Article : Google Scholar
|
|
5
|
Ziegler G1, Prinz V, Albrecht MW,
Harhausen D, Khojasteh U, Nacken W, Endres M, Dirnagl U, Nietfeld W
and Trendelenburg G: Mrp-8 and -14 mediate CNS injury in focal
cerebral ischemia. Biochim Biophys Acta. 1792:1198–1204. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vogl T, Tenbrock K, Ludwig S, Leukert N,
Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg
C, et al: Mrp8 and Mrp14 are endogenous activators of Toll-like
receptor 4, promoting lethal, endotoxin-induced shock. Nat Med.
13:1042–1049. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harja E, Bu DX, Hudson BI, Chang JS, Shen
X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, et al:
Vascular and inflammatory stresses mediate atherosclerosis via RAGE
and its ligands in apoE−/− mice. J Clin Invest.
118:183–194. 2008. View
Article : Google Scholar
|
|
8
|
Björk P, Björk A, Vogl T, Stenström M,
Liberg D, Olsson A, Roth J, Ivars F and Leanderson T:
Identification of human S100A9 as a novel target for treatment of
autoimmune disease via binding to quinoline-3-carboxamides. PLoS
Biol. 7:e972009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hofmann MA, Drury S, Fu C, Qu W, Taguchi
A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al: RAGE
mediates a novel proinflammatory axis: A central cell surface
receptor for S100/calgranulin polypeptides. Cell. 97:889–901. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donato R: Intracellular and extracellular
roles of S100 proteins. Microsc Res Tech. 60:540–551. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roth J, Vogl T, Sorg C and Sunderkötter C:
Phagocyte-specific S100 proteins: A novel group of proinflammatory
molecules. Trends Immunol. 24:155–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghavami S, Rashedi I, Dattilo BM, Eshraghi
M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C and Los M:
S100A8/A9 at low concentration promotes tumor cell growth via RAGE
ligation and MAP kinase-dependent pathway. J Leukoc Biol.
83:1484–1492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Neumar RW, Nolan JP, Adrie C, Aibiki M,
Berg RA, Böttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC,
et al: Post-cardiac arrest syndrome: Epidemiology, pathophysiology,
treatment, and prognostication A consensus statement from the
International Liaison Committee on Resuscitation (American Heart
Association, Australian and New Zealand Council on Resuscitation,
European Resuscitation Council, Heart and Stroke Foundation of
Canada, InterAmerican Heart Foundation, Resuscitation Council of
Asia, and the Resuscitation Council of Southern Africa); the
American Heart Association Emergency Cardiovascular Care Committee;
the Council on Cardiovascular Surgery and Anesthesia; the Council
on Cardiopulmonary, Perioperative, and Critical Care; the Council
on Clinical Cardiology; and the Stroke Council. Circulation.
118:2452–2483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schiopu A and Cotoi OS: S100A8 and S100A9:
DAMPs at the crossroads between innate immunity, traditional risk
factors, and cardiovascular disease. Mediators Inflamm.
2013:8283542013. View Article : Google Scholar
|
|
15
|
Ryckman C, Vandal K, Rouleau P, Talbot M
and Tessier PA: Proinflammatory activities of S100: Proteins
S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and
adhesion. J Immunol. 170:3233–3242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Viemann D, Strey A, Janning A, Jurk K,
Klimmek K, Vogl T, Hirono K, Ichida F, Foell D, Kehrel B, et al:
Myeloid-related proteins 8 and 14 induce a specific inflammatory
response in human microvascular endothelial cells. Blood.
105:2955–2962. 2005. View Article : Google Scholar
|
|
17
|
Bierhaus A, Humpert PM, Morcos M, Wendt T,
Chavakis T, Arnold B, Stern DM and Nawroth PP: Understanding RAGE,
the receptor for advanced glycation end products. J Mol Med (Berl).
83:876–886. 2005. View Article : Google Scholar
|
|
18
|
Sunahori K, Yamamura M, Yamana J, Takasugi
K, Kawashima M, Yamamoto H, Chazin WJ, Nakatani Y, Yui S and Makino
H: The S100A8/A9 heterodimer amplifies proinflammatory cytokine
production by macrophages via activation of nuclear factor kappa B
and p38 mitogen-activated protein kinase in rheumatoid arthritis.
Arthritis Res Ther. 8:R692006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boyd JH, Kan B, Roberts H, Wang Y and
Walley KR: S100A8 and S100A9 mediate endotoxin-induced
cardiomyocyte dysfunction via the receptor for advanced glycation
end products. Circ Res. 102:1239–1246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Turovskaya O, Foell D, Sinha P, Vogl T,
Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, et
al: RAGE, carboxylated glycans and S100A8/A9 play essential roles
in colitis-associated carcinogenesis. Carcinogenesis. 29:2035–2043.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
O'Neill LA and Kaltschmidt C: NF-kappa B:
A crucial transcription factor for glial and neuronal cell
function. Trends Neurosci. 20:252–258. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mattson MP: NF-kappaB in the survival and
plasticity of neurons. Neurochem Res. 30:883–893. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu H, Li Z, Zhu X, Lin R and Chen L:
Salidroside reduces cell mobility via NF-kappa B and MAPK signaling
in LPS-induced BV2 microglial cells. Evid Based Complement Alternat
Med. 2014:3838212014. View Article : Google Scholar
|
|
24
|
Jeong YH, Kim Y, Song H, Chung YS, Park SB
and Kim HS: Anti-inflammatory effects of α-galactosylceramide
analogs in activated microglial: Involvement of the p38 MAPK
signaling pathway. PLoS One. 9:e870302014. View Article : Google Scholar
|
|
25
|
Yuan L, Wu Y, Ren X, Liu Q, Wang J and Liu
X: Isoorientin attenuates lipopolysaccharide-induced
proinflammatory responses through down-regulation of ROS-related
MAPK/NF-κB signaling pathway in BV-2 microglial. Mol Cell Biochem.
386:153–165. 2014. View Article : Google Scholar
|
|
26
|
Li L, Wu Y, Wang Y, Wu J, Song L, Xian W,
Yuan S, Pei L and Shang Y: Resolvin D1 promotes the
interleukin-4-induced alternative activation in BV-2 microglial
cells. J Neuroinflammation. 11:722014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manderson AP, Kay JG, Hammond LA, Brown DL
and Stow JL: Subcompartments of the macrophage recycling endosome
direct the differential secretion of IL-6 and TNFalpha. J Cell
Biol. 178:57–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu J, Qu C, Lu X and Zhang S: Activation
of microglial by histamine and substance P. Cell Physiol Biochem.
34:768–780. 2014. View Article : Google Scholar
|
|
29
|
Ehlermann P, Eggers K, Bierhaus A, Most P,
Weichenhan D, Greten J, Nawroth PP, Katus HA and Remppis A:
Increased proinflammatory endothelial response to S100A8/A9 after
preactivation through advanced glycation end products. Cardiovasc
Diabetol. 5:62006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ishihara K, Namura T, Murayama H, Arai S,
Totani M and Ikemoto M: Possibility of formation of the
S100A8/A9-proinflammatory cytokine complexes in vivo in acute
inflammation and their functional roles. Rinsho Byori. 57:324–331.
2009.In Japanese. PubMed/NCBI
|
|
31
|
Cesaro A, Anceriz N, Plante A, Pagé N,
Tardif MR and Tessier PA: An inflammation loop orchestrated by
S100A9 and calprotectin is critical for development of arthritis.
PLoS One. 7:e454782012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee Y, Jang S, Min JK, Lee K, Sohn KC, Lim
JS, Im M, Lee HE, Seo YJ, Kim CD, et al: S100A8 and S100A9 are
messengers in the crosstalk between epidermis and dermis modulating
a psoriatic milieu in human skin. Biochem Biophys Res Commun.
423:647–653. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chimenti MS, Ballanti E, Perricone C,
Cipriani P, Giacomelli R and Perricone R: Immunomodulation in
psoriatic arthritis: Focus on cellular and molecular pathways.
Autoimmun Rev. 12:599–606. 2013. View Article : Google Scholar
|
|
34
|
McColl BW, Allan SM and Rothwell NJ:
Systemic infection, inflammation and acute ischemic stroke.
Neuroscience. 158:1049–1061. 2009. View Article : Google Scholar
|
|
35
|
Aliprantis AO, Yang RB, Weiss DS, Godowski
P and Zychlinsky A: The apoptotic signaling pathway activated by
Toll-like receptor-2. EMBO J. 19:3325–3336. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Karikó K, Weissman D and Welsh FA:
Inhibition of Toll-like receptor and cytokine signaling - a
unifying theme in ischemic tolerance. J Cereb Blood Flow Metab.
24:1288–1304. 2004. View Article : Google Scholar
|
|
37
|
Kielian T: Toll-like receptors in central
nervous system glial inflammation and homeostasis. J Neurosci Res.
83:711–730. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mishra BB, Mishra PK and Teale JM:
Expression and distribution of Toll-like receptors in the brain
during murine neurocysticercosis. J Neuroimmunol. 181:46–56. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lehnardt S, Massillon L, Follett P, Jensen
FE, Ratan R, Rosenberg PA, Volpe JJ and Vartanian T: Activation of
innate immunity in the CNS triggers neurodegeneration through a
Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA.
100:8514–8519. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kilic U, Kilic E, Matter CM, Bassetti CL
and Hermann DM: TLR-4 deficiency protects against focal cerebral
ischemia and axotomy- induced neurodegeneration. Neurobiol Dis.
31:33–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marsh BJ, Williams-Karnesky RL and
Stenzel-Poore MP: Toll-like receptor signaling in endogenous
neuroprotection and stroke. Neuroscience. 158:1007–1020. 2009.
View Article : Google Scholar :
|
|
42
|
Heizmann CW, Ackermann GE and Galichet A:
Pathologies involving the S100 proteins and RAGE. Subcell Biochem.
45:93–138. 2007. View Article : Google Scholar
|
|
43
|
Hermani A, De Servi B, Medunjanin S,
Tessier PA and Mayer D: S100A8 and S100A9 activate MAP kinase and
NF-kappaB signaling pathways and trigger translocation of RAGE in
human prostate cancer cells. Exp Cell Res. 312:184–197. 2006.
View Article : Google Scholar
|
|
44
|
Eggers K, Sikora K, Lorenz M, Taubert T,
Moobed M, Baumann G, Stangl K and Stangl V: RAGE-dependent
regulation of calcium-binding proteins S100A8 and S100A9 in human
THP-1. Exp Clin Endocrinol Diabetes. 119:353–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fisher CJ Jr, Agosti JM, Opal SM, Lowry
SF, Balk RA, Sadoff JC, Abraham E, Schein RM and Benjamin E:
Treatment of septic shock with the tumor necrosis factor
receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study
Group. N Engl J Med. 334:1697–1702. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Clark MA, Plank LD, Connolly AB, Streat
SJ, Hill AA, Gupta R, Monk DN, Shenkin A and Hill GL: Effect of a
chimeric antibody to tumor necrosis factor-alpha on cytokine and
physiologic responses in patients with severe sepsis - a
randomized, clinical trial. Crit Care Med. 26:1650–1659. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rammes A, Roth J, Goebeler M, Klempt M,
Hartmann M and Sorg C: Myeloid-related protein (MRP) 8 and MRP14,
calcium-binding proteins of the S100 family, are secreted by
activated monocytes via a novel, tubulin-dependent pathway. J Biol
Chem. 272:9496–9502. 1997. View Article : Google Scholar : PubMed/NCBI
|