Introduction
Osteoarthritis (OA) is a chronic degenerative joint
disorder that causes pain, tenderness and limitation of movement
(1). OA is a disease affecting
the articular cartilage, in which the molecular biological
characteristics are an aberrant expression of the genes involved in
the synthesis and degradation of cartilage (2). The pathogenesis of OA is suspected
to involve several risk factors, including age, obesity, prior
joint injury, gender and genetic predisposition (3). Inflammation is a characteristic
feature of OA. Inflammatory mediators, such as cytokines
[interleukin (IL)-6 and tumor necrosis factor-α (TNF-α)], lipid
derivatives (leptin, adiponectin and visfatin) and reactive oxygen
species can be produced and activate the cells of the joint tissues
(4).
The nuclear factor-κB (NF-κB) proteins belong to a
family of ubiquitously expressed transcription factors that play a
significant role in most inflammatory responses (5). The NF-κB family includes 5 members:
RelA (p65), RelB, c-Rel, NF-κB1 and NF-κB2. Earlier studies have
revealed that NF-κB is associated with the pathogenesis of OA. The
NF-κB pathway acts as the central regulator of catabolic actions,
mediating the crucial events in the inflammatory responses of
chondrocytes, and leading to extracellular matrix damage and
cartilage erosion (6). For
example, the adenovirus-mediated delivery of p65 siRNA to rats with
OA has been shown to attenuate cartilage destruction (7). p65 activates human SRY-box 9 (SOX9)
promoter activity in chondrogenic cells (8). Therefore, NF-κB signaling plays a
vital role both in the pro-inflammatory stress-related responses of
chondrocytes and in the control of their differentiation
program.
Leptin is an ubiquitous 16-kDa pleiotropic protein
produced predominantly in white adipose tissue (9). Leptin is involved in various
physiological processes, such as immune responses, inflammatory
diseases, cardiovascular functions and respiratory pathophysiology
(10,11). Leptin is regarded as the new
regulator of bone growth via the induction of collagen synthesis
and the proliferation of osteoblasts (12). Leptin and the leptin receptor Ob-R
are produced by articular cartilage and the expression of these two
factors is upregulated through NF-κB activation in patients with OA
(13). Previous studies have
demonstrated that the overexpression of leptin is directly
associated with the degree of OA (14,15).
MicroRNAs (miRNAs or miRs) are a group of small
(approximately 22 nucleotides in length), non-coding RNAs and are
regarded as crucial post-transcriptional gene regulators (16). Studies have demonstrated that
miRNAs are involved in the progression of OA. miR-222 has been
shown to control OA pathogenesis by targeting histone deacetylase-4
(17). The reduced functions of
miR-370 and miR-373 have also been shown to result in the promotion
of cell apoptosis in OA-affected chondrocytes (18). miR-27 has previously been reported
to be decreased in OA-affected chondrocytes (1). The software predicated that miR-27
could target the 3′UTR of leptin. However, whether miR-27 plays an
important role in the progression of OA by regulating leptin and
the underlying mechanisms have not yet been determined. Thus, the
aim of this study was to evaluate the exact effects of miR-27 and
leptin in the progression of OA and to explore the underlying
mechanisms.
Materials and methods
Cell culture
The CH8 cells were purchased from Shanghai Bioleaf
Biotech Co., Ltd. (Shanghai, China). The cells were cultured in
Dulbecco's modified Eagle's medium/Nutrient F-12 Ham (DMEM/F12)
with 10% fetal bovine serum (FBS) (both from Sigma Chemical Co.,
St. Louis, MO, USA) in a humidified incubator with an atmosphere of
95% air-5% CO2 at 37°C. For in vitro experiments,
the CH8 cells were exposed to IL-1β (the final concentration was 10
μg/l), and untreated CH8 cells were considered as the
control group. Human articular cartilage was obtained from patients
with OA following total knee replacement surgery. Twenty cartilage
tissues from patients with OA and 20 normol control tissues were
collected. The chondrocytes were extracted according to a
previously described method (19). Briefly, after surgical removal,
the tissues were collected, and were frozen in liquid nitrogen, and
stored at −80°C. The chondrocytes were minced and digested in 0.15%
(w/v) collagenase in DMEM containing 10% FBS, 100 U/ml penicillin
and 100 μg/ml streptomycin (Sigma Chemical Co.) for 16 h at
37°C. Primary OA chondrocytes used in the experiments were at 80%
confluence.
RNA extraction and real-time PCR
Total RNA was extracted from the cells and tissues
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First Strand
cDNA was synthesized using the MMLV Reverse Transcriptase kit
(Takara, Dalian, China). Real-time PCR was performed using the SYBR
Premix Ex Taq™ kit (Takara). All the primers used in this study
were synthesized by Sangon Biotech (Shanghai, China). Each
individual sample was run in triplicate wells and conducted in the
ABI 7500 Real-time PCR system (Applied Biosystems, Carlsbad, CA,
USA). The reactions were initially denatured at 95°C for 30 sec
followed by 40 cycles at 95°C for 10 sec and 60°C for 60 sec. The
relative degrees of expression of the genes tested were calculated
using the 2−ΔΔCq method. 18s RNA was selected as the
reference gene.
Isolation and culture of primary rat
chondrocytes
All the animal experiment protocols were approved by
the Institutional Animal Care and Use Committee of the Second
Hospital of Lanzhou University, Lanzhou, China. After
experimentation, the mice were euthanized. Sprague-Dawley rats (8
weeks, 210–250 g) were purchased from Better Biotec hnology Co.,
Ltd. (Nangjing, China). A rat model of OA was established by
subjecting the rats to anterior cruciate ligament transection
(ACLT) in the right knees. A total of 60 rats were randomly divided
into 6 groups as follows: the normal control group (NC, n=10), the
OA model group (OA, n=10), the OA model injected with miR-27
lentivirus overexpression vector (OA + pre-lenti-miR-27, n=10), the
OA model injected with lentivirus overexpression vector control (OA
+ pre-lenti-control, n=10), the OA model injected with lentivirus
inhibitor vector (OA + inhibitor-lenti-miR-27, n=10), the OA model
injected with miR-27 lentivirus inhibitor vector control (OA +
inhibitor-lenti-control, n=10). The rats in each group were
euthanized on the 14th day after the injection of miR-27 lentivirus
vector. Primary rat chondrocytes were isolated as previously
described (20). Brefily,
articular cartilages were removed under sterile conditions. The
slices were then cultured in DMEM/F12 (containing 10% FCS, 100
μg/ml streptomycin, 100 U/ml penicillin) after being cut
into small sections. The cells were then maintained at 37°C for 24
h. The undigested cartilage was removed and the chondrocyte cells
were centrifuged at 2,000 × g for 5 min. The supernatants were
collected for testing by western blot analysis and enzyme- linked
immunosorbent assay (ELISA).
Transfection
The CH8 cells were transfected with 80 μM of
the miR-27 mimic, miR-27 inhibitor and corresponding control using
Lipofectamine 2000 reagent (Invitrogen). After 48 h, the cells were
harvested for RNA isolation and western blot analysis.
Western blot analysis
The chondrocytes were extracted using protein lysis
buffer supplemented with a protease inhibitor cocktail. The
chondrocytes were then placed on ice for 30 min and the cells were
then centrifuged at 12,000 × g for 10 min. The total proteins (30
mg) were electrophoresed and transferred onto polyvinylidene
difluoride (PVDF) membranes (Millipore, Darmstadt, Germany). The
membranes were then probed with primary antibodies specific for
type-II collagen (1:5,000 dilution; Cat. no. ab34712; Abcam,
Cambridge, UK), type-X collagen (1:300 dilution; Cat. no. ab58632),
glycosaminoglycan (GAG) (1:1,500 dilution; Cat. no. ab100970) and
aggrecan (ACAN) (1:100 dilution; Cat. no. ab3778), matrix
metalloproteinase (MMP)-9 (1:1,000 dilution; Cat. no. ab73734),
MMP-13 (1:3,000 dilution; Cat. no. ab39012); p65 (sc-8008) and
p-IκBα (sc-52943) (1:1,000 dilution; Santa Cruz Biotechnology,
Santa Cruz, CA). Following incubation at 4°C overnight, the
appropriate HRP-conjugated secondary antibody (1:2,000 dilution;
Cat. no. ab6721, Abcam) was added for 1 h of incubation at room
temperature. The immunoreactive proteins were visualized using an
ECL system (Amersham Biosciences, Amersham, UK).
MTT assay
After the CH8 cells (4×104 cells/well)
were cultured overnight, the cells were transfected with the miR-27
mimic, miR-27 inhibitor and corresponding controls for 24, 48 and
72 h using Lipofectamine. Subsequently, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
was added for a further 4 h of incubation. The blue formazan
crystals of viable cells were solubilized in 150 μl dimethyl
sulfoxide (DMSO). The absorbance was measured at 490 nm using a
microplate reader. The experiments were repeated 3 times.
Bioinformatics analysis
TargetScanHuman7.0 software was used to predict the
target gene of miR-27 (http://www.targetscan.org/vert_71/).
Luciferase reporter assay
The CH8 cells were transfected with 0.25 μg
of the p-MiR-report plasmid (Ambion, Austin, TX, USA) containing
the 3′-untranslated region (3′-UTR) of leptin. A mutated 3′-UTR of
leptin was introduced into the potential miR-27 binding site using
the Nested PCR method. The missing sites of the mutant were from
700 to 725. The CH8 cells were then transfected with the reporter
vectors containing the wild-type or mutant of leptin 3′-UTR and
miR-27 mimic, inhibitor and corresponding controls. Luciferase
activity was measured using a dual-luciferase reporter assay system
(Promega, Madison, WI, USA) following 48 h of transfection.
ELISA
The culture supernatants were used to detect the
levels of IL-6, IL-8 and leptin. The levels of IL-6 and IL-8 were
measured using IL-6, IL-8 specific ELISA kits (Sigma Chemical Co.)
according to the manufacturer's instructions. Leptin was measured
using the human leptin ELISA kit (Sigma Chemical Co.).
Statistical analysis
Statistical analysis was performed using the
Student's unpaired t-test (SPSS release 19.0; SPSS, Inc., Chicago,
IL, USA). Data are expressed as the means ± SD.
Results
miR-27 expression is decreased and that
of leptin is increased in chondrocytes from patients with OA
To examine the effects of miR-27 and leptin on the
progression of OA, we initially measured the degrees of miR-27 and
leptin expression in the human articular cartilage from patients
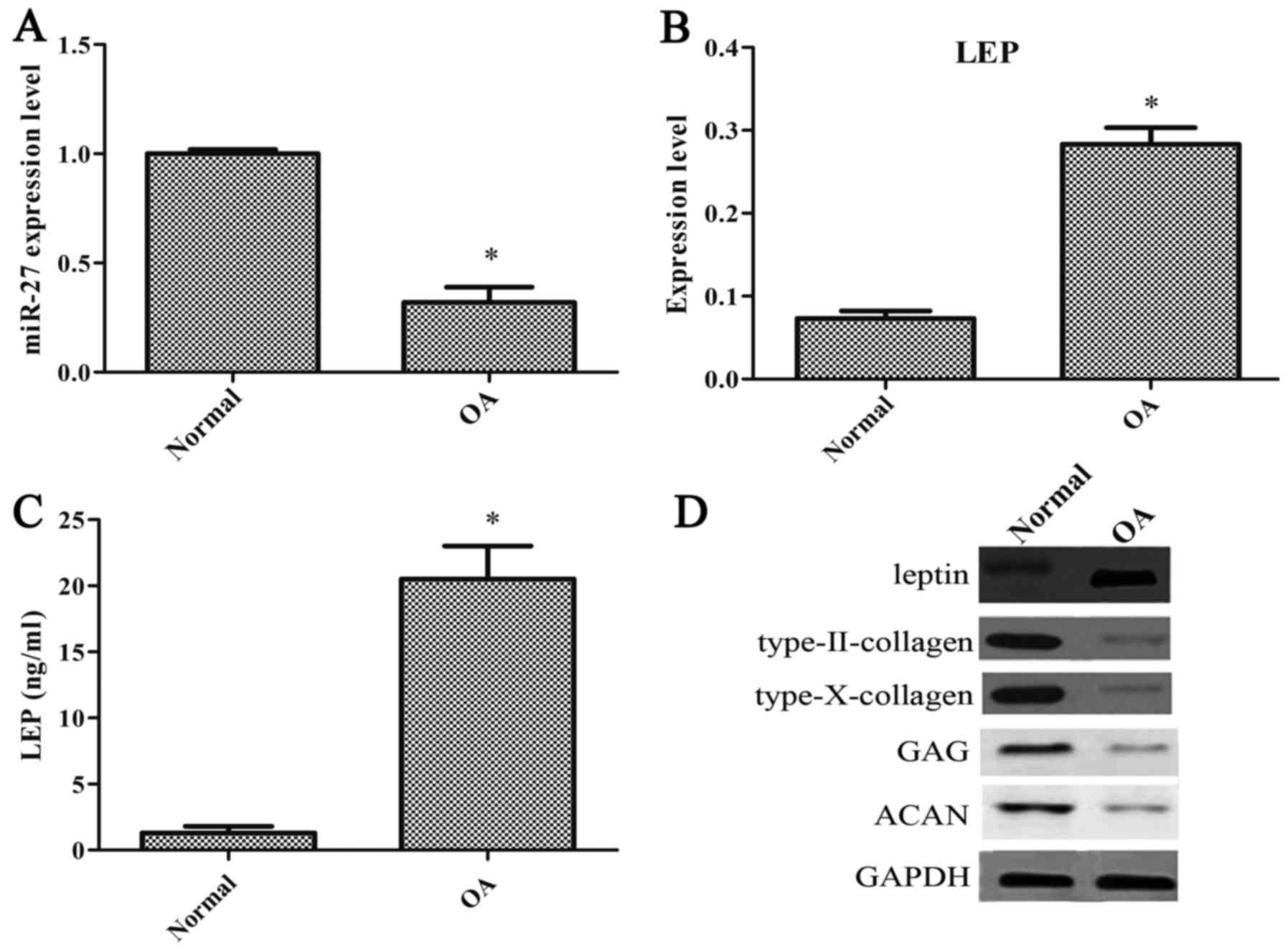
with OA and normal healthy patients. The degree of miR-27
expression was significantly decreased in the OA-affected
chondrocytes compared with the normal chondrocytes (Fig. 1A). The degree of leptin expression
was notably increased in the OA-affected chondrocytes (Fig. 1B). Compared with the normal
chondrocytes, the concentration of leptin was significantly
increased in the OA-affected chondrocytes (Fig. 1C). The levels of type-II collagen,
type-X collagen, GAG and ACAN were also decreased in the
OA-affected chondrocytes (Fig.
1D).
miR-27 enhances the viability of the CH8
cells and induces chondrogenesis
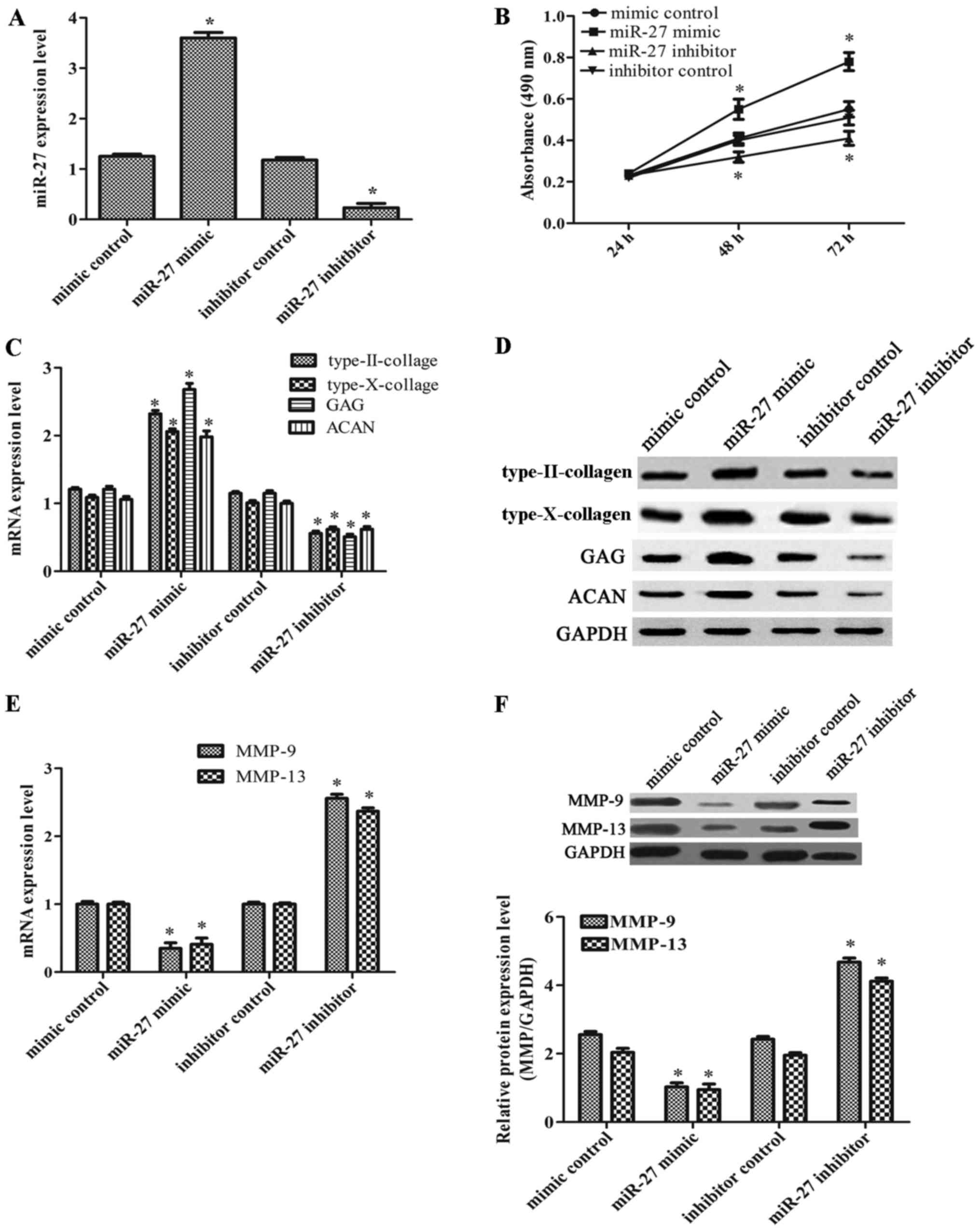
As miR-27 expression was markedly decreased in
OA-affected chondrocytes, we thus considered it possible that
miR-27 may act as an inhibitor of OA. Therefore, we further
investigated the effects of miR-27 on cell viability. The CH8 cells
that were exposed to IL-1β were transfected with miR-27 mimic,
miR-27 inhibitor and their corresponding controls. The transfection
efficiency was very high compared with the corresponding controls
(Fig. 2A). As shown in Fig. 2B, cell viability in the group
transfected with the miR-27 mimic was markedly increased, while it
was significantly decreased in the group transfected with the
miR-27 inhibitor. In order to determine whether miR-27 plays a
positive role in chondrogenesis, we also investigated the
expression levels of type-II collagen, type-X collagen, ACAN and
GAG. As shown in Fig. 2C and D,
we found that infection with the miR-27 mimic induced an increase
in the expression of type-II collagen, type-X collagen, GAG and
ACAN; however, infection with the miR-27 inhibitor led to a marked
decrease in these expression levels. In addition, transfection with
miR-21 mimic markedly decreased the degrees of MMP-9 and MMP-13
expression, whereas these expression levels were increased in the
group of the miR-27 inhibitor (Fig.
2E and F). On the whole, our data demonstrated that miR-27
increased the viability of the CH8 cells and induced
chondrogenesis.
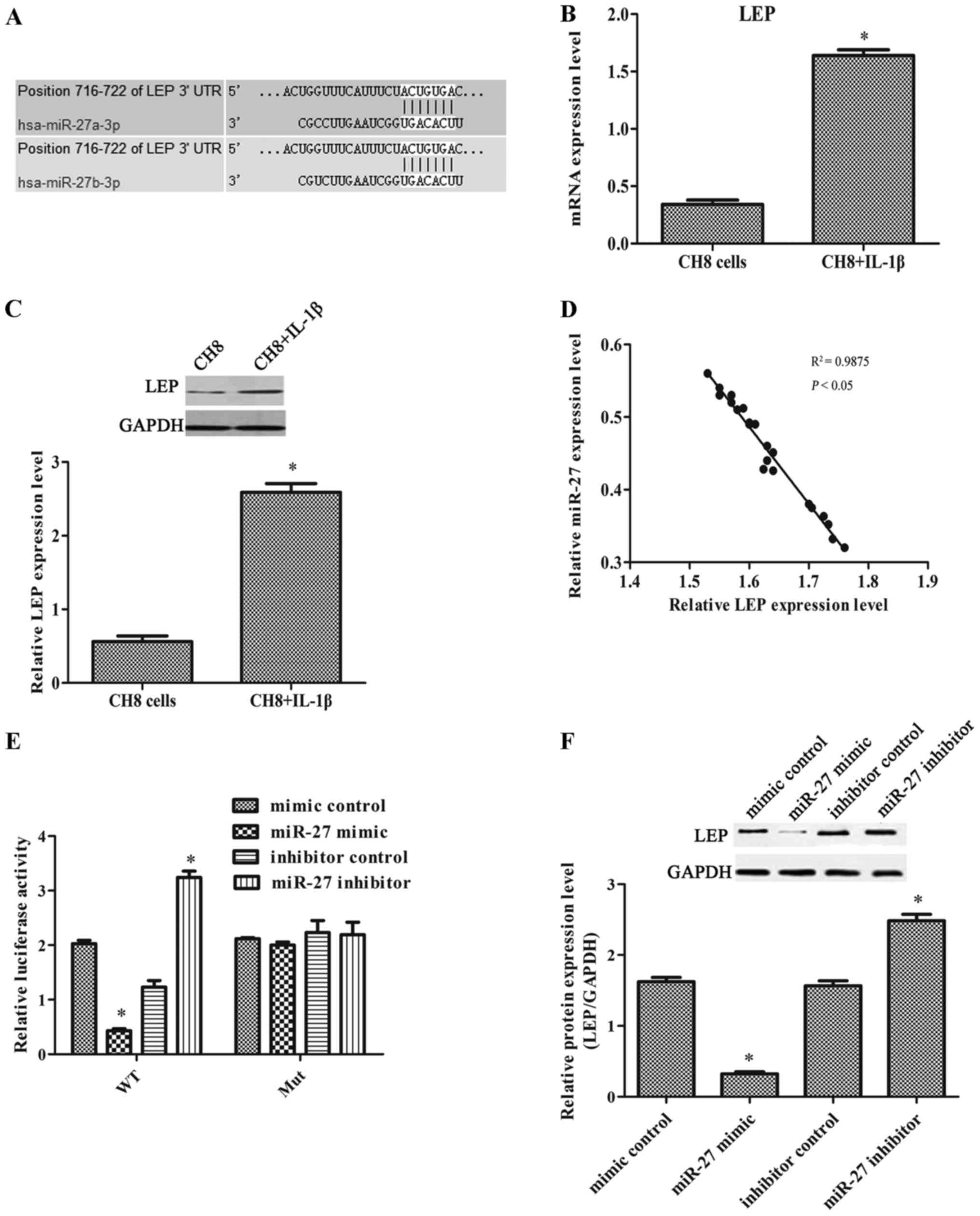
miR-27 directly targets leptin
The results of the analysis using TargetScan Human
7.0 revealed that leptin may be the target of miR-27 (Fig. 3A). The results of real-time PCR
and western blot analysis revealed that leptin expression was
significantly increased in the IL-1β-exposed CH8 cells compared
with the control cells (Fig. 3B and
D). This result was consistent with leptin expression in human
OA chondrocytes. As shown in Fig.
3D, we found that miR-27 expression inversely correlated with
leptin expression. The relative luciferase activity was markedly
decreased when the cells were transfected with the wild-type leptin
3′-UTR and miR-27 mimic, and significantly increased when the cells
were transfected with the wild-type leptin 3′-UTR and miR-27
inhibitor (Fig. 3E). The results
indicated that leptin was the direct target of miR-27. The results
of western blot analysis also confirmed that when the IL-1β-exposed
cells were transfected with the miR-27 mimic, the level of leptin
expression was markedly decreased. When the IL-1β-exposed cells
were transfected with the miR-27 inhibitor, leptin expression was
markedly increased (Fig. 3F).
These results indicated that miR-27 suppressed leptin expression
post-transcriptionally.
miR-27 increases the immunomodulatory
activity and inhibits the activation of the NF-κB pathway in a rat
model of OA
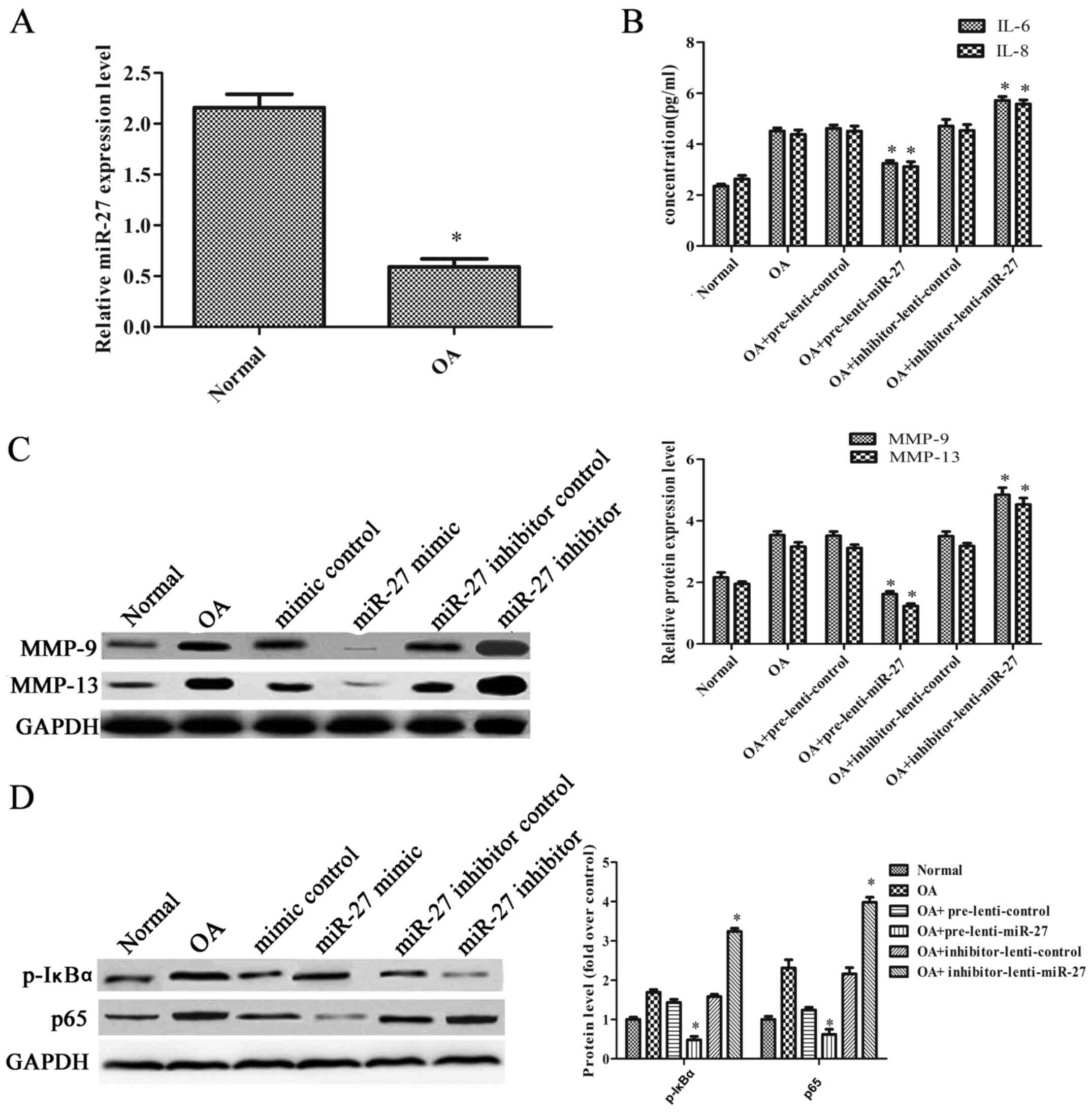
To examine the effect of miR-27 in vivo, a
rat model of OA was established by performing ACLT on the right
knees of the rats. The rats with OA were injected with the
overexpression or inhibitor vectors of the miR-27 lentivirus and
their corresponding controls. As shown in Fig. 4A, the expression level of miR-27
in the articular cartilage was significantly decreased in the rats
with OA. The levels of IL-6 and IL-8 were markedly decreased when
the rats with OA were injected with the miR-27 lentivirus
overexpression vector. However, the levels of IL-6 and IL-8 were
markedly increased when the rats with OA were injected with the
miR-27 lentivirus inhibitor vector (Fig. 4B). In addition, the expression
levels of MMP-9 and MMP-13 were notably decreased when the rats
with OA were injected with the miR-27 lentivirus overexpression
vector, and they were significantly increased when the rats with OA
were injected with the miR-27 lentivirus inhibitor vector (Fig. 4C). The expression of p-IκBα was
decreased 2.98-fold compared with the control when the rats with OA
were injected with the miR-27 lentivirus overexpression vector, and
was increased 2.05-fold when the rats with OA were injected with
the miR-27 lentivirus inhibitor vector. The expression of p65 was
decreased 2.00-fold compared when the rats with OA were injected
with the miR-27 lentivirus vector overexpression, and increased
1.84-fold when the rats with OA were injected with the miR-27
lentivirus inhibitor vector (Fig.
4D). These results suggested that miR-27 increased the
immunomodulatory activity and inhibited the NF-κB pathway in the
rats with OA.
Discussion
A previous study confirmed that miR-27 was
downregulated in human OA-affected chondrocytes (1). In this study, we also verified that
miR-27 expression was decreased both in vivo and in
vitro. Leptin was predicted to be a target of miR-27. Leptin
has been proven to strongly stimulate the anabolic functions of
chondrocytes and to play a vital role in the pathophysiology of OA
(21). Moreover, our results
revealed that the degree of leptin expression inversely correlated
with miR-27 in CH8 cells or human osteoarthritis tissue. Therefore,
we first proposed an assumption that the miR-27-leptin regulatory
pathway may control the progression of osteoarthritis.
A number of studies have proven that miR-27 is
involved in the regulation of cell proliferation. For example,
miR-27b overexpression has been shown to inhibit the growth of
neuroblastoma cells by targeting peroxisome proliferator-activated
receptor γ (PPARγ) (22). miR-27a-3p and miR-24-3p have been
shown to increase the proliferation of glioma cells (23). miR-27a and miR-27b have been shown
to increase the viability of endothelial cells (24). Our results also demonstrated a
significant increase in cell viability when the cells were
transfected with the miR-27 mimic. However, when the cells were
transfected with the miR-27 inhibitor, CH8 cell proliferation was
markedly decreased. Furthermore, the degrees of
chondrogenesis-related protein expression displayed similar
effects. The levels of type-II collagen, type-X collagen, GAG and
ACAN were all increased in response to miR-27 overexpression.
Therefore, it was suggested that miR-27 increased the viability of
CH8 cells and induced chondrogenesis.
We further confirmed that miR-27 played protective a
role in OA by targeting leptin. The results demonstrated that
leptin was upregulated in the OA-affected chondrocytes. The results
of luciferase activity assay indicated that leptin was the direct
target of miR-27. The results of western blot analysis also
indicated that miR-27 mimic suppressed leptin expression. Over the
years, leptin has been recognized as a cytokine-like factor with
pleiotropic actions both in the immune response and inflammation
(25,26). For instance, leptin has been shown
to promote MMP-1 and MMP-3 production in human OA cartilage
(27). Leptin also induces the
proliferation of osteoarthritis-related subchondral osteoblasts
(28). Moreover, low leptin
levels promote chondrocyte proliferation and proteoglycan
synthesis, and correspondingly the overproduction of leptin-induced
nitric oxide synthase, which accelerates cartilage degradation
(29). Therefore, it was
suggested that miR-27 acts as an inhibitor of OA through the
downregulation of leptin expression.
Earlier studies have confirmed that leptin activates
the NF-κB pathway in B lymphomas (30). Leptin enhanced the production of
IL-6 and IL-8 through the activation of NF-κB in OA cartilage
(13). In this study, when the
cells were transfected with the miR-27 mimic, leptin expression was
decreased, thus resulting in the inhibition of NF-κB, and the
downregulation of IL-6, IL-8, MMP-9 and MMP-13. Some miRNAs have
been reported to negatively regulate NF-κB activation and the
production of downstream pro-inflammatory cytokines (5,31).
For example, miR-30c-2-3p negatively regulates NF-κB signaling, and
downregulates IL-8, IL-6 in breast cancer (32). miR-148a has been shown to inhibit
NF-κB activation and decrease the expression of ILs and MMPs in the
calcification of the aortic valve (33). Our results indicated that miR-27
increased the immunomodulatory activity and inhibited the
activation of the NF-κB pathway by targeting leptin in a rat model
of OA.
In conclusion, in the present study, we demonstrate
that miR-27 inhibits the progression of OA by targeting leptin. The
overexpression of miR-27 exerted anti-inflammatory effects by
inhibiting the NF-κB signaling pathway, suggesting that miR-27 may
act as a potential leptin inhibitor for the treatment of OA.
References
|
1
|
Akhtar N, Rasheed Z, Ramamurthy S,
Anbazhagan AN, Voss FR and Haqqi TM: MicroRNA-27b regulates the
expression of matrix metalloproteinase 13 in human osteoarthritis
chondrocytes. Arthritis Rheum. 62:1361–1371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vonk LA, Kragten AH, Dhert WJ, Saris DB
and Creemers LB: Overexpression of hsa-miR-148a promotes cartilage
production and inhibits cartilage degradation by osteoarthritic
chondrocytes. Osteoarthritis Cartilage. 22:145–153. 2014.
View Article : Google Scholar
|
|
3
|
Kerkhof HJ, Bierma-Zeinstra SM, Arden NK,
Metrustry S, Castano-Betancourt M, Hart DJ, Hofman A, Rivadeneira
F, Oei EH, Spector TD, et al: Prediction model for knee
osteoarthritis incidence, including clinical, genetic and
biochemical risk factors. Ann Rheum Dis. 73:2116–2121. 2014.
View Article : Google Scholar
|
|
4
|
Berenbaum F, Eymard F and Houard X:
Osteoarthritis, inflammation and obesity. Curr Opin Rheumatol.
25:114–118. 2013. View Article : Google Scholar
|
|
5
|
Zhang D, Cao X, Li J and Zhao G: MiR-210
inhibits NF-κB signaling pathway by targeting DR6 in
osteoarthritis. Sci Rep. 5:127752015. View Article : Google Scholar
|
|
6
|
Marcu KB, Otero M, Olivotto E, Borzí RM
and Goldring MB: NF-kappaB signaling: Multiple angles to target OA.
Curr Drug Targets. 11:599–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen LX, Lin L, Wang HJ, Wei XL, Fu X,
Zhang JY and Yu CL: Suppression of early experimental
osteoarthritis by in vivo delivery of the adenoviral
vector-mediated NF-kappaBp65-specific siRNA. Osteoarthritis
Cartilage. 16:174–184. 2008. View Article : Google Scholar
|
|
8
|
Ushita M, Saito T, Ikeda T, Yano F,
Higashikawa A, Ogata N, Chung U, Nakamura K and Kawaguchi H:
Transcriptional induction of SOX9 by NF-kappaB family member RelA
in chondrogenic cells. Osteoarthritis Cartilage. 17:1065–1075.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang P, Zhong ZH, Yu HT and Liu B:
Significance of increased leptin expression in osteoarthritis
patients. PLoS One. 10:e01232242015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bassi M, Furuya WI, Zoccal DB, Menani JV,
Colombari E, Hall JE, da Silva AA, do Carmo JM and Colombari DS:
Control of respiratory and cardiovascular functions by leptin. Life
Sci. 125:25–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wollman L, Powell G, Levine R and Fregosi
R: Leptin acutely inhibits respiratory function in neonatal rats
(712.18). FASEB J. 28(1 Supplement): 712.182014.
|
|
12
|
Bartell SM, Rayalam S, Ambati S, Gaddam
DR, Hartzell DL, Hamrick M, She JX, Della-Fera MA and Baile CA:
Central (ICV) leptin injection increases bone formation, bone
mineral density, muscle mass, serum IGF-1, and the expression of
osteogenic genes in leptin-deficient ob/ob mice. J Bone Miner Res.
26:1710–1720. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vuolteenaho K1, Koskinen A, Kukkonen M,
Nieminen R, Päivärinta U, Moilanen T and Moilanen E: Leptin
enhances synthesis of proinflammatory mediators in human
osteoarthritic cartilage - mediator role of NO in leptin-induced
PGE2, IL-6, and IL-8 production. Mediators Inflamm.
2009:3458382009. View Article : Google Scholar
|
|
14
|
Ku JH, Lee CK, Joo BS, An BM, Choi SH,
Wang TH and Cho HL: Correlation of synovial fluid leptin
concentrations with the severity of osteoarthritis. Clin Rheumatol.
28:1431–1435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Honsawek S and Chayanupatkul M:
Correlation of plasma and synovial fluid adiponectin with knee
osteoarthritis severity. Arch Med Res. 41:593–598. 2010. View Article : Google Scholar
|
|
16
|
Li ZC, Han N, Li X, Li G, Liu YZ, Sun GX,
Wang Y, Chen GT and Li GF: Decreased expression of microRNA-130a
correlates with TNF-α in the development of osteoarthritis. Int J
Clin Exp Pathol. 8:2555–2564. 2015.
|
|
17
|
Song J, Jin EH, Kim D, Kim KY, Chun CH and
Jin EJ: MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during
osteoarthritis pathogenesis. BBA Clin. 3:79–89. 2014. View Article : Google Scholar
|
|
18
|
Song J, Kim D, Chun CH and Jin EJ: miR-370
and miR-373 regulate the pathogenesis of osteoarthritis by
modulating one-carbon metabolism via SHMT-2 and MECP-2,
respectively. Aging Cell. 14:826–837. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hautier A, Salentey V, Aubert-Foucher E,
Bougault C, Beauchef G, Ronzière MC, De Sobarnitsky S, Paumier A,
Galéra P, Piperno M, et al: Bone morphogenetic protein-2 stimulates
chondrogenic expression in human nasal chondrocytes expanded in
vitro. Growth Factors. 26:201–211. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang JG, Xia C, Zheng XP, Yi TT, Wang XY,
Song G and Zhang B: 17β-Estradiol promotes cell proliferation in
rat osteoarthritis model chondrocytes via PI3K/Akt pathway. Cell
Mol Biol Lett. 16:564–575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dumond H, Presle N, Terlain B, Mainard D,
Loeuille D, Netter P and Pottie P: Evidence for a key role of
leptin in osteoarthritis. Arthritis Rheum. 48:3118–3129. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JJ, Drakaki A, Iliopoulos D and Struhl
K: MiR-27b targets PPARγ to inhibit growth, tumor progression and
the inflammatory response in neuroblastoma cells. Oncogene.
31:3818–3825. 2012. View Article : Google Scholar
|
|
23
|
Xu W, Liu M, Peng X, Zhou P, Zhou J, Xu K,
Xu H and Jiang S: miR-24-3p and miR-27a-3p promote cell
proliferation in glioma cells via cooperative regulation of MXI1.
Int J Oncol. 42:757–766. 2013.
|
|
24
|
Urbich C, Kaluza D, Frömel T, Knau A,
Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN,
Hergenreider E, et al: MicroRNA-27a/b controls endothelial cell
repulsion and angiogenesis by targeting semaphorin 6A. Blood.
119:1607–1616. 2012. View Article : Google Scholar
|
|
25
|
Coppari R and Bjørbæk C: Leptin revisited:
Its mechanism of action and potential for treating diabetes. Nat
Rev Drug Discov. 11:692–708. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terlain B, Presle N, Pottie P, Mainard D
and Netter P: Leptin: a link between obesity and osteoarthritis?
Bull Acad Natl Med. 190:1421–1437. 1475–1477. 2006.
|
|
27
|
Koskinen A, Vuolteenaho K, Nieminen R,
Moilanen T and Moilanen E: Leptin enhances MMP-1, MMP-3 and MMP-13
production in human osteoarthritic cartilage and correlates with
MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp
Rheumatol. 29:57–64. 2011.PubMed/NCBI
|
|
28
|
Mutabaruka MS, Aissa MA, Delalandre A,
Lavigne M and Lajeunesse D: Research article Local leptin
production in osteoarthritis subchondral osteoblasts may be
responsible for their abnormal phenotypic expression. Arthritis Res
Ther. 12:R202010. View
Article : Google Scholar
|
|
29
|
Stannus OP, Jones G, Quinn SJ, Cicuttini
FM, Dore D and Ding C: Research article the association between
leptin, interleukin-6, and hip radiographic osteoarthritis in older
people: A cross-sectional study. Arthritis Res Ther. 12:R952010.
View Article : Google Scholar
|
|
30
|
Lam QLK, Wang S, Ko OKH, Kincade PW and Lu
L: Leptin signaling maintains B-cell homeostasis via induction of
Bcl-2 and Cyclin D1. Proc Natl Acad Sci USA. 107:13812–13817. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qi J, Qiao Y, Wang P, Li S, Zhao W and Gao
C: microRNA-210 negatively regulates LPS-induced production of
proinflammatory cytokines by targeting NF-κB1 in murine
macrophages. FEBS Lett. 586:1201–1207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shukla K, Sharma AK, Ward A, Will R,
Hielscher T, Balwierz A, Breunig C, Münstermann E, König R,
Keklikoglou I, et al: MicroRNA-30c-2-3p negatively regulates NF-κB
signaling and cell cycle progression through downregulation of
TRADD and CCNE1 in breast cancer. Mol Oncol. 9:1106–1119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carrion K, Patel V, Dyo J, Holland A,
Gallegos T, Hardiman G, Mohamed S, Leire E, Nigam S, Nizet V and
Nigam V: miR-148a is a novel repressor of NF-κB signaling in aortic
valve calcification. Circulation. 128:A160672013.
|