Introduction
In recent years, epigenetics has become a hot topic
in cancer research. The balance of methylation and demethylation in
epigenetic modification affects gene expression and cellular
activity. Studies have demonstrated that aberrant histone lysine
methylation in cancer is associated not only with the repression of
chromatin related to specific genes, but also with the repression
of large chromosomal regions (1).
Epigenetic alterations in lysine-specific demethylase 1 (LSD1; also
known as KDM1A) have been shown to play a key role in
carcinogenesis (2). As LSD1
belongs to the flavin-dependent amine oxidase family,
LSD1specifically catalyzes the demethylation of mono- and
dimethylated histone H3 lysine 4 (H3K4) and H3 lysine 9 (H3K9)
through a redox process. LSD1 is overexpressed in many solid tumors
such as breast, colon, neuroblastoma, bladder, small cell lung and
prostate cancers (3–7) and plays an important role in
leukemia (8), attracting steadily
increasing attention as a major target for epigenetic drug
discovery (9–11). To date, only few studies have
implicated LSD1 in mantle cell lymphoma (MCL) and T-cell acute
lymphoblastic leukemia (T-ALL) (12).
MCL is a type of non-Hodgkin's lymphoma (NHL),
comprising approximately 6% of all NHL cases. MCL cells generally
overexpress cyclin D1 due to a t(11;14)(q13;q32) chromosomal
translocation, which plays an important role in the pathogenesis of
the disease. These cases exhibit a higher proliferative activity
and a more aggressive clinical evolution (13). Conventional therapy leads to
complete remission in only 10–15% of patients. T-ALL, which
accounts for 25% of cases of adult ALL, is characterized by the
malignant clonal expansion of immature T-cell progenitors (14). These are aggressive malignancies
that do not respond well to chemotherapy and have a poorer
prognosis than their B cell counterparts.
In this study, we investigated the state of LSD1 and
the histone methylation of H3K4me1 and H3K4me2 in MCL. We also
examined the correlation of LSD1 and H3K4me1 and H3K4me2 with Ki67,
which is prognostic marker in MCL. Furthermore, we silenced LSD1
using small interfering RNA (siRNA) and measured consequent histone
modification, gene transcription, cell proliferation and apoptosis
in JeKo-1 and MOLT-4 cells.
Materials and methods
Patients and specimens
Surgical specimens from 30 patients with MCL
obtained at the Zhangzhou Affiliated Hospital of Fujian Medical
University, Zhangzhou, China between January 2005 and December
2010, were retrospectively collected for analysis. Lymphoid tissue
samples were collected from all participants after obtaining
written informed consent. This study was approved by the Ethics
Committee of Zhangzhou Affiliated Hospital of Fujian Medical
University. Diagnosis was made according to the WHO classification.
In all 30 cases of MCL, cyclin D1 was overexpressed. Samples of
patients with hyperplastic lymphadenitis (n=30) were used as
controls. None of the patients had received radiotherapy or
chemotherapy prior to surgery.
Antibodies
Antibodies for rabbit against human monomethylated
H3K4 (cat. no. 07-436), dimethylated H3K4 (cat. no. 07-030) and
trimethylated H3K4 (cat. no. 07-992), rabbit against human
acetylation H3 (cat. no. 07-677-I), and rabbit against human LSD1
(cat. no. 07-705) were purchased from Upstate Biotechnology (Lake
Placid, NY, USA); antibodies for cyclin D1 (EP12) and Ki67 (MIB1)
were from (Dako, Glosrup, Denmark). Antibodies for Bcl-2 (cat. no.
sc-578), Bax (cat. no. sc-20067), pro-caspase-3 (cat. no.
sc-22139), C-myc (cat. no. sc-4084), p15 (cat. no. sc-271791),
DNMT1 (cat. no. sc-514784) and β-actin (cat. no. sc-8432) were from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Tissue microarray (TMA) construction and
immunohistochemistry
The archival formalin-fixed and paraffin
wax-embedded tissue blocks of 30 MCL and 30 hyperplastic
lymphadenitis samples were removed by surgery. A representative
tumor paraffin block (donor block) was collected from each case,
and two tissue cores (2 mm in diameter) were obtained using a
trephine apparatus. Trephinated paraffin tissue cores were then
arranged in a new recipient paraffin block (tissue array block).
Cores containing tumors in >50% of the area were considered
adequate. Immunohistochemical staining was performed using
commercial polyclonal rabbit antibodies to histone
demethytransferase LSD1, and mono- and dimethyl histone H3K4, Ki67
and cyclin D1. Tissue array blocks were sectioned at a thickness of
4 µm and mounted on pre-coated glass slides. The sections
were deparaffinized and hydrated prior to immunohistochemistry. The
immunohistochemical SP method was performed according to the
manufacturer's instructions. Tissues positive for all the purchased
antibodies were used as positive controls. Negative control
staining was carried out by substituting non-immune rabbit and
phosphate-buffered saline (PBS) for the primary antibodies. The
positive control exhibited brown color in the nuclei. Chevallier's
semi-quantity system analysis was used in the calculation of the
results of immunohistochemistry. For LSD1, mono and dimethylated
histone H3K4, the results are presented as the sum of scores
presenting color density and the percentage of stained cells.
According to color density, non-stained cells were scored as 0,
slightly stained were scored as 1, intermediate-stained were scored
as 2 and strongly stained were scored as 3. When the number of
positive cells was <25, 25–50, or50–75, or >75%, the
immunoreactivity was scored as 0, 1+, 2+, 3+and 4+, respectively.
The two scores of presenting color density and the number of
positive cells were added for each slide. The sum that was 0 was
negative, 1–2 was presented as +, 3–4 as ++, 5–6 as +++, and 7 as
++++. If the sum of the two scores was <2, it was considered
negative staining and >2 was considered positive staining. The
scores of each patient group were averaged.
The Ki67 index was defined as the percentage of
Ki67- positive tumor cells in representative areas of the lymphoma.
To count the number of Ki67-positive cells, 2 representative areas
were selected. A representative area was defined not to contain
residual germinal centers, hot spots of proliferation or
proliferating T-cells. Hot spots of proliferation were areas of
tumor cells of <2 high-power fields in size (field of vision at
×400 magnification), which proliferate higher than the rest of the
tumor. Counting was performed by one observer. The positive cells
among 500 cells were counted using an eyepiece with a grid in a 40×
objective. The Ki67 index was calculated as the percentage of
positive cells by averaging the values obtained for the 2 areas
(count-Ki67 index) with a semi-quantitative evaluation system,
consisting of 4 levels: <10, 10–30, 30–50 and>50% labeled
cells. Negative control staining was obtained by omitting the
primary antibodies.
Cell culture
The human MCL JeKo-1 (MCL) and MOLT-4(acute
lymphoblastic leukemia) cell lines used in this study were
purchased from the Shanghai Institute of Cell Bank (Shanghai,
China). The cells were cultured in 10% fetal bovine serum, and
RPMI-1640 containing 2 mM L-glutamine under 37°C, saturated
humidity and 5% CO2. The cells were subcultured every
3–5 days. The cells were seeded and treated with the desired siRNA
concentrations in different time points.
siRNA and transfection
The cells were seeded with 5×104 cells in
96-well plates, and incubated for 2–4 days in standard medium in
the presence of 10–20 nmol/l siRNA directed against LSD1. The siRNA
LSD1 sequences used in this study were as follows: siRNA LSD1
(sense, 5′-CCACGAGUCAAACCUUUAUTT-3′ and antisense,
5′-AUAAAGGUUUGACUCGUGGTT-3′), which was synthesized by Shanghai
GenePharma Co., Ltd. (Shanghai, China); the cells were transfected
using Lipofectamine™ 2000 according to the manufacturer's
instructions (Invitrogen, Carlsbad, CA, USA). Inducible JeKo-1 and
MOLT-4 cells (1×105 cells/well) were seeded onto 24-well
plates (Costar Life Science, Acton, MA, USA) and transiently
transfected with the desired concentrations of LSD1 siRNA. All
experiments were conducted in triplicate using independent
cultures. Both total RNA and protein were extracted at 24 h after
transfection.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from samples of
1×106 cells using TRIzol reagent (Invitrogen). The
quantity and quality of the RNA samples were measured by absorbance
at 260 and 280 nm. RNA samples with an A260, A280 ratio of 1.8–2.0
were stored at −80°C until use. Copy DNA synthesis was performed
using an avian myeloblastosis virus reverse transcriptase kit,
according to the manufacturer's instructions (Promega, Madison, WI,
USA). β-actin was used as an internal control. The primers used in
the PCR amplifications were: LSD1 forward,
5′-GTTGGAGAGTAGCCTCAAATGTC-3′ and reverse,
5′-TGACCGGATGACTTCTCAAGA-3′; and β-actin forward,
5′-GAGACCTTCAAGACCCCAGCC-3 and reverse,
5′-TCGGGGCATCGGAACCGCTCA-3′. In addition, the amplicon size was 155
basepairs (bp), 404 bp for LSD1 and β-actin, respectively. The PCR
reaction conditions were: 95°C for30 sec, 60°C for 30 sec, 72°C for
40 sec, and was repeated 32 cycles. PCR-amplified products were
electrophoresed on 1.5% (w/v) agarose gels with 1 µg/ml
ethidium bromide. The experiments were repeated twice.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bro mide (MTT)
growth inhibition assay
To examine the effect of the silencing of LSD1 by
siRNA on cell growth, cell viability assay was determined using
MTT. The JeKo-1 and Motl-4 cells were seeded at a density of
1×105 cells/well in 96-well culture dishes (Costar Life
Science) and treated with siRNA directed against LSD1. This was
followed by transfection (n=5) with the indicated concentrations of
LSD1 siRNA at different time points in the JeKo-1 and MOLT-4 cells.
Cell proliferation was measured by MTT assay (0.5 mg/ml;
Sigma-Aldrich, St. Louis, MO, USA), which was accomplished as
indicated. The spectrophotometric absorbance of the samples was
determined using the Ultra Multifunctional Microplate Reader
(Tecan, Durham, NC, USA) at 492 and 630 nm for absorbance (OD
value). The inhibitionn ratio was also calculated. The experiment
was repeated in triplicate. Cell proliferation rate (%) =
(Aexperiment − Ablank)/(Acontrol −
Ablank) ×100%.
Evaluation of apoptosis by flow
cytometry
Apoptosis assay was performed according to the
manufacturer's instructions (BD Biosciences, San Jose, CA, USA). A
total of 1×106 cells/well were seeded in 6-well plates
(Costar Life Science) with sterile cover glass placed at the bottom
of each well, and then transfected with negative control siRNA, or
40, 80 and 120 nmol/l of LSD1 siRNA for 24 h, while at at the
logarithmic phase (for JeKo-1 cells), and at 30, 60 and 120 nmol/l
LSD1 siRNA for 24 h (for MOLT-4 cells). The cells were then
resuspended in binding buffer and stained with Annexin
V-FITC/propidium iodide (PI) following the manufacturer's
instructions (BD Biosciences) by flow cytometry (Epics-XL; Beckman
Coulter, Inc., Brea, CA, USA). A total of 10,000 events/sample were
acquired on the FACScan. Fluorescent commissions were collected
through 5.30 and 5.70 nm pass filters for FITC and PI,
respectively. The measurements were performed independently at
least 3 times with similar results.
Western blot analysis for the detection
of the protein expression of LSD1, monomethylated H3K4,
dimethylated H3K4, trimethylated H3K4 and Act-H3, and
apoptosis-related proteins
Total protein lysis and western blot analysis were
performed according to the instructions of the manufacturer
(Pierce, Rockford, IL, USA). Briefly, the JeKo-1 andMOLT-4 cells
were plated on culture dishes and transfected with LSD1 siRNA at
the indicated concentrations for the JeKo-1 and MOLT-4 cells for 24
h. The control cells were incubated in medium with
Na2CO3 using same the time points. Following
transfection with siRNA LSD1, total proteins were prepared from
each culture condition with lysis buffer (Pierce) containing
freshly prepared protease inhibitors. Protein concentration was
then measured using BCA protein assay (Pierce). Cell extracts were
subjected to 12% SDS-PAGE and electrophoretically transferred onto
nitrocellulose membranes. The membranes were incubated with
monoclonal anti-mono-, di- and trimethylated histone H3K4,
anti-histone acetylated H3, LSD1 (Upstate Biotechnology), Bcl-2,
Bax, pro-caspase-3, C-myc, p15, DNMT1 and cyclin D1 (Santa Cruz
Biotechnology, Inc.) antibodies. After being washed with TBS, the
membranes were incubated with secondary antibodies conjugated with
peroxidase [goat anti-rabbit (cat. no. sc-3837) and goat anti-mouse
(cat. no. sc-395758) antibodies; Santa Cruz Biotechnology, Inc.].
The signal was then detected using the chemiluminescent detection
system (Pierce) and analyzed by a color image analysis system
(AlphaDigiDoc; Alpha Innotech).
Statistical analysis
All statistical calculations were performed using
SPSS version 18.0 for Windows software (SPSS, Inc., Chicago, IL,
USA). Data are presented as the means ± SD. The t test was carried
out to compare the levels of the parameters between 2 groups.
Differences between the values were assessed for statistical
significance using the Chi-square, Wilcoxon rank sum test, one-way
analysis of variance (ANOVA) and repeated measure ANOVA to evaluate
correlations between Ki67 and LSD1, and H3K4me1 and H3K4me2.
Agreement measure was calculated by Cohen's kappa statistic κ. The
statistical level of significance was set at the 5% level
(p<0.05).
Results
High expression of LSD1 and lower
expression of methylated H3K4me1 and H3K4me2 in MCL
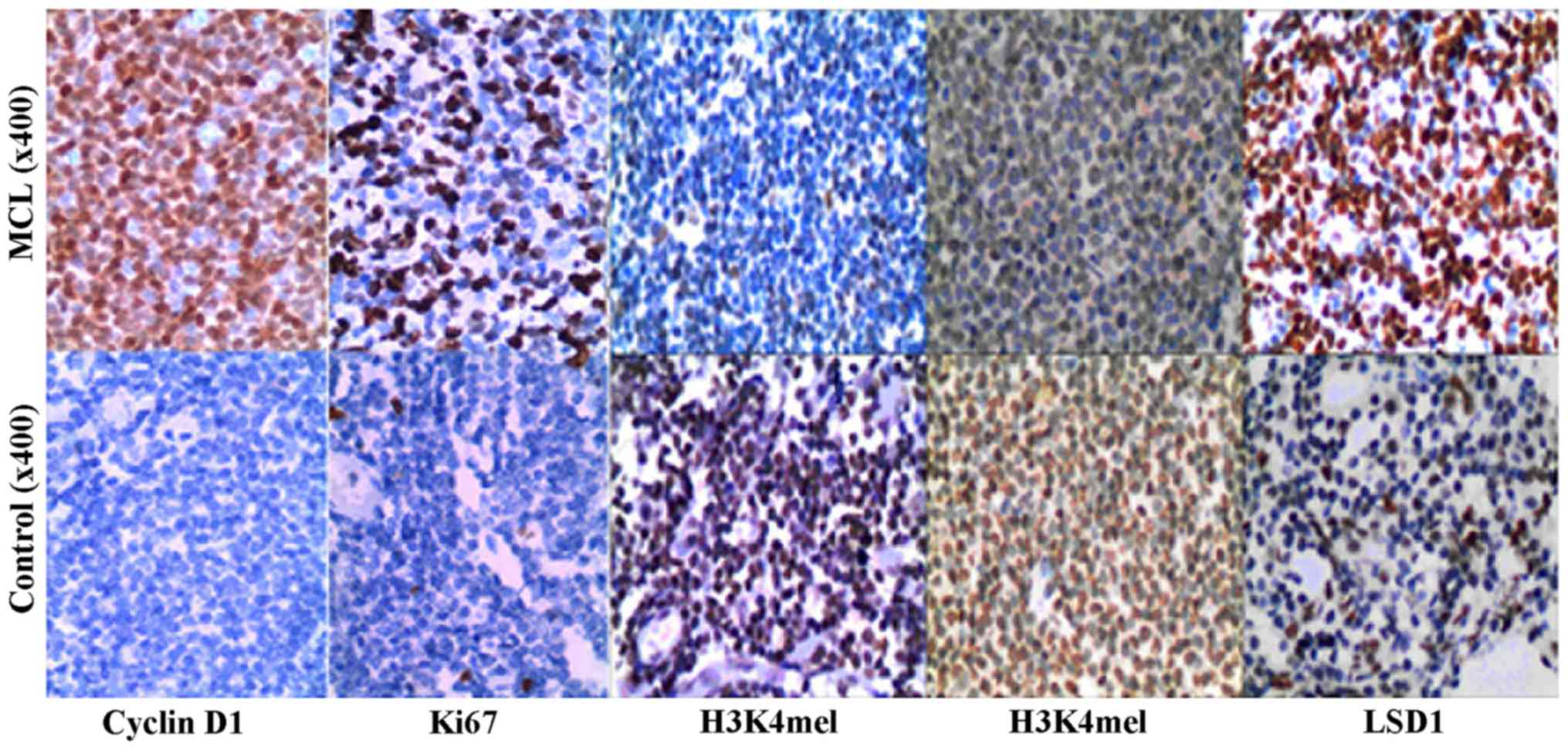
LSD1 expression is upregulated in MCL cancer
tissues. We first examined the expression levels of LSD1 in 30 MCL
and 30 hyperplastic lymphadenitis (controls) by TMA staining. A
high LSD1 expression was detected in the nuclei of malignant cells,
while less staining was observed in the non-neoplastic tissues
(Fig. 1). Specifically, high
levels of LSD1 expression (score, 3+ to 4+) were observed in 15 out
of the 30 (50.0%) cases of MCL, and in 17% (5 out of 30) of the
controls, indicating a significant elevation of LSD1 expression in
MCL (p<0.01) (Table I). The
expression of H3K4me1 in the MCL cases was 33% (10/30), lower than
that in the controls (71%; 23/30; p<0.01). The expression of
H3K4me2 in the MCL cases was 10% (3/30), lower than that in the
controls (47%; 14/30; p<0.01) (Table I). Thus, we hypothesized that LSD1
may play an important role in the pathogenesis of MCL. In addition,
the expression of cyclin D1 in the MCL cases was 100% positive, but
that in the hyperplastic lymphadenitis cases was 100% negative.
 | Table IExpression of LSD1, H3K4me1 and
H3K4me2 in MCL cases. |
Table I
Expression of LSD1, H3K4me1 and
H3K4me2 in MCL cases.
| MCL cases (n=30)
| Rate (%) | Control cases
(n=30)
| Rate (%) | χ2 | P-value |
|---|
| − | + | 2+ | 3+ | 4+ | − | + | 2+ | 3+ | 4+ |
|---|
| LSD1 | 0 | 5 | 10 | 12 | 3 | 50.0 | 2 | 7 | 16 | 4 | 1 | 17 | 7.5 | 0.0062 |
| H3K4me1 | 3 | 3 | 14 | 9 | 1 | 33.3 | 0 | 1 | 6 | 19 | 4 | 71 | 11.38 | 0.0007 |
| H3K4me2 | 16 | 6 | 5 | 3 | 0 | 10.0 | 1 | 8 | 7 | 12 | 2 | 47 | 9.93 | 0.0016 |
Correlation of methylated H3K4me1 and
H3K4me2, and LSD1 with Ki67 in MCL
To evaluate the clinical significance of LSD1
overexpression in MCL, we examined whether the expression levels of
LSD1, H3K4me1 and H3K4me2 correlated with Ki67, which is a poor
prognostic factor in MCL. The Ki67 labeling index was 100% in MCL
[≤10, 66.7% (20/30; 11–30%, 16.7% (5/30); 31–50%, 6.7 (2/30); and
>50%, 10.0% (3/30)]. The mean index was 12.53±23.70%. In
hyperplastic lymphadenitis, the Ki67 score was as follows: ≤10,
93.3% (28/30); and 11–30%, only 6.7%. The mean index was
1.97±2.70%, p<0.05 (Fig. 1 and
Table II). To evaluate the
correlation of LSD1, H3K4me1 and H3K4me2 with Ki67, we examined the
correlation between LSD1, H3K4me1 and H3K4me2 with Ki67 by
statistical analysis. The data indicated that the Ki67 labeling
index positively correlated with LSD1 (κ=0.667, p<0.01). There
were no significant correlations between H3K4me1 and H3K4me2, and
Ki67 (κ=−0.182, p>0.05, κ=−0.200, p>0.05) (Table III). This suggested that LSD1
may be one of the prognostic factors in MCL.
| Table IIExpression of Ki67 in MCL cases. |
Table II
Expression of Ki67 in MCL cases.
| Ki67
| P-value |
|---|
| ≤10% | 11–30% | 31–50% | >50% | Mean ± SD |
|---|
| MCL, n (%) | 20 (66.7) | 5 (16.7) | 2 (6.7) | 3 (10) | 12.53±23.70 | <0.05 |
| Control, n (%) | 28 (93.3) | 2 (6.7) | 0 | 0 | 1.97±2.70 | |
| Table IIICorrelation of LSD1, H3K4me1 and
H3K4me2 with Ki67 in MCL. |
Table III
Correlation of LSD1, H3K4me1 and
H3K4me2 with Ki67 in MCL.
| Ki67 | LSD1
| H3K4me1
| H3K4me2
|
|---|
| + | − | + | − | + | − |
|---|
| + | 10 | 0 | 2 | 8 | 0 | 10 |
| − | 5 | 15 | 8 | 12 | 3 | 17 |
| κ | 0.667 | −0.200 | −0.182 |
| P-value | <0.01 | >0.05 | >0.05 |
Silencing efficiency by transfection of
JeKo-1 and MOLT-4cells with LSD1 siRNA
At 24 h following transfection with the indicated
concentrations of LSD1 siRNA into the JeKo-1 and MOLT-4 cells,
green fluorescence in the transfected cells was observed by an
inverted fluorescence microscope, and transfection efficiency was
calculated, which was 90±4.34 in the JeKo-1 and 64±2.18% in the
MOLT-4 cells (n=5) (Fig. 2A). The
amplification of LSD1 mRNA was decreased in a
concentration-dependent manner. The gray value (to β-actin)
indicated that the amplification of LSD1 was 1.02±0.17 at 40
nmol/l, 0.89±0.13 at 80 nmol/l and 0.13±0.04 at 120 nmol/l in the
JeKo-1 cells, compared to the controls (1.02±0.17,
χ2=18.61; p<0.05) (Fig.
2B). Compared to the controls, the protein expression of LSD1
deceased in a concentration-dependent manner. Following
transfection with 40, 80 and 120 nmol/l LSD1 siRNA into the JeKo-1
cells for 24 h, the protein expression of LSD1 was decreased 0.38-,
0.21- and 0.15-fold respectively. Following transfection with siRNA
LSD1 at 120 nmol/l for 24 h, LSD1 expression was decreased by
85.56% (Fig. 2C and D). The gray
value (to β-actin) indicated that the amplification of LSD1 was
0.302±0.083 at 30 nmol/l, 0.153±0.082 at 60 nmol/land 0.091±0.024
at 120 nmol/l in the MOLT-4 cells, compared to the controls
(0.967±0.124, p<0.05) (Fig.
2B). Following transfection with siRNA LSD1 at 120 nmol/l for
24 h, LSD1 expression was decreased by 67.67% (Fig. 2C and D).
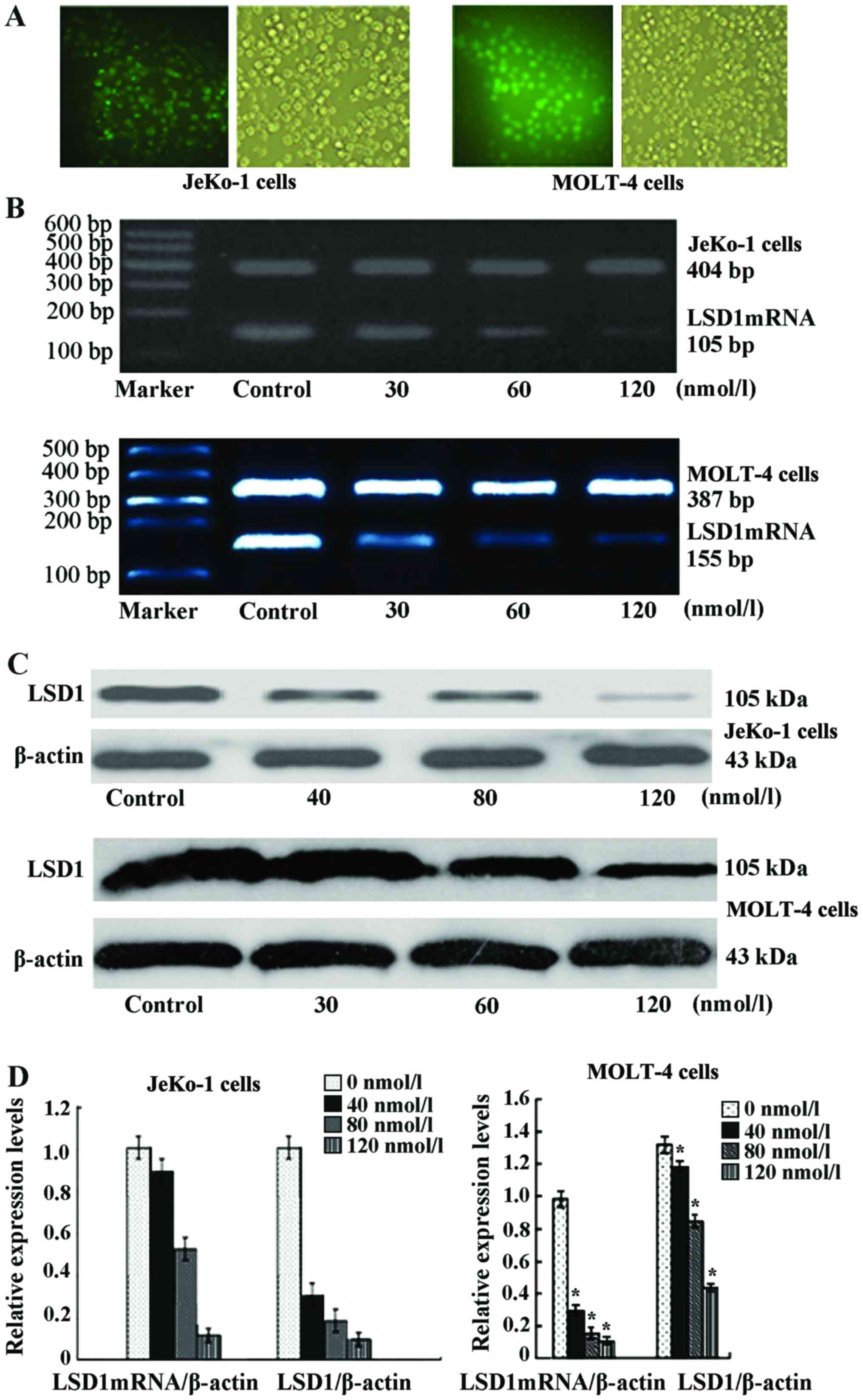 | Figure 2(A) JeKo-1 cells (left panel) were
transfected with lysine-specific demethylase 1 (LSD1) small
interfering RNA (siRNA) and after 24 h the transfection efficiency
was examined under a fluorescence microscope. The transfection
efficiency was 90±4.34% (n=5). MOLT-4 cells (right panel) were
transfected with LSD1 siRNA and after 24 h, the transfection
efficiency was examined under a fluorescence microscope. The
transfection efficiency was 64±2.18% (n=5). (B) mRNA expression of
LSD1 following transfection with various concentrations of siRNA in
JeKo-1 and MOLT-4 cells. At 24 h after transfection, the
amplification of LSD1 mRNA was decreased in a
concentration-dependent manner. The amplification of LSD1 was
1.02±0.17 at 40 nmol/l, 0.89±0.13 at 80 nmol/l, and 0.13±0.04 at
120 nmol/l in the JeKo-1 cells, compared to the controls
(1.02±0.17, χ2=18.61, p<0.05; upper panel). Compared
to the controls, the protein expression of LSD1 was deceased in
concentration-dependent manner. The amplification of LSD1 was
0.302±0.083 at 30 nmol/l, 0.153±0.082 at 60 nmol/l and
0.091±0.024at 120 nmol/l in the MOLT-4 cells, compared to the
controls (0.967±0.124, p<0.05; lower panel). (C) Following
transfecton with siRNA LSD1 at the indicated concentrations for 24
h, the protein expression of LSD1 was decreased in a
concentration-dependent manner in the Jako-1 and MOLT-4 cells. (D)
Relative protein density (*p<0.05 vs. 0 nmol/l). |
Silencing of LSD1 inhibits cell
proliferation and promotes the apoptosis of JeKo-1 and MOLT-4
cells
The inhibition of LSD1 impaired the proliferation
and induced the apoptosis of MCL cells in vitro. We
performed a knockdown experiment using siRNA targeting LSD1, to
further investigate the roles of LSD1 in the proliferation and
apoptosis of JeKo-1 and MOLT-4 cells. The silencing of LSD1
significantly suppressed the proliferation of the JeKo-1 and MOLT-4
cells. The cells were transfected with various concentrations of
LSD1 siRNA for 24 h. We detected the OD value of JeKo-1 and MOLT-4
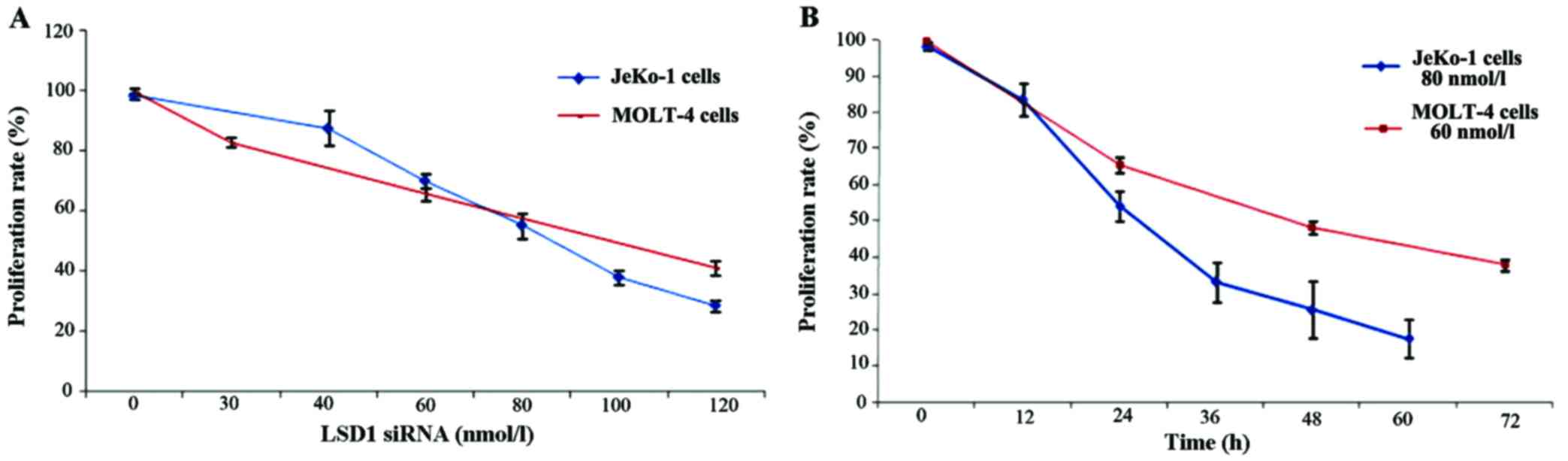
cells by MTT assay to generate cell growth curves. In the JeKo-1
cell line, approximately 87.64±5.73% proliferation was observed
following transfection with LSD1 siRNA at 40 nmol/l, 70.23±2.57% at
60 nmol/l, 55.41±4.24% at 80 nmol/l, 38.15±2.24% at 100 nmol/land
28.72±1.82% at 120 nmol/l, compared to 98.42±1.12% in the controls
(0 nmol/l) (p<0.01). In the MOLT-4 cell line, approximately,
83.02±1.69% cell proliferation was observed following transfection
with LSD1 siRNA at 30 nmol/l, 65.72±2.16% at 60nmol/l, 41.15±2.23%
at 120nmol/l, compared to 99.65±1.21% in the controls (0 nmol/l)
(p<0.01). Cell proliferation decreased along with siRNA LSD1
transfection in a concentration-dependent manner (Fig. 3A). In the JeKo-1 cells, following
transfection with LSD1 siRNA at 80 nmol/l, the cell proliferation
rate was 83.56±4.51, 54.14±4.15, 33.15±5.50, 25.42±7.85 and
17.50±5.32% at 12, 24, 36, 48 and 60 h, respectively. In the MOLT-4
cells, following transfection with LSD1 siRNA at 60 nmol/l, the
cell proliferation rate was 65.72±2.16, 48.26±1.92 and 37.86±1.66%,
at 24, 48 and 72 h, respectively. LSD1 siRNA induced the loss of
cell viability in a time-dependent manner (Fig. 3B).
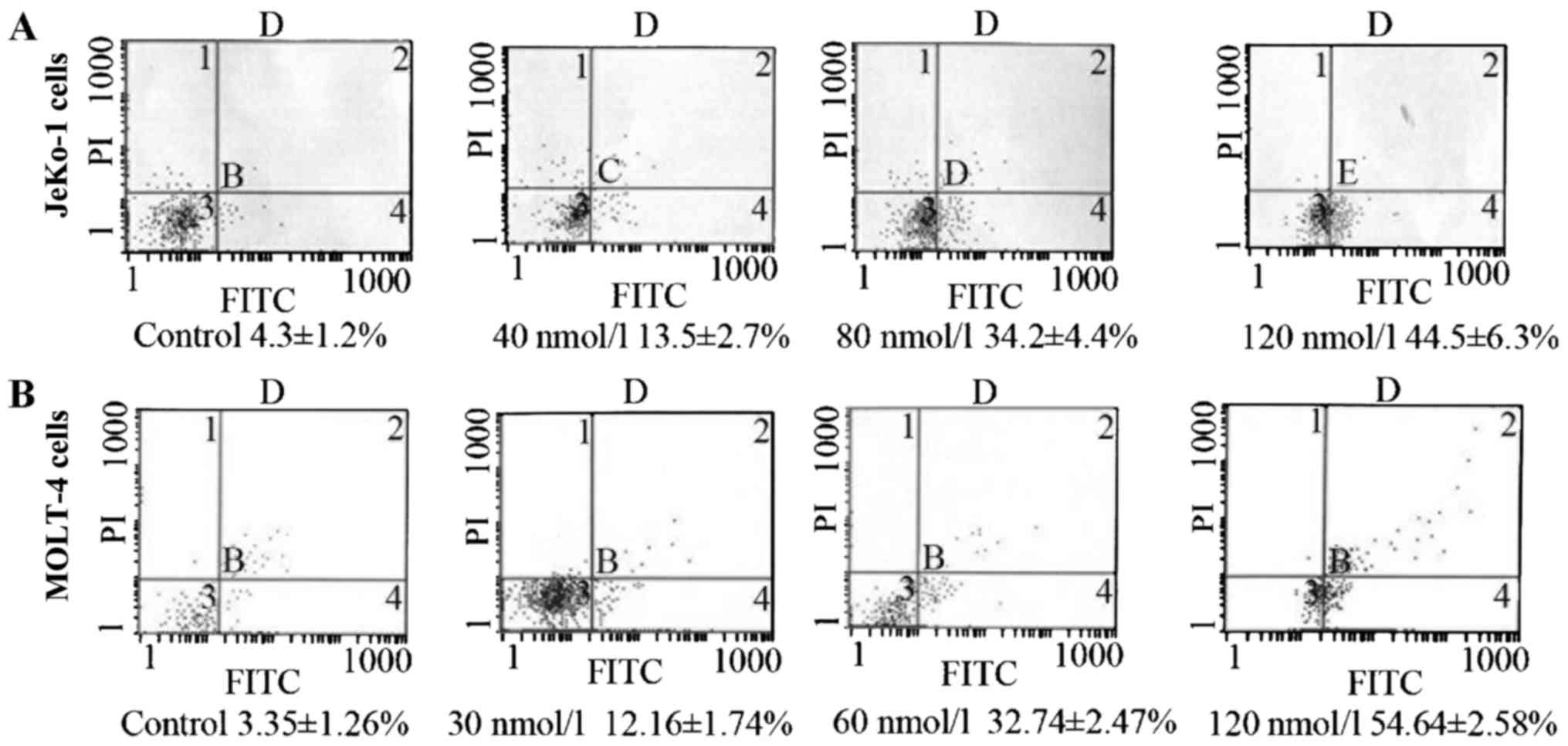
To verify whether the LSD1 siRNA-induced
cytotoxicity was due to the induction of apoptosis, Annexin V-FITC
labeling was performed following 24 h. In the JeKo-1 cells, the
apoptotic rate was 13.5±2.7, 34.2±4.4 and 44.5±6.3% following
transfection with siRNA LSD1 at 40, 80 and 120 nmol/l for 24 h,
respectively, compared to the untreated cells (4.3±1.2%, F=33.41,
p<0.01) (Fig. 4A). In the
MOLT-4 cells, the apoptotic rate was 12.16±1.74, 32.74±2.47 and
54.64±2.58% following transfection with siRNA LSD1 at 30, 60 and
120 nmol/l for 24 h, respectively, compared to the untreated cells
(3.35±1.26%, p<0.05) (Fig.
4B). The apoptotic rate was increased in a
concentration-dependent manner. We further investigated the
expression of the apoptosis-associated proteins, pro-caspase-3,
Bcl-2, C-myc and Bax. Our results revealed that the silencing of
depletion of LSD1 induced the break-up of pro-caspase-3, Bcl-2 and
C-myc significantly, and the upregulation of Bax. The silencing of
the LSD1 gene with LSD1 siRNA in the JeKo-1 cells at 40, 80 and 120
nmol/l for 24 h decreased Bcl-2 expression 0.59-, 0.69- and
0.89-fold, respectively. The expression of pro-caspase-3 was
decreased 0.13-, 0.25- and 0.52-fold, respectively. The expression
of C-myc was also decreased 0.40-, 0.49- and 0.73-fold,
respectively. Bax was increased 1.80-, 5.43- and 7.32-fold,
respectively. A simlar effect was observed in the MOLT-4 cells. In
the MOLT-4 cells, siRNA LSD1 also decreased DNMT1 expression, and
increased p15 expression (Fig.
5).
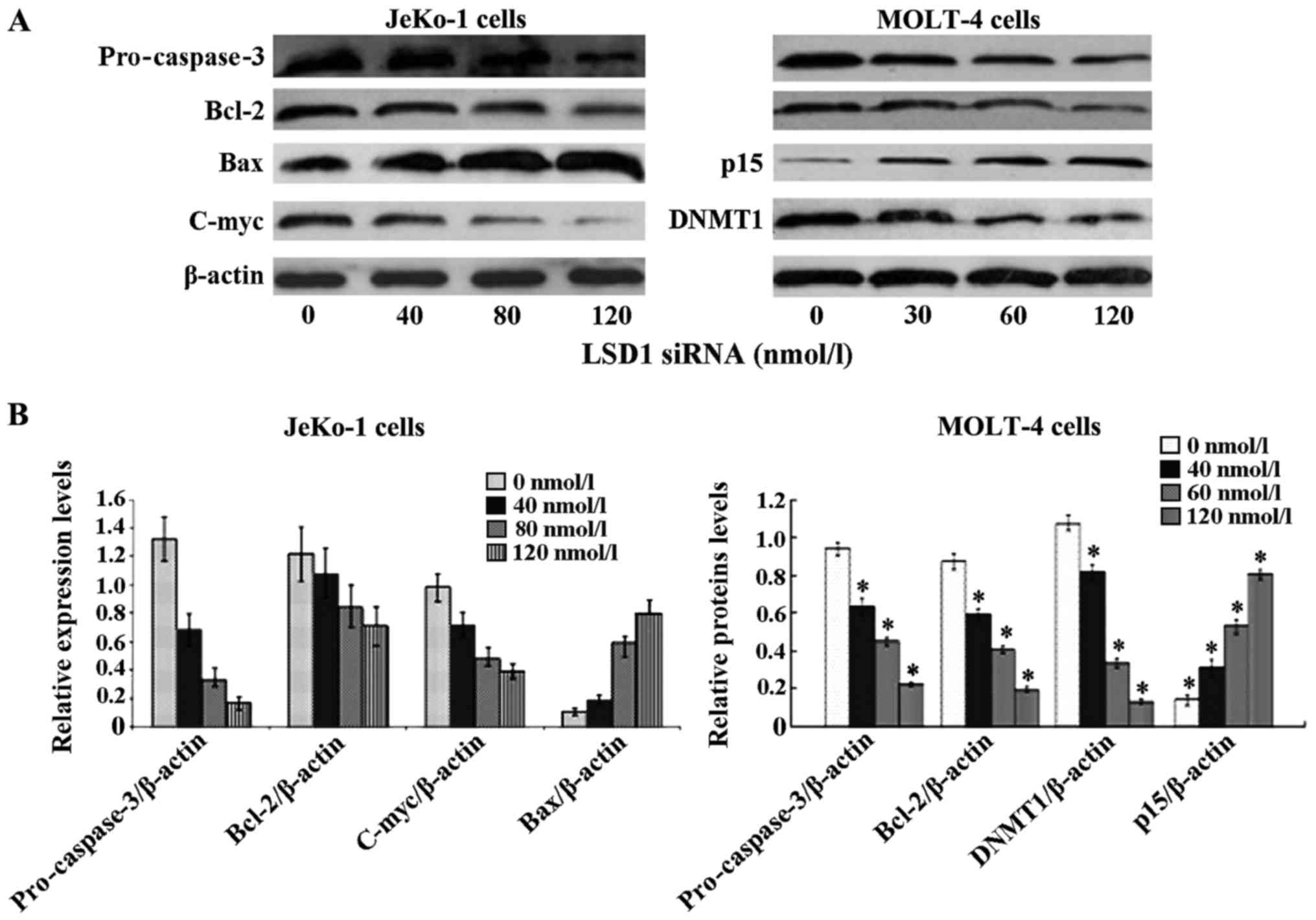 | Figure 5Lysine-specific demethylase 1 (LSD1)
small interfering RNA (siRNA) -induced apoptosis is
mitochondria-mediated and caspase-dependent. JeKo-1and MOLT-4 cells
were treated for 24 h with siRNA LSD1 at the indicated
concentrations. Following transfection with siRNA LSD1, cytosolic
proteins were isolated and separated on a 12% SDS gel, transferred
onto PVDF membranes and blotted with pro-caspase-3, Bcl-2, C-myc,
Bax, DNMT1 and p15 antibodies. The proteins were determined by
immunoblotting using appropriate antibody. β-actin was used as a
loading control. (A) siRNA LSD1 signifiantly decreased the levels
of Bcl-2, pro-caspase-3 and C-myc, and upregulated Bax in JeKo-1
cells. In MOLT-4 cells, siRNA LSD1 decreased the protein expression
of Bcl-2, pro-caspase-3 and DNMT1, and increased that of p15. (B)
Relative protein density (*p<0.05 vs. 0 nmol/l). |
Silencing of the LSD1 gene modulates
histone methylation and acetylation, and downregulates cyclin
D1
To examine our hypothesis, siRNA LSD1 was used to
silence LSD1 gene expression, and this was found to upregulate
methylated H3K4me1 and H3K4me2 in the JeKo-1 and MOLT-4 cells. The
effect on histone lysine methylation was examined by
immunocytochemistry using an antibody against H3K4me1 and H3K4me2.
Following transfection of the JeKo-1 cells with LSD1 siRNA at 40,
80 and 120 nmol/l for 24 h, the expression of H3K4me1 was increased
1.18-, 1.66- and 2.01-fold, respectively compared to the untreated
cells. The expression of H3K4me2 was increased 1.22-, 1.73- and
2.03-fold, respectively compared to the untreated cells. However,
the expression of histone trimethylated H3K4me3 was not altered
significnatly (Fig. 6). The
effects of siRNA LSD1 in the MOLT-4 cells were similar as those
observed in the JeKo-1 cells (Fig.
6). These data indicate that LSD1 is probably a H3K4me1 and
H3K4me2 demethylase. LSD1 siRNA increased the histone acetylation
of H3 in the JeKo-1 and MOLT-4 cells. At 24 h following
transfection with LSD1 siRNA, the expression of histone acetylation
H3 increased 1.25-, 1.42- and 1.56-fold. As MCL is characterized by
carrying the t(11;14) (q13;q32) translocation that leads to the
overexpression of cyclin D1, we further examined the effects of
siRNA LSD1 on cyclin D1. The expression of cyclin D1 was decreased
0.79-, 0.5- and 0.49-fold following transfection with siRNA LSD1 at
the indicated concentrations (Fig.
6).
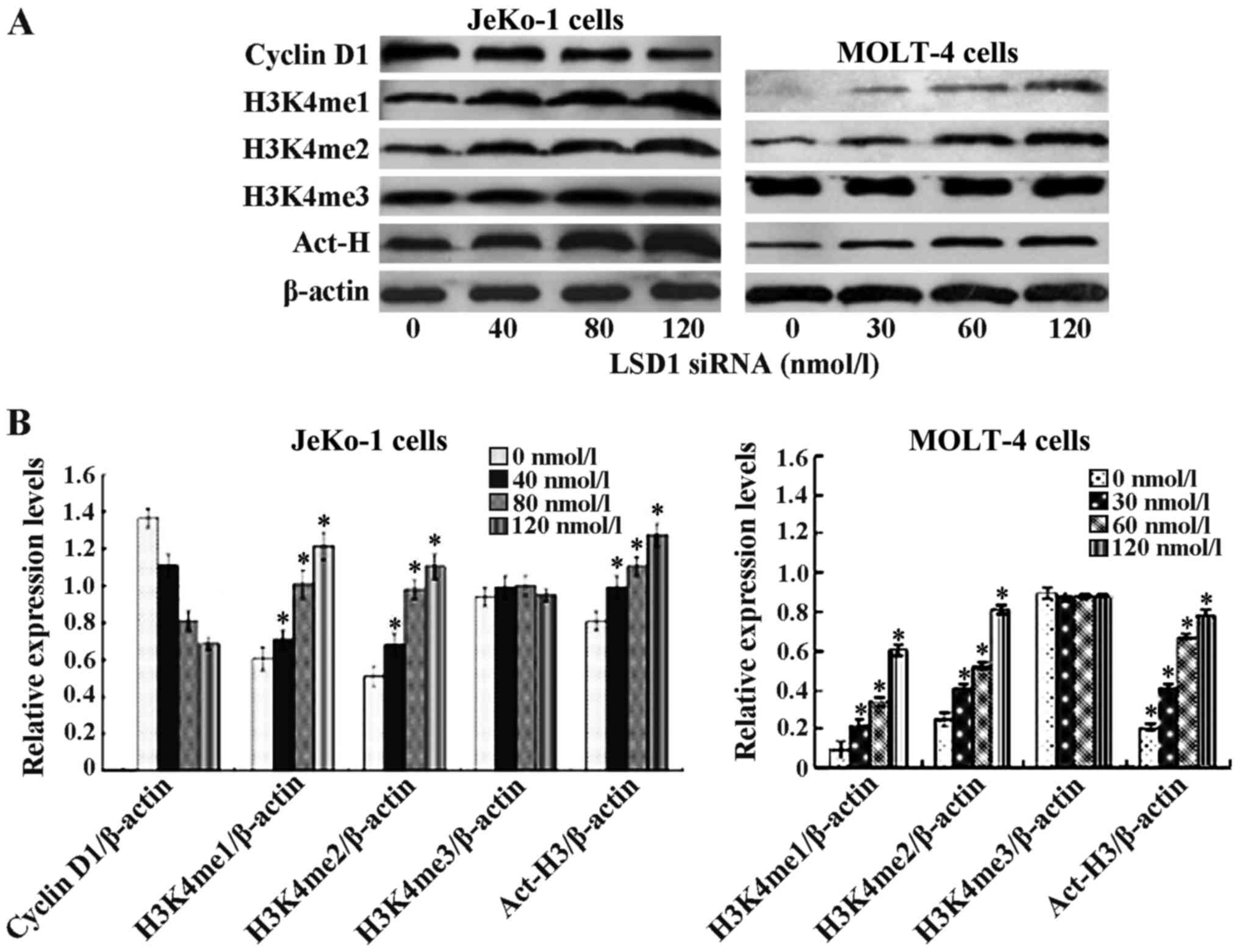 | Figure 6Lysine-specific demethylase 1 (LSD1)
small interfering RNA (siRNA) modulated histone methylation and
acetylation. Effect of LSD1 siRNA atvarious concentrations on
histone modulation in JeKo-1 and MOLT-4 cells was determined by
immunoblotting. Following 24 h LSD1 siRNA treatment, total proteins
were isolated and separated on a 12% SDS gel, transferred onto PVDF
membranes and blotted with mono-, di-, trimethylation H3K4, Act-H3,
and cyclin D1 antibodies. β-actin was used as a loading control.
(A) LSD1 siRNA at 40, 80, 120 nmol/l and at 30, 60, 120 nmol/l
upregulated histone mono-, di-methylated H3K4, acetylated H3 in
concentration-dependent manner in JeKo-1 and MOLT-4 cells,
respectively. No changes were observed in acetylated
tri-methylation H3K4. Cyclin D1 expression decreased in a
concentration-dependent manner in JeKo-1 cells. (B) Relative
protein density (*p<0.05 vs. 0 nmol/l). |
Discussion
LSD1 was the first histone demethylase to be
identified, which belongs to the flavin-dependent amine oxidase
family. It is composed of 3 domains, including an N-terminal SWIRM
domain, a conserved motif shared by many chromatin regulatory
complexes, an amine oxidase domain (AO domain) and a C-terminal
Tower domain (15–17). LSD1 cooperates with CoREST, CtBP24
corepressor complex, demethylates histone H3K4 and H3K9 through
this interaction (18–20) and regulates the expression of its
target gene by this epigenetic modification.
Epigenetic aberrations, including DNA methylation,
histone modifications and microRNA dysregulation are now well
established in the pathogenesis in many types of cancer, including
ovarian cancer, some leukemia, breast, small-cell lung, colorectal,
prostate, neuroblastoma and bladder cancers (3,10,21), and are thought to be one of the
most important mechanisms of tumorigenesis (5,22).
In ovarian cancer, colon cancer and prostate cancer, their gradual
accumulation is associated with an advanced disease stage and grade
(6,23–26). Our previous study showed that
JARID1B, another histone demethylase, was highly expressed in MCL,
resulting in the downregulation of histone methylation
H3K4me31 and histone H3 was hypomethylated at H3K4, and
hypermethylated at H3K9 in leukemia patients (26,27). In this study, we provide evidence
that LSD1 is highly expressed, and H3K4me1 and H3K4me2 are
expressed at low levels in MCL. A strong point of this study was
the finding that the expression of LSD1 significantly positively
correlated with Ki67 (κ=0.667, p<0.01), which is a poor
prognostic marker in MCL. It may related to the function that LSD1
promotes metastasis in various types of cancer cells (28–31).
The aberrant overexpression of LSD1 in MCL may make
it a good candidate as a therapeutic molecular target. LSD1
promotes the proliferation, migration and invasion of various types
of cancer cells (19,31). In this study, siRNA LSD1 was
transfected into JeKo-1 and MOLT-4 cells to silence the LSD1 gene,
which resulted in a decrease in LSD1 protein and mRNA expression,
and in the suppression of cell proliferation and the induction of
apoptosis. A previous study also demonstrated that the inhibition
of LSD1 impaired proliferation and invasiveness, and induced the
apoptosis of colon cancer cells in vitro (32). LSD1 is required for cell
proliferation in both a p53-dependent and -independent manner, and
a deficiency inLSD1 can lead to a partial cell-cycle arrest in the
G2/M phase and sensitizes cells to growth suppression induced by
DNA damage or murine double minute 2 (MDM2) inhibition (33). Thge methylation of p15 increases
the risk of methylation in p53, and vice versa, indicating the
possible synergistic epigenetic disruption of different phases of
the cell cycle or between the cell cycle and apoptosis (34). Through the enhancement of cell
cycle progression, LSD1 promotes the growth of cancer cells,
whereas the inhibition of LSD1 suppresses the G1 to S phase
progression and even induces cell apoptosis (35,36). Upto 88% of adult acute myelogenous
leukemias or acute lymphocytic leukemias have specific methylation
of the p15INK4b CpG island. In this study, we
demonstrated that the silencing of LSD1 in MOLT-4 cells decreased
DNMT1 and increased p15 expression, resulted in decreased cell
proliferation and increased cell apoptosis. Our results revealed
that siRNA against LSD1 decreased the expression of pro-caspase-3,
Bcl-2 and C-myc and induced cell apoptosis. We demonstrated that
LSD1 siRNA decreased cyclin D1 expression, which characterizes 98%
of MCL cases (37). The cyclin D1
promoter contains a CpG island which can be potentially regulated
by DNA methylation (38).
It is unclear whether histone methylation is also
regulated by enzymes with opposing activities. LSD1, also known as
AOF2 or KDM1A, was the first identified FAD-dependent histone
demethylase capable of specifically demethylating mono- and
dimethylated lysine 4 of histone H3 (H3K4me1 and H3K4me2) (39). In this study, we silenced LSD1,
leading to an increase in histone methylated H3K4me1 and H3K4me2
and the histone acetylation of H3. However, the silencing of LSD1
did not affect the methylation of H3K4me3. Our previous study
demonstrated that JARID1B, improved H3K4me3 (26). However, the mechanisms through
which LSD1 promotes the acetyltion of H3 are unknown. It has been
demonstrated that LSD1 is typically found in association with
HDAC1/2, Co-REST, BHC80 and BRAF35 (39). LSD1 has been proposed to
demethylate its histone substrate that requires the intimate
collaboration between LSD1 and HDAC1/2 (17,40,41). Treatment with zinc-dependent class
I/II HDAC inhibitors has been shown to markedly diminish the
activity of LSD1 in breast cancer cells. HDAC inhibitor and LSD1
inhibitor cooperate to increase both histone methylation and
acetylation, indicating the synergistic effects of the combination
of DNA methyltransferase and HDAC inhibitors in re-expressing
epigenetically silenced genes in cancer cells, and leading to
clinical responses in patients with leukemia (42). In breast cancer cells, it leads to
significant synergy in growth inhibition when used in combination
(43). These observations
indicate that histone demethylation is an important component of
the activity of HDAC inhibitors.
In conclusion, in this study, we demonstrate that
epigenetic aberration occurs in MCL, in which is LSD1 overexpressed
and the methylation of H3K4me1 and H3K4me2 is downregulated. LSD1
may thus be a prognostic factor, as it positively correlated with
Ki67. LSD1 may be a marker of carcinogenesis. The silencing of the
LSD1 gene led to the downregulation of LSD1, and the upregulation
of H3K4me1 and H3K4me2. It also led to the accumulation of histone
acetylation H3, and the inhibition of DNMT1 but increased p15,
which are associated with the 5′ regions of virtually all active
genes and positively correlate with transcription rates. The
silencing of the LSD1 gene induced cell apoptosis and inhibited
cell proliferation. LSD1 may thus be an important alternative
target for the treament of MCL.
Acknowledgments
This study was partly supported by a Grant-in-Aid
from the Foundation of Science and Technology of Fujian Medical
University, Fujian, China (no. FZS08018), Science Research
Foundation of Ministry of Health, United Fujian Provincial Health,
and Education Project for Tackling the Key Research, P.R. China
(WKJ2008-2-55), Science Research Foundation of Fujian, P.R. China
(2012J01420), Medical Innovations Research Foundation of Fujian
Province, P.R. China (2012-CX-32) and the major project funded by
R&D institutions of Fujain (2012I2004).
References
|
1
|
Seligson DB, Horvath S, McBrian MA, Mah V,
Yu H, Tze S, Wang Q, Chia D, Goodglick L and Kurdistani SK: Global
levels of histone modifications predict prognosis in different
cancers. Am J Pathol. 174:1619–1628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lim S, Metzger E, Schüle R, Kirfel J and
Buettner R: Epigenetic regulation of cancer growth by histone
demethylases. Int J Cancer. 127:1991–1998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hayami S, Kelly JD, Cho HS, Yoshimatsu M,
Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, et al:
Overexpression of LSD1 contributes to human carcinogenesis through
chromatin regulation in various cancers. Int J Cancer. 128:574–586.
2011. View Article : Google Scholar
|
|
4
|
Kahl P, Gullotti L, Heukamp LC, Wolf S,
Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J,
Metzger E, et al: Androgen receptor coactivators lysine-specific
histone demethylase 1 and four and a half LIM domain protein 2
predict risk of prostate cancer recurrence. Cancer Res.
66:11341–11347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lim S, Janzer A, Becker A, Zimmer A,
Schüle R, Buettner R and Kirfel J: Lysine-specific demethylase 1
(LSD1) is highly expressed in ER-negative breast cancers and a
biomarker predicting aggressive biology. Carcinogenesis.
31:512–520. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schulte JH, Lim S, Schramm A, Friedrichs
N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L,
Kuhfittig-Kulle S, et al: Lysine-specific demethylase 1 is strongly
expressed in poorly differentiated neuroblastoma: implications for
therapy. Cancer Res. 69:2065–2071. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh MM, Manton CA, Bhat KP, Tsai WW,
Aldape K, Barton MC and Chandra J: Inhibition of LSD1 sensitizes
glioblastoma cells to histone deacetylase inhibitors. Neuro-oncol.
13:894–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schenk T, Chen WC, Göllner S, Howell L,
Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et
al: Inhibition of the LSD1 (KDM1A) demethylase reactivates the
all-trans-retinoic acid differentiation pathway in acute myeloid
leukemia. Nat Med. 18:605–611. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Culhane JC and Cole PA: LSD1 and the
chemistry of histone demethylation. Curr Opin Chem Biol.
11:561–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rotili D and Mai A: Targeting histone
demethylases: a new avenue for the fight against cancer. Genes
Cancer. 2:663–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pui CH, Relling MV and Downing JR: Acute
lymphoblastic leukemia. N Engl J Med. 350:1535–1548. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Deng C, Hu X, Patel B, Fu X, Qiu Y,
Brand M, Zhao K and Huang S: Dynamic interaction between TAL1
oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and
leukemogenesis. Oncogene. 31:5007–5018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alinari L, Christian B and Baiocchi RA:
Novel targeted therapies for mantle cell lymphoma. Oncotarget.
3:203–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marks DI and Rowntree C: Management of
adults with T-cell lymphoblastic leukemia. Blood. 129:1134–1142.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Forneris F, Binda C, Vanoni MA, Mattevi A
and Battaglioli E: Histone demethylation catalysed by LSD1 is a
flavin-dependent oxidative process. FEBS Lett. 579:2203–2207. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi Y and Whetstine JR: Dynamic regulation
of histone lysine methylation by demethylases. Mol Cell. 25:1–14.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang M, Culhane JC, Szewczuk LM, Gocke CB,
Brautigam CA, Tomchick DR, Machius M, Cole PA and Yu H: Structural
basis of histone demethylation by LSD1 revealed by suicide
inactivation. Nat Struct Mol Biol. 14:535–539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee MG, Wynder C, Cooch N and Shiekhattar
R: An essential role for CoREST in nucleosomal histone 3 lysine 4
demethylation. Nature. 437:432–435. 2005.PubMed/NCBI
|
|
19
|
Gatta R and Mantovani R: NF-Y substitutes
H2A-H2B on active cell-cycle promoters: recruitment of CoREST-KDM1
and fine-tuning of H3 methylations. Nucleic Acids Res.
36:6592–6607. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X
and Song Y: Over-expression of LSD1 promotes proliferation,
migration and invasion in non-small cell lung cancer. PLoS One.
7:e350652012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei SH, Balch C, Paik HH, Kim YS, Baldwin
RL, Liyanarachchi S, Li L, Wang Z, Wan JC, Davuluri RV, et al:
Prognostic DNA methylation biomarkers in ovarian cancer. Clin
Cancer Res. 12:2788–2794. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsai HC and Baylin SB: Cancer epigenetics:
linking basic biology to clinical medicine. Cell Res. 21:502–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suikki HE, Kujala PM, Tammela TL, van
Weerden WM, Vessella RL and Visakorpi T: Genetic alterations and
changes in expression of histone demethylases in prostate cancer.
Prostate. 70:889–898. 2010.PubMed/NCBI
|
|
24
|
Balch C, Fang F, Matei DE, Huang TH and
Nephew KP: Minireview: epigenetic changes in ovarian cancer.
Endocrinology. 150:4003–4011. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Asadollahi R, Hyde CA and Zhong XY:
Epigenetics of ovarian cancer: from the lab to the clinic. Gynecol
Oncol. 118:81–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su H, Ma X, Huang Y, Han H, Zou Y and
Huang W: JARID1B deletion induced apoptosis in JeKo-1 and HL-60
cell lines. Int J Clin Exp Pathol. 8:171–183. 2015.PubMed/NCBI
|
|
27
|
Zou Y, Ma X, Huang Y, Hong L and Chiao JW:
Effect of phenylhexyl isothiocyanate on aberrant histone H3
methylation in primary human acute leukemia. J Hematol Oncol.
5:36–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Willmann D, Lim S, Wetzel S, Metzger E,
Jandausch A, Wilk W, Jung M, Forne I, Imhof A, Janzer A, et al:
Impairment of prostate cancer cell growth by a selective and
reversible lysine-specific demethylase 1 inhibitor. Int J Cancer.
131:2704–2709. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin L, Hanigan CL, Wu Y, Wang W, Park BH,
Woster PM and Casero RA Jr: Loss of Lysine-Specific Demethylase 1
(LSD1)suppresses growth and alters gene expression of human colon
cancer cells in a p53 and DNA methyltransferase 1
(DNMT1)independent manner. Biochem J. 449:459–468. 2013. View Article : Google Scholar
|
|
30
|
Zhao ZK, Dong P, Gu J, Chen L, Zhuang M,
Lu WJ, Wang DR and Liu YB: Overexpression of LSD1 in hepatocellular
carcinoma: a latent target for the diagnosis and therapy of
hepatoma. Tumour Biol. 34:173–180. 2013. View Article : Google Scholar
|
|
31
|
Cho HS, Suzuki T, Dohmae N, Hayami S,
Unoki M, Yoshimatsu M, Toyokawa G, Takawa M, Chen T, Kurash JK, et
al: Demethylation of RB regulator MYPT1 by histone demethylase LSD1
promotes cell cycle progression in cancer cells. Cancer Res.
71:655–660. 2011. View Article : Google Scholar
|
|
32
|
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y,
Liu S, Zhang Y and Yan ZS: LSD1-mediated epigenetic modification
contributes to proliferation and metastasis of colon cancer. Br J
Cancer. 109:994–1003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scoumanne A and Chen X: The
lysine-specific demethylase 1 is required for cell proliferation in
both p53-dependent and -independent manners. J Biol Chem.
282:15471–15475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bodoor K, Haddad Y, Alkhateeb A, Al-Abbadi
A, Dowairi M, Magableh A, Bsoul N, Ghabkari A and Bodoor K: DNA
hypermethylation of cell cycle (p15 and p16) and apoptotic (p14,
p53DAPK and TMS1) genes in peripheral blood of leukemia patients.
Asian Pac J Cancer Prev. 15:75–84. 2014. View Article : Google Scholar
|
|
35
|
Wang J, Hevi S, Kurash JK, Lei H, Gay F,
Bajko J, Su H, Sun W, Chang H, Xu G, et al: The lysine demethylase
LSD1 (KDM1) is required for maintenance of global DNA methylation.
Nat Genet. 41:125–129. 2009. View
Article : Google Scholar
|
|
36
|
Wen L, Chen Y, Zeng LL, Zhao F, Li R, Liu
Y and Zhang C: Triptolide induces cell-cycle arrest and apoptosis
of human multiple myeloma cells in vitro via altering expression of
histone demethylase LSD1 and JMJD2B. Acta Pharmacol Sin.
33:109–119. 2012. View Article : Google Scholar
|
|
37
|
Sabattini E, Bacci F, Sagramoso C and
Pileri SA: WHO classification of tumours of haematopoietic and
lymphoid tissues in2008: an overview. Pathologica. 102:83–87.
2010.PubMed/NCBI
|
|
38
|
Kitazawa S, Kitazawa R and Maeda S:
Transcriptional regulation of rat cyclin D1 gene by CpG methylation
status in promoter region. J Biol Chem. 274:28787–28793. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi Y, Lan F, Matson C, Mulligan P,
Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation
mediated by the nuclear amine oxidase homolog LSD1. Cell.
119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi YJ, Matson C, Lan F, Iwase S, Baba T
and Shi Y: Regulation of LSD1 histone demethylase activity by its
associated factors. Mol Cell. 19:857–864. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lan F, Collins RE, De Cegli R, Alpatov R,
Horton JR, Shi X, Gozani O, Cheng X and Shi Y: Recognition of
unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene
repression. Nature. 448:718–722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gore SD, Baylin S, Sugar E, Carraway H,
Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, et al:
Combined DNA methyltransferase and histone deacetylase inhibition
in the treatment of myeloid neoplasms. Cancer Res. 66:6361–6369.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang Y, Vasilatos SN, Boric L, Shaw PG
and Davidson NE: Inhibitors of histone demethylation and histone
deacetylation cooperate in regulating gene expression and
inhibiting growth in human breast cancer cells. Breast Cancer Res
Treat. 131:777–789. 2012. View Article : Google Scholar
|