Introduction
Acute respiratory distress syndrome (ARDS) is a
severe medical condition, which is associated with a high mortality
rate, and is characterized by noncardiogenic pulmonary edema and
hypoxemia (1,2). Although ARDS is not a classified
pulmonary vascular disease, disruptions in endothelial barrier
integrity and dysfunction of endothelial barrier permeability are
considered the pathological hallmarks of ARDS, particularly at the
very early stage of ARDS development (3,4).
It is well known that the development of ARDS is
associated with various predisposing risk factors, among which
obesity has been identified in recent epidemiological studies
(5–8). Obesity, particularly visceral
obesity, has been reported to impair systemic vasculature and is
involved in the initiation and progression of cardiovascular
diseases (9,10). Adipose tissue is considered a
significant endocrine organ that is capable of crosstalk with
peripheral organs via diverse bioactive molecules known as
adipokines, which participate in the progression of diverse
vascular disorders (11–13). Although different from the
well-documented effects of obesity on cardiovascular disease, the
relationship between obesity and ARDS has proven to be considerably
complex; clinical and experimental data have focused on pertinent
physiological alterations in obesity and have highlighted the
pivotal role of pulmonary vascular 'priming' and neutrophil
functional impairment at baseline (8,14).
Since persistent, low-grade inflammation as a result of fat
accumulation impairs systemic blood vessels and contributes to the
development of various obesity-associated vascular diseases,
emerging evidence has suggested that obesity alters ARDS
pathogenesis by 'priming' the pulmonary endothelial barrier for
insult and injury, as well as amplifying the early inflammatory
response, thus lowering the threshold required to initiate ARDS via
dysregulated adipokine production in obesity. Previous studies have
demonstrated that circulating adipokine levels are associated with
the initiation and progression of ARDS (15–18).
Visceral adipose tissue-derived serine proteinase
inhibitor (vaspin) is identified as a favorable adipokine that is
secreted from visceral white adipose tissues, which is associated
with glucose tolerance and chronic inflammation (19,20). Vaspin has been reported to
counteract the pathogenesis of various obesity-related vascular
complications via its anti-inflammatory and anti-apoptotic
properties. In a human study, serum vaspin levels were demonstrated
to be lower in patients with coronary artery disease (CAD) compared
with in the control individuals; this tendency was confirmed in
control individuals with higher systolic blood pressure compared
with in control individuals with normal blood pressure, thus
indicating that vaspin may be a predictor of CAD (21). Furthermore, a previous study
reported that lower fasting vaspin levels were correlated with
ischemic vascular events in the last 3 months in patients with
carotid stenosis compared to those with asymptomatic stenosis
(22). Decreased serum vaspin
levels were also observed in patients with ankylosing spondylitis
and were associated with flow-mediated dilation levels, thus
indicating that vaspin may be a marker for detecting early stage
atherosclerosis in patients with ankylosing spondylitis (23). Vaspin is believed to serve a local
and endocrine role in the initiation and development of vascular
disorders by affecting endothelial cells (ECs), thus disrupting
vascular homeostasis. A previous study suggested that vaspin, as a
ligand for the cell surface 78 kDa glucose-regulated protein
(GRP78)/voltage-dependent anion channel complex in ECs, exerted
beneficial effects on diabetic vascular complications by promoting
proliferation and inhibiting apoptosis under the diabetic milieu
via a protein kinase B (Akt)-dependent mechanism (24). It has also been demonstrated that
vaspin may significantly attenuate methylglyoxal-induced cell death
and reactive oxygen species (ROS) generation in human umbilical
vein ECs (HUVECs) (25).
Treatment with vaspin also significantly decreased tumor necrosis
factor (TNF)-α-induced activation of nuclear factor (NF)-κB, as
well as the expression of the adhesion molecules intercellular cell
adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1
(VCAM-1), E-selectin and monocyte chemotactic protein-1 (MCP-1) in
human aortic ECs (26).
Furthermore, vaspin is capable of preventing apoptosis induced by
free fatty acids through upregulation of the phosphoinositide
3-kinase (PI3K)/Akt signaling pathway in vascular ECs, suggesting
the beneficial effects of vaspin on obesity-associated vascular
diseases (27). A recent study
also indicated that vaspin was able to inhibit the progression of
atherosclerotic plaques in apolipoprotein E−/− mice by
suppressing endoplasmic reticulum stress-induced macrophage
apoptosis (28).
Collectively, these data suggested that vaspin, as a
pleiotropic adipokine capable of exerting anti-inflammatory,
anti-apoptotic and antioxidant effects on ECs, may exert a
favorable role against the progression of obesity-associated
vascular complications. Although the pathogenesis of ARDS is
considerably complex, hyperpermeability of the pulmonary EC barrier
is a pathological hallmark of ARDS at the very early stage, in
which obesity, inflammation, apoptosis and ROS serve important
roles. Therefore, it may be hypothesized that vaspin contributes to
the protection of ARDS via its endothelial-protective effects.
Nevertheless, to the best of our knowledge, no previous studies
have assessed the effects of vaspin on pulmonary ECs, specifically
in the setting of ARDS.
Therefore, the present study aimed to investigate
the effects of vaspin on lipopolysaccharide (LPS)-induced ARDS
in vivo and in vitro, and to further explore the
molecular basis and potential mechanisms underlying these effects.
This study aimed to provide a novel insight into the crosstalk
between vaspin and ARDS, particularly focusing on the pulmonary EC
barrier.
Materials and methods
Chemicals and reagents
LPS (Escherichia coli LPS serotype 0111:B4),
sodium pentobarbital, Evans blue dye and dihydroethidium (DHE),
goat serum (G9023) and bovine serum albumin (A2153) were purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Recombinant
human vaspin (rh-vaspin) protein was purchased from GeneTex, Inc.
(Irvine, CA, USA). The following primary antibodies were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA):
Anti-ICAM-1 (cat. no. 4915), anti-NF-κB Rel (cat. no. 8242),
anti-phosphorylated (p)-NF-κB Rel (Ser536; E1Z1T; cat. no. 13346),
anti-Akt (cat. no. 4685) and anti-p-Akt (Ser473; cat. no. 4060),
anti-rabbit IgG, HRP-linked antibody (cat. no. 7074) and Alexa
Fluor 488-labeled anti-rabbit secondary antibodies (cat. no. 4412).
Anti-vascular endothelial (VE)-cadherin (ab205336) and anti-NADPH
oxidase antibodies (ab133303) were purchased from Abcam (Cambridge,
UK). Anti-GAPDH (cat. no. AP0063), anti-β-catenin (cat. no.
BS6879), anti-glycogen synthase kinase (GSK)-3β (cat. no. BS1402)
and anti-p-GSK3β (Ser9; cat. no. BS4084P) antibodies were purchased
from Bioworld Technology (Nanjing, China). Adenoviral vectors (Ad)
expressing β-galactosidase (Ad-β-gal) and full-length vaspin
(Ad-vaspin) were constructed by Shanghai GeneChem Co., Ltd.
(Shanghai, China) (primer sequences available upon request).
Ad-β-gal was used as a control.
LPS-induced ARDS model in mice
C57BL/6 mice (weighing 20–25 g; 10 weeks old; male;
of specific-pathogen-free grade; Department of Laboratory Animal
Center, Chongqing Medical University, Chongqing, China) were housed
under light-controlled conditions (12-h light/dark cycle) at room
temperature (25°C) with 60% humidity and were granted ad
libitum access to food and water. All animal experimental
protocols were implemented in accordance with the instructions of
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals (29). The
present study was approved by the Ethics Committee of the Second
Affiliated Hospital of Chongqing Medical University (Chongqing,
China). Mice were randomly assigned to 4 groups as follows: the
control group not pre-treated with Ad-vaspin (n=24), the control
group pre-treated with Ad-vaspin (n=24), the ARDS group not
pre-treated with Ad-vaspin and the ARDS group treated with
Ad-vaspin (n=24). Mice were anesthetized with sodium pentobarbital
(50 mg/kg, i.p.) prior to exposure of the trachea and right
internal jugular vein (IJV). Subsequently, 3×107 PFU
Ad-vaspin or Ad-β-gal per mouse was injected into the IJV for 3
days prior to LPS or vehicle (PBS) intratracheal instillation. Mice
were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). As
described previously (18), to
establish a mouse model of ARDS, mice were intratracheally
instilled with 5 mg/kg LPS (E. coli LPS serotype 0111:B4) in
50 µl sterile PBS, or PBS alone (control group) using an
18-G catheter. The mice were sacrificed 4 h after the LPS injection
and lung tissues were harvested and stored at −80°C until further
analysis.
Lung histological evaluation
Left lung lobes were harvested, fixed in 3.7%
paraformaldehyde (24 h at room temperature), embedded in paraffin
wax and cut into 5-µm sections. Subsequently, the sections
were stained with hematoxylin and eosin. Histological lung injury
in each mouse was evaluated in 5 random fields (×200 and ×400
magnification) using an inverted microscope (TE2000-U; Nikon
Corporation, Tokyo, Japan). A standardized scoring system,
published by the American Thoracic Society (30), was used to assess histological
lung injury in mice.
ELISA
Lung homogenates were used to determine the levels
of TNF-α (MTA00B), interleukin (IL)-6 (M6000B) and IL-10 (M1000B)
using commercially available ELISA kits (R&D Systems, Inc.,
Minneapolis, MN, USA) according to the manufacturer's protocols. In
cultured cells, basal levels of secretion were determined by
measuring TNF-α, IL-6 and IL-10 concentration in cell lysates. The
effectiveness of the adenoviral vector expression system was
confirmed by measuring mean plasma vaspin concentrations in the
treated mice using a commercial ELISA kit (DSA120; R&D Systems,
Inc.).
Immunohistochemistry
Mouse left lung tissue sections from each group were
deparaffinized with xylene, rehydrated in gradient ethanol and
incubated in 3% H2O2 at 37°C for 15 min.
Subsequently, the sections were rinsed three times in PBS (10
min/wash). Antigen retrieval was performed by immersing the slices
in citrate buffer in a microwave at 96°C for 30 min. Tissues were
blocked with goat serum albumin in an incubator at room temperature
for 1 h and the sections were then incubated with anti-ICAM-1
primary antibodies (1:50 dilution) at 4°C overnight. Sections were
washed a further three times with PBS (10 min/wash) and were
incubated with a biotin-labeled secondary antibody (1:1,000;
ZB-2010; ZSBIO Biotech Co., Ltd., Beijing, China) at 37°C for 30
min, and were then stained with DAB. Sections were counterstained
with hematoxylin, dehydrated in gradient ethanol, vitrified with
xylene and sealed with neutral resins. Images were captured using
an inverted microscope (TE2000-U; Nikon Corporation).
Measurement of Evans blue-dyed albumin
(EBDA) concentrations in lung tissue
Pulmonary capillary permeability was assessed by
determining EBDA concentrations. The right IJV of mice was injected
with EBDA (30 mg/kg). Then mice were sacrificed and lungs free of
blood were excised, weighed and homogenized in 1 ml PBS, after
which they were extracted in 2 ml formamide (24 h, 60°C) and
centrifuged at 5,000 x g for 30 min at 20°C. The absorbance of the
supernatants was measured by spectrophotometry at 620 and 740 nm,
plotted against a standard curve, normalized, and converted to
µg EBDA/g lung tissue.
Analysis of BALF
A trimmed 18-G catheter was inserted into the
trachea. A syringe was connected to the catheter, and 1 ml of
sterile normal saline was infused into the airway. Four hours after
LPS administration, BALF was collected by the intra-tracheal
instillation of 1 ml of sterile normal saline followed by repeated
aspiration 3 times and centrifugation at 500 x g for 10 min at 4°C.
According to manufacturer's instructions, the protein
concentrations in the BALF supernatants were determined using a
bicinchoninic acid protein assay (BCA) kit (KeyGen Biotech Co.,
Ltd., Nanjing, China).
Wet/dry (W/D) lung weight ratio
The right upper lung lobes were harvested and
weighed to determine wet lung weight. Subsequently, tissues were
dried in an oven at 80°C for 24 h and were weighed again to
calculate the W/D ratios.
EC culture
Human pulmonary microvascular ECs (HPMECs) were
cultured in EC medium (both ScienCell Research Laboratories, Inc.,
San Diego, CA, USA) supplemented with 10% fetal bovine serum (FBS;
cat. no. 0025), 1% endothelial cell growth supplement (ECGS; cat.
no. 1052) and 1% penicillin/streptomycin (P/S; cat. no. 0503) (all
from ScienCell Research Laboratories, Inc.) in a 5% CO2
incubator at 37°C. Cells between passages 4 and 10 were grown as a
monolayer and starved (1% serum) for 6 h prior to each treatment.
Human serum concentrations of vaspin are reported to range between
0.1 and 100 ng/ml; therefore, 10 ng/ml vaspin was used in the
present study. Cells were pretreated with rh-vaspin (10 ng/ml) or
PBS as a control for 24 h, after which they were washed with PBS
and exposed to LPS or vehicle (PBS) at 100 ng/ml for 2 h. Cell
lysates were collected for subsequent analysis at the indicated
time intervals.
EC monolayer permeability assay
Permeability was determined based on the
paracellular permeability of 70 kDa fluorescein isothiocyanate
(FITC)-dextran into the lower chamber as described previously
(18). Briefly, HPMECs were grown
on 0.4 µm Transwell inserts at 1×105 cells per
well. Following the indicated time interval for each treatment, 0.5
ml FITC-dextran (1 mg/ml) was added to the upper wells, and 1.5 ml
medium was added to the bottom chamber. Following 1 h incubation in
the dark, 50 µl medium was aspirated and absorbance was
measured using a luminometer (BioTek Instruments, Inc., Winooski,
VT, USA) at an excitation wavelength of 488 nm and an emission
wavelength of 520 nm. The basal FITC-dextran permeability for
unstimulated monolayers was set at 100%.
EC viability assay
Cell viability was measured using Cell Counting
kit-8 (CCK-8). Cell suspensions of each group were seeded in
96-well plates at 2×104 cells/well and were
preincu-bated at 37°C in a humidified atmosphere containing 5%
CO2. Subsequently, 10 µl CCK-8 solution was added
to each well, and the plates were incubated for 2 h in an
incubator. The absorbance of each well was measured using a
microplate reader at 450 nm (BioTek Instruments, Inc.) at room
temperature (25°C). Cell viability was calculated using the
following equation: Viability = (ODtest group −
ODblank group) / (ODcontrol group −
ODblank group) × 100%; where OD refers to optical
density.
TdT-mediated dUTP nick end labeling
(TUNEL) staining
According to the manufacturer's protocol, TUNEL
staining was conducted using an in situ cell death detection
kit (Roche Diagnostics GmbH, Mannheim, Germany) to detect apoptosis
of HPMECs, as described previously (18). DAPI (Nanjing KeyGen Biotech Co.,
Ltd.) was used to stain nuclei. In situ apoptosis detection
(Abcam, Cambridge, MA, USA) was used to assess the apoptotic rate
of paraffin-embedded lung sections according to the manufacturer's
protocols. DAB reacted with the horseradish peroxidase
(HRP)-labeled sample to generate a dark brown signal at the site of
DNA fragmentation. TUNEL-positive cells were counted in 5 randomly
selected fields (×400 magnification) under an inverted microscope
(TE2000-U; Nikon Corporation).
Annexin V-FITC/propidium iodide (PI)
staining
Following the indicated treatments, cells were
collected and resuspended in 500 µl 1X Annexin V binding
buffer, after which they were incubated with 5 µl Annexin
V-FITC and 5 µl PI for 15 min at room temperature in the
dark. Flow cytometry (FCM; BD Biosciences, Franklin Lakes, NJ, USA)
was performed to detect apoptotic cells.
Western blot analysis
Radioimmunoprecipitation assay buffer was used to
extract total protein, and a Membrane and Cytoplasmic Protein
Extraction kit (both Nanjing KeyGen Biotech Co., Ltd.) was used to
extract protein from cells and the left lung tissues of mice from
each treatment group according to the manufacturer's protocols.
Bicinchoninic acid kit was used to measure protein concentration.
Equivalent amounts of protein (30 µg) were separated by
SDS-PAGE and were electrotransferred to polyvinylidene fluoride
membranes. Subsequently, the membranes were blocked with 5% dry
milk/bovine serum albumin (BSA) at room temperature (25°C) for 1 h,
and were immunoblotted with anti-NF-κB Rel (1:1,000),
anti-phospho-NF-κB Rel (Ser536; 1:1,000), anti-β-catenin (1:500),
anti-VE-cadherin (1:1,000), anti-NADPH oxidase antibody (1:500),
anti-GSK-3β (1:500), anti-phospho-GSK-3β (Ser9; 1:500) and
anti-GAPDH (1:8,000) primary antibodies overnight at 4°C, followed
by incubation with the corresponding HRP-conjugated secondary
antibodies (1:5,000). Protein bands were detected according to an
enhanced chemiluminescence (ECL) method (EMD Millipore, Billerica,
MA, USA) using a Bio-Rad Gel Imaging system and were analyzed with
Quantity One software version 4.4.0 (both Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The expression levels were determined by
measuring the corresponding band intensities.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's protocol. The relative RNA
levels were quantified using a NanoDrop 2000 spectrophotometer
(NanoDrop; Thermo Fisher Scientific, Wilmington, DE, USA).
Subsequently, 1 µg RNA was used as a template for the
generation of cDNA using a HiScript First Strand cDNA synthesis kit
(Vazyme, Piscataway, NJ, USA). The reverse transcription reaction
conditions were 25°C for 5 min, 42°C for 15 min, and 85°C for 5
min. Gene expression levels of TNF-α, IL-6, VE-cadherin and
β-catenin were detected using a HiScript® II One Step
qRT-PCR SYBR®-Green kit (Vazyme) and StepOne Real-Time
PCR apparatus (Applied Biosystem; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Polymerase chain reaction conditions were as
follows: pre-denaturation at 95°C for 30 sec, 40 cycles of
denaturation at 95°C for 5 sec, annealing at 55°C for 30 sec, and
polymerization at 72°C for 30 sec. Relative gene expression levels
were normalized to GAPDH using CFX manager softwareversion 3.0
(Bio-Rad Laboratories, Inc.) according to the comparative Cq (ΔΔCq)
method (31). The primer
sequences are presented in Table
I.
 | Table IPolymerase chain reaction primer
sequences. |
Table I
Polymerase chain reaction primer
sequences.
| Gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| GADPH |
AGGTCGGTGTGAACGGATTTG |
TGTAGACCATGTAGTTGAGGTCA |
| TNF-α |
CCCTCACACTCAGATCATCTTCT |
GCTACGACGTGGGCTACAG |
| IL-6 |
TAGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
| VE-cadherin |
CACTGCTTTGGGAGCCTTC |
GGGGCAGCGATTCATTTTTCT |
| β-catenin |
CACAAGCAGAGTGCTGAAGGTG |
GATTCCTGAGAGTCCAAAGACAG |
Immunofluorescence staining
Coverslips (3 from each group) of the HPMECs
(1×105 cells per well) were fixed with 3.7%
paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked
with PBS containing 5% goat serum and incubated with anti-NF-κB p65
antibodies (1:400) at 4°C overnight. Subsequently, coverslips were
incubated with Alexa Fluor 488-labeled secondary antibodies (1:500)
for 1 h in the dark. Coverslips were rinsed three times with PBS,
and the nuclei were stained with DAPI (Nanjing KeyGen Biotech Co.,
Ltd.) for 5 min. Images were captured by inverted microscopy
(TE2000-U; Nikon Corporation) after washing.
Fluorescent phalloidin for F-actin
staining
HPMECs (1×105 cells per well) were seeded
on coverslips (3 from each group) and were fixed in 3.7%
paraformaldehyde in PBS. The coverslips were then rinsed with PBS,
permeabilized with 0.1% Triton X-100 in PBS, preincubated with PBS
containing 1% BSA and stained with 200 µl
Fluorescent-iFluor™ 594 phalloidin solution (Nanjing KeyGen Biotech
Co., Ltd.) for 30 min at room temperature. Finally, coverslips were
washed, sealed and images were captured by inverted microscopy
(TE2000-U; Nikon Corporation).
Detection of ROS production
Intracellular ROS production was detected using the
superoxide indicator dihydroethidium (DHE; D1168; Invitrogen;
Thermo Fisher Scientific, Inc.). HPMECs were pretreated with
rh-vaspin (10 ng/ml) or PBS as a control for 24 h, and were then
exposed to PBS or LPS (100 ng/ml) for 4 h. HPMECs were washed twice
with PBS, incubated in fresh culture medium without FBS, and were
incubated with DHE (10 µM dissolved in dimethyl sulfoxide)
for 15 min at 37°C. Once the DHE probe is oxidized by ROS to
2-hydroxyethidium, it intercalates within DNA, resulting in the
fluorescent red staining of cell nuclei. Images of the cells were
captured using an inverted fluorescence microscope (TE2000-U; Nikon
Corporation) from three different fields of view. Fluorescence
intensity was measured using ImageJ software (National Institutes
of Health, Bethesda, MD, USA), averages were calculated and were
normalized to the control.
Lucigenin assay
Following exposure of HPMECs to PBS or LPS (100
ng/ml) for 4 h in the absence or presence of rh-vaspin (100 ng/ml)
for 24 h, total cell lysates were harvested. Nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase (NOX) activity was
determined using a lucigenin assay. As a chemiluminescent probe,
lucigenin is used to indicate the presence of superoxide anion
radicals in cells. The reaction was performed in a total volume of
200 µl assay buffer containing 10 µM lucigenin
(L-6868; NSC-151912; MedChem Express, Monmouth Junction, NJ, USA),
1 mM NADPH, and 15 µg cell lysates. Following incubation for
30 min at 37°C in the dark, absorbance was measured using a
luminometer (BioTek Instruments, Inc.) at an excitation wavelength
of 540 nm and an emission wavelength of 605 nm. Samples were well
mixed and chemiluminescence was continuously measured for 30 min.
Chemiluminescence of relative light units per second (RLU/sec) was
obtained every 10 sec and the results were calculated as area under
the curve and normalized to the control.
Statistical analysis
Data are presented as the mean ± standard deviation.
Results are representative of at least three independent
experiments performed in triplicate. Unpaired Student's t-test was
performed for comparisons between two independent groups. One-way
analysis of variance (ANOVA) followed by the Student-Newman-Keuls
post hoc test was performed to compare continuous variables with
normal distribution from three or more independent groups, and to
detect significant differences between particular groups.
Kruskal-Wallis ANOVA followed by Mann-Whitney U test was performed
to compare continuous variables with abnormal distribution from
three or more independent groups, and to detect significant
differences between particular groups. P<0.05 (95% confidence
interval) was considered to indicate a statistically significant
difference at. All statistical analyses were conducted using
GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
Vaspin attenuates LPS-induced lung injury
and pulmonary inflammation in vivo and in vitro
In order to explore the effects of vaspin on lung
histological injury in an LPS-induced mouse model of ARDS, lung
histopathological examination was conducted. Mice were systemically
instilled with Ad-vaspin or Ad-β-gal as control (3×107
PFU/mouse for 3 days) and were subjected to intratracheal injection
with LPS (5 mg/kg) to establish a mouse model of ARDS or with PBS
as a control. Plasma vaspin levels were enhanced to 99.0±7.9 ng/ml
on day 3 after Ad-vaspin injection, which could not be detected in
the control mice treated with Ad-β-gal (data not shown). Mice that
were injected with LPS developed ARDS as early as 4 h after LPS
insult, as indicated by histopathological alterations in the lungs,
including increased inflammatory cell infiltration, thickened
alveolar septum, intra-alveolar and interstitial edema fluid, and
patchy areas of hemorrhage (Fig.
1A). These histopathological alterations were significantly
attenuated following administration of Ad-vaspin (Fig. 1A and B). Since lung injury in ARDS
is associated with an exaggerated inflammatory response, the
present study further examined the effects of vaspin on pulmonary
inflammation. Consistent with the results of lung injury
assessment, expression levels of the proinflammatory cytokines
TNF-α (Fig. 1C) and IL-6
(Fig. 1D) were reduced in
Ad-vaspin-pretreated mice compared with in Ad-β-gal-pretreated mice
4 h post-LPS insult, whereas expression of the anti-inflammatory
cytokine IL-10 (Fig. 1E) was
increased. In addition, the expression levels of the adhesion
molecule, ICAM-1 (Fig. 1F), were
reduced in Ad-vaspin-pretreated mice compared with in
Ad-β-gal-pretreated mice 4 h post-LPS insult. These findings
suggested that vaspin may exert an anti-inflammatory effect on lung
injury in a mouse model of LPS-induced ARDS.
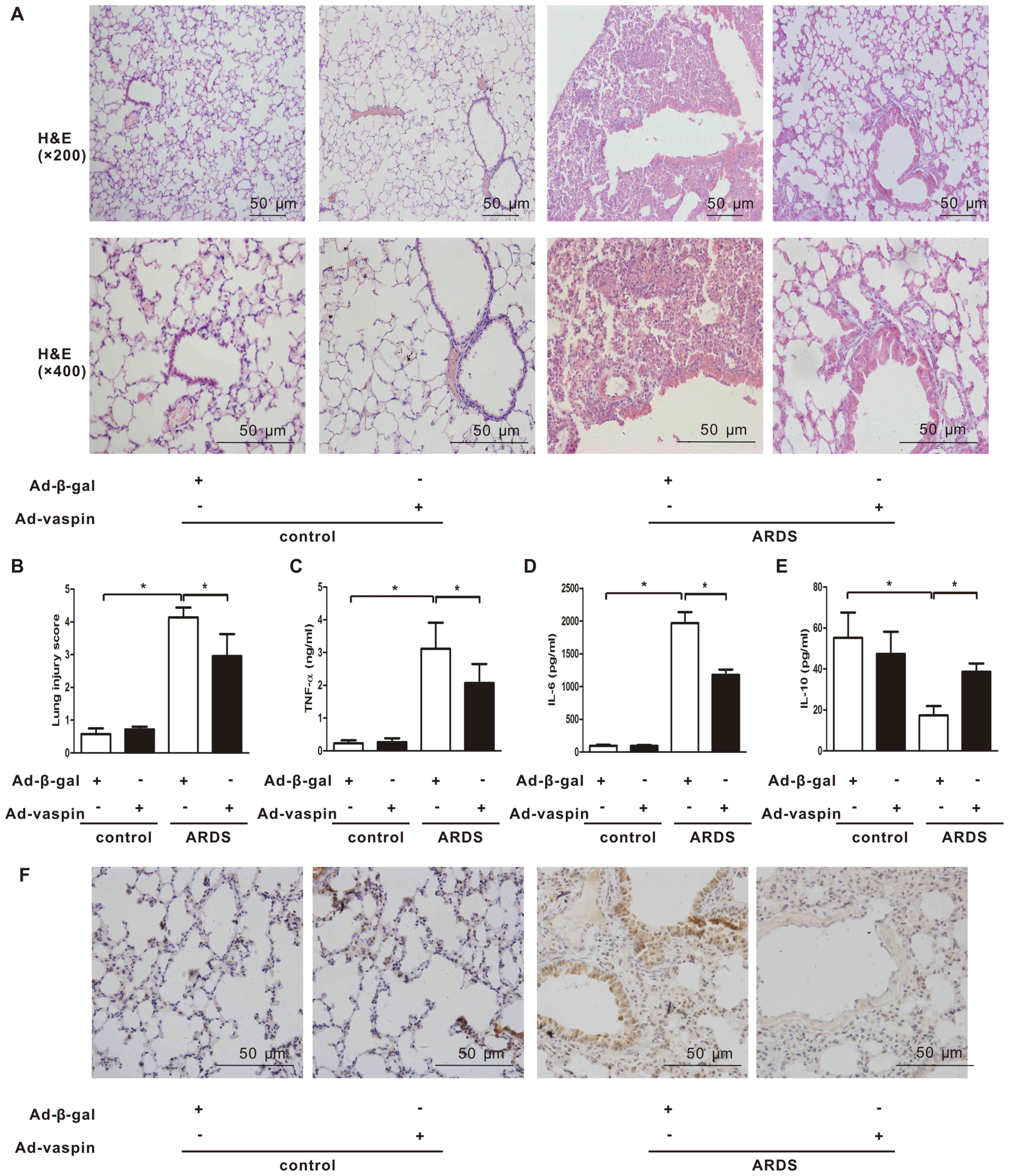 | Figure 1Vaspin ameliorates LPS-induced lung
injury and pulmonary inflammation in vivo. Mice were
systemically instilled with Ad-vaspin or Ad-β-gal as a control
(3×107 PFU/mouse) for 3 days and were subjected to
intratracheal injection with LPS (5 mg/kg) in order to establish a
mouse model of ARDS. Mice were intratracheally injected with PBS as
a control. A total of 4 h after LPS/PBS injection, lung lobes were
harvested. (A) Lung hispathological alterations were assessed by
hematoxylin and eosin staining (n=5 mice from each group assessed
in triplicate; magnification, ×200 and ×400). (B) Lung injury
scores were used to analyze lung histopathological alterations (n=5
mice from each group assessed in triplicate). Expression levels of
(C) TNF-α, (D) IL-6 and (E) IL-10 were measured in lung homogenates
by ELISA (n=5 mice from each group assessed in triplicate). (F)
Expression levels of ICAM-1 were detected by immunohistochemistry
(n=5 mice from each group assessed in triplicate; magnification,
×400). Data are presented as the mean ± standard deviation.
*P<0.05. Ad-β-gal, adenoviral vector expressing
β-galactosidase; Ad-vaspin, adenoviral vector expressing vaspin;
ARDS, acute respiratory distress syndrome; ICAM-1, intercellular
adhesion molecule-1; IL, interleukin; LPS, lipopolysaccharide;
TNF-α, tumor necrosis factor-α.. |
Vaspin has previously been reported to exert
anti-inflammatory effects on various types of vascular ECs
(24–27). Therefore, it may be hypothesized
that vaspin mediates mitigation of inflammation in pulmonary ECs.
To assess the anti-inflammatory effects of vaspin in vitro,
the mRNA expression levels of inflammatory cytokines (TNF-α and
IL-6) and endothelial-specific adhesion markers (VCAM-1 and
E-selectin) were analyzed in HPMECs 2 h after LPS insult.
Consistent with the LPS-mediated inflammatory response in lung
tissue, the expression levels of TNF-α (Fig. 2A), IL-6 (Fig. 2B), VCAM-1 (Fig. 2C) and E-selectin (Fig. 2D) were significantly increased in
HPMECs following LPS administration, which were reversed by
pretreatment with rh-vaspin. The observed anti-inflammatory effects
of vaspin were further confirmed by examining the activation of
NF-κB, which is a pivotal inflammatory mediator; the results
indicated that the phosphorylation and nuclear translocation of the
NF-κB Rel subunit were reduced following treatment of HPMECs with
rh-vaspin 2 h after LPS insult (Fig.
2E and F). Taken together, these findings indicated that vaspin
may exert a protective role during the early stage of LPS-induced
ARDS via the suppression of EC inflammation.
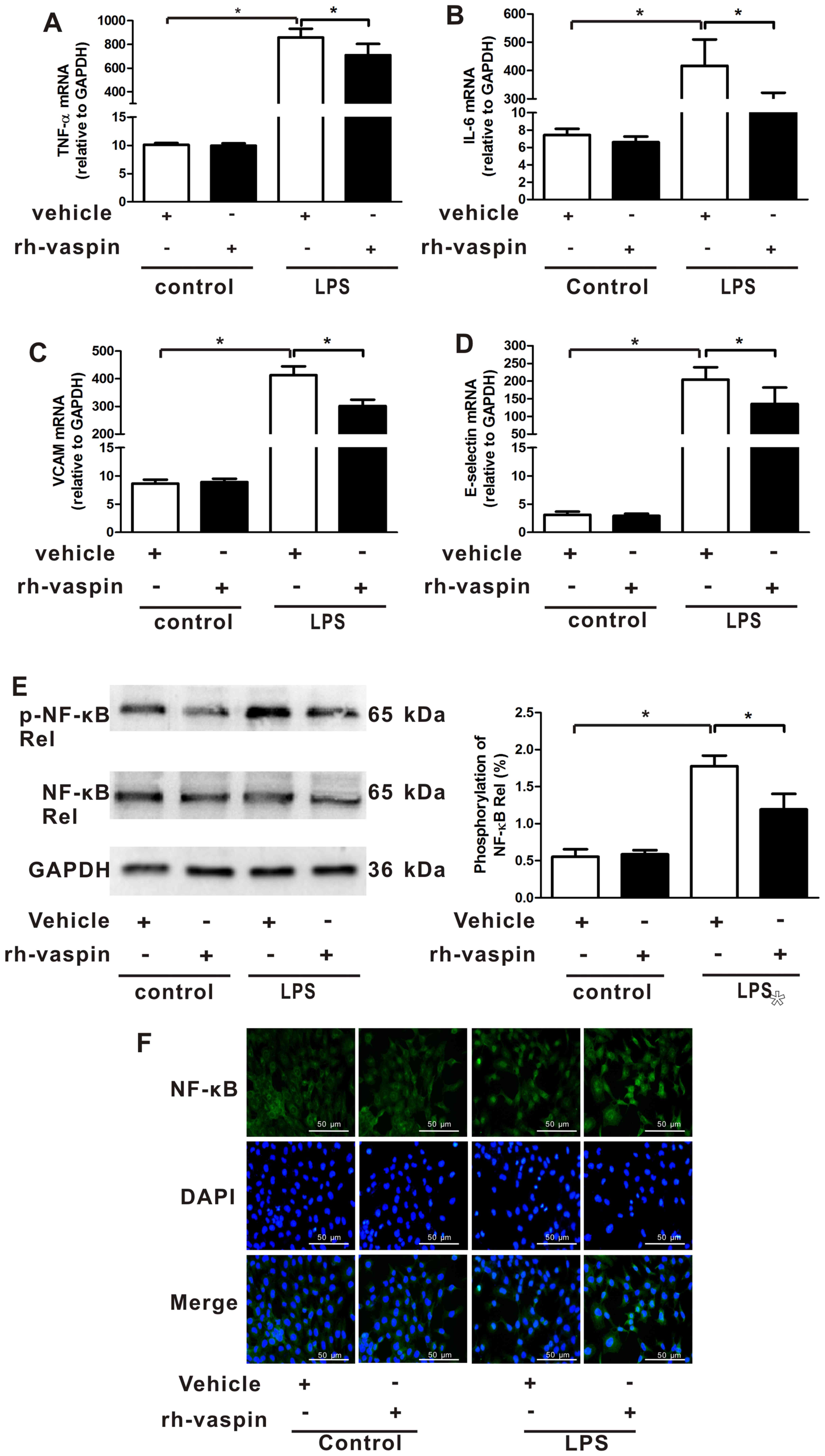 | Figure 2Vaspin attenuates LPS-induced
endothelial cell inflammation in vitro. HPMECs were
pretreated with rh-vaspin (10 ng/ml) or PBS as a control for 24 h,
and were then exposed to LPS (100 ng/ml) or vehicle (PBS) for 2 h.
mRNA expression levels of (A) TNF-α, (B) IL-6, (C) VCAM-1 and (D)
E-selectin in the HPMECs lysate were assessed by quantitative
polymerase chain reaction (n=5 cultures from each group assessed in
triplicate). (E) Expression levels of p-NF-κB Rel subunit measured
by western blot analysis (n=5 cultures from each group assessed in
triplicate). The phosphorylation levels of NF-κB Rel subunit were
normalized to total NF-κB Rel signals. (F) Nuclear translocation of
the NF-κB Rel subunit was measured by immunofluorescence (n=3
cultures from each group assessed in triplicate). Data are
presented as the mean ± standard deviation. *P<0.05.
HPMECs, human pulmonary microvascular endothelial cells; IL,
interleukin; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB;
p-NF-κB, phosphorylated-NF-κB; rh-vaspin, recombinant human vaspin;
TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion
molecule 1. |
Vaspin restores the pulmonary EC barrier
following LPS insult in vivo and in vitro
Pulmonary microvascular hyperpermeability is a
common feature of ARDS; therefore, it was assessed in vitro
and in vivo in the present study (Fig. 3). To address whether vaspin
modulates pulmonary endothelial barrier function in vivo,
total bronchoalveolar lavage fluid (BALF) protein concentrations,
EBDA extravasation and lung W/D ratios were analyzed in mice with
LPS-induced ARDS. Lung histological damage was observed to be
associated with pulmonary microvascular hyperpermeability following
LPS instillation, as manifested by increases in BALF protein
concentrations (Fig. 3A), EBDA
extravasation (Fig. 3B) and W/D
ratios (Fig. 3C) at 4 h after LPS
treatment. The time point selected to evaluate the effects of
vaspin on ARDS was based on its coincidence with the histological
onset of lung injury in mice. Pretreatment with Ad-vaspin
significantly attenuated pulmonary microvascular hyperpermeability
in a murine model of ARDS (Fig.
3A–C). In addition, to further confirm the ability of vaspin to
mitigate LPS-induced EC hyperpermeability in vitro, HPMECs
were cultured in the presence or absence of rh-vaspin (10 ng/ml)
for 24 h, and the influx of FITC-dextran was measured. Treatment
with a physiological concentration of rh-vaspin prevented
LPS-induced increases in the influx of FITC-dextran (Fig. 3E). These data indicated that
vaspin may significantly attenuate LPS-challenged pulmonary
microvascular hyperpermeability in vivo and EC barrier
dysfunction in vitro.
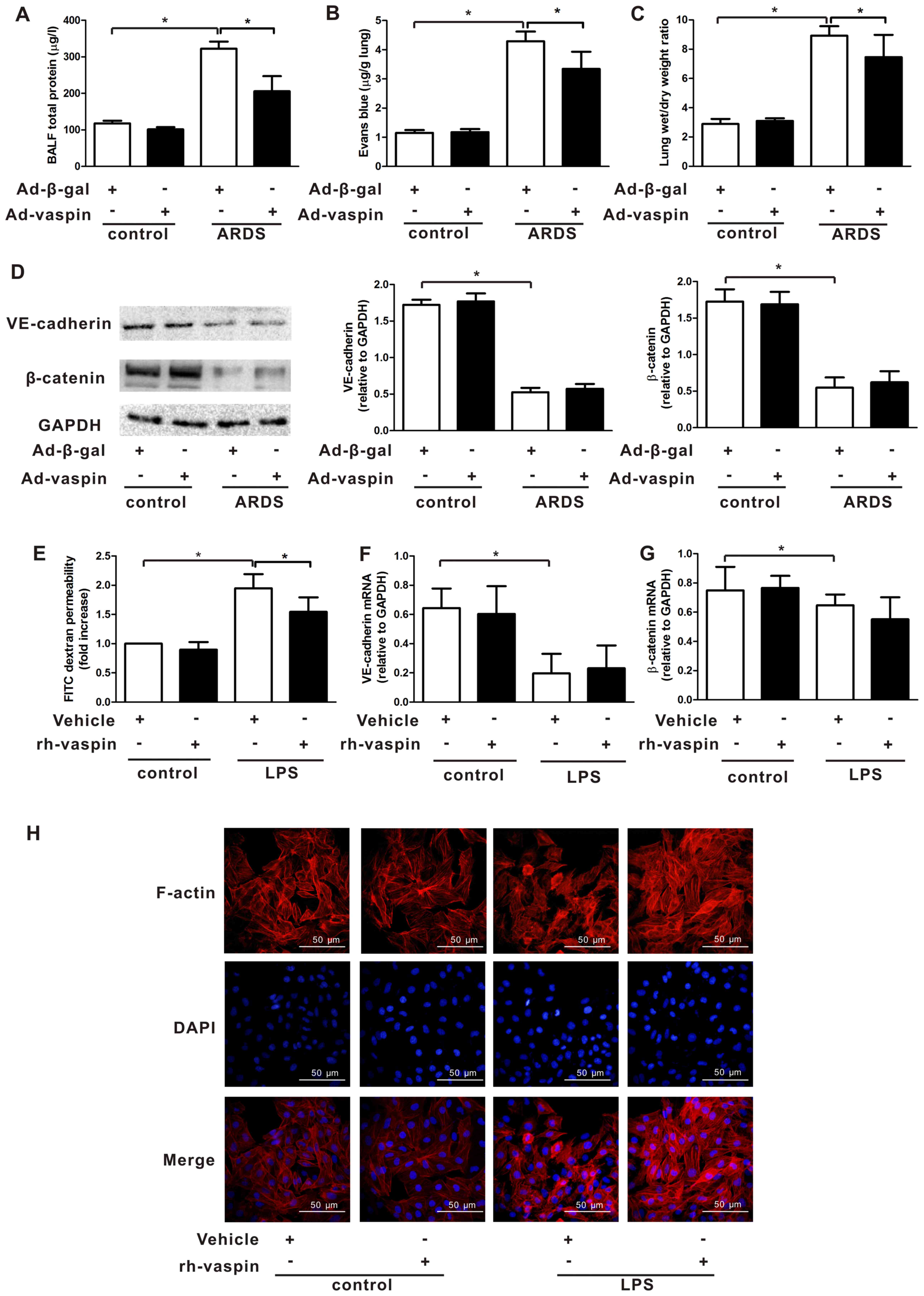 | Figure 3Vaspin reverses pulmonary endothelial
cell barrier dysfunction in vivo and in vitro, with
no effect on pulmonary endothelial cell adherens junctions and the
actin cytoskeleton following LPS insult. Mice were systemically
instilled with Ad-vaspin or Ad-β-gal as a control (3×107
PFU/mouse, 3 days) and were subjected to an intratracheal injection
with LPS (5 mg/kg) to establish a mouse model of ARDS. Mice were
intratracheally injected with PBS as a control. (A) Total BALF
protein concentrations, (B) Evans blue dyed-albumin extravasation
and (C) wet/dry ratios in a murine model of ARDS were measured 4 h
post-LPS injection (n=5 mice from each group assessed in
triplicate). (D) Expression levels of VE-cadherin and β-catenin
were detected in lung tissues by western blot analysis (n=5 mice
from each group assessed in triplicate). Relative protein
expression levels were semi-quantified by measuring corresponding
band intensities, and values were expressed relative to GADPH.
HPMECs were pretreated with rh-vaspin (10 ng/ml) or PBS as a
control for 24 h, and were then exposed to PBS or LPS (100 ng/ml)
for 4 h. (E) Influx of FITC-dextran was measured by permeability
assay (n=5 cultures from each group assessed in triplicate). mRNA
expression levels of (F) VE-cadherin and (G) β-catenin in the
HPMECs lysate were assessed by quantitative polymerase chain
reaction (n=5 cultures from each group assessed in triplicate). (H)
Actin cytoskeleton distribution was assessed by phalloidin staining
(n=3 cultures from each group assessed in triplicate). Data are
presented as the mean ± standard deviation. *P<0.05.
Ad-β-gal, adenoviral vector expressing β-galactosidase; Ad-vaspin,
adenoviral vector expressing vaspin; ARDS, acute respiratory
distress syndrome; BALF, bronchoalveolar lavage fluid; FITC,
fluorescein isothiocyanate; HPMECs, human pulmonary microvascular
endothelial cells; LPS, lipopolysaccharide; rh-vaspin, recombinant
human vaspin; VE-cadherin, vascular endothelial-cadherin. |
Vaspin has no effect on pulmonary EC
adherens junctions (AJs), actin cytoskeleton and EC differentiation
following LPS insult in vivo and in vitro
The stabilization of interendothelial AJs and the
actin cytoskeleton are essential for a restrictive pulmonary EC
barrier; lung endothelium permeability can increase due to
alterations in AJs and the endothelial cyto-skeleton. Therefore, to
further address the effects of vaspin on vascular homeostasis, the
expression levels of VE-cadherin and β-catenin, which are two
important AJ proteins of ECs, were evaluated in the lung tissue of
mice. Notably, analysis of AJs expression did not detect a
difference in the Ad-vaspin-pretreated mice compared with those
treated with LPS alone (Fig. 3D).
In addition, LPS caused a reduction in the mRNA expression levels
of AJs (Fig. 3F and G), as well
as cell retraction, F-actin reorganization and stress fiber
formation in HPMECs, as determined by phalloidin staining (Fig. 3H). However, consistent with the
results of the in vivo experiments, AJs gene expression and
distribution of the actin cytoskeleton in vaspin-treated HPMECs
were not altered compared with the controls treated with LPS alone
(Fig. 3F–H). Collectively, these
findings indicated that vaspin may exert beneficial effects on
pulmonary microvascular hyperpermeability via restoring EC barrier
function; however, these effects may not depend on the ability of
vaspin to regulate interendothelial AJs or the endothelial
cytoskeleton but may be dependent on other mechanisms associated
with endothelial barrier integrity.
Vaspin improves pulmonary EC survival and
suppresses pulmonary EC apoptosis following LPS insult in vitro and
in vivo
Vaspin has been reported to inhibit the apoptosis of
ECs, including human aortic ECs, HUVECs and smooth muscle cells
(SMCs). Furthermore, the function of the pulmonary endothelial
barrier depends on its integrity (24,25,27). Focusing on the beneficial effects
of vaspin on the function and integrity of pulmonary ECs at the
cellular level, HPMECs were treated with LPS in the presence or
absence of rh-vaspin (10 ng/ml, pretreatment for 24 h). CCK-8,
TUNEL staining and FCM analyses were performed to ascertain whether
vaspin modulates the survival and apoptosis of pulmonary ECs
following exposure to LPS (Fig.
4). In HPMECs, pretreatment with rh-vaspin significantly
promoted pulmonary EC survival and suppressed pulmonary EC
apoptosis under LPS stimulus compared with those pretreated with
PBS, as determined by increased cell viability (Fig. 4B), a reduction in the number of
TUNEL-positive cells (Fig. 4A and
C) and reduced apoptotic rate, as determined by FCM (Fig. 4A and D). In situ apoptotic
detection was further performed to verify the effects of vaspin on
apoptosis in lung tissues. Compared with in the control group,
Ad-vaspin markedly reduced the number of TUNEL-positive cells in
lung tissues post-LPS insult (Fig.
4E). These results suggested that vaspin exerts a protective
role on HPMECs, at least partially via its prosurvival and
anti-apoptotic properties.
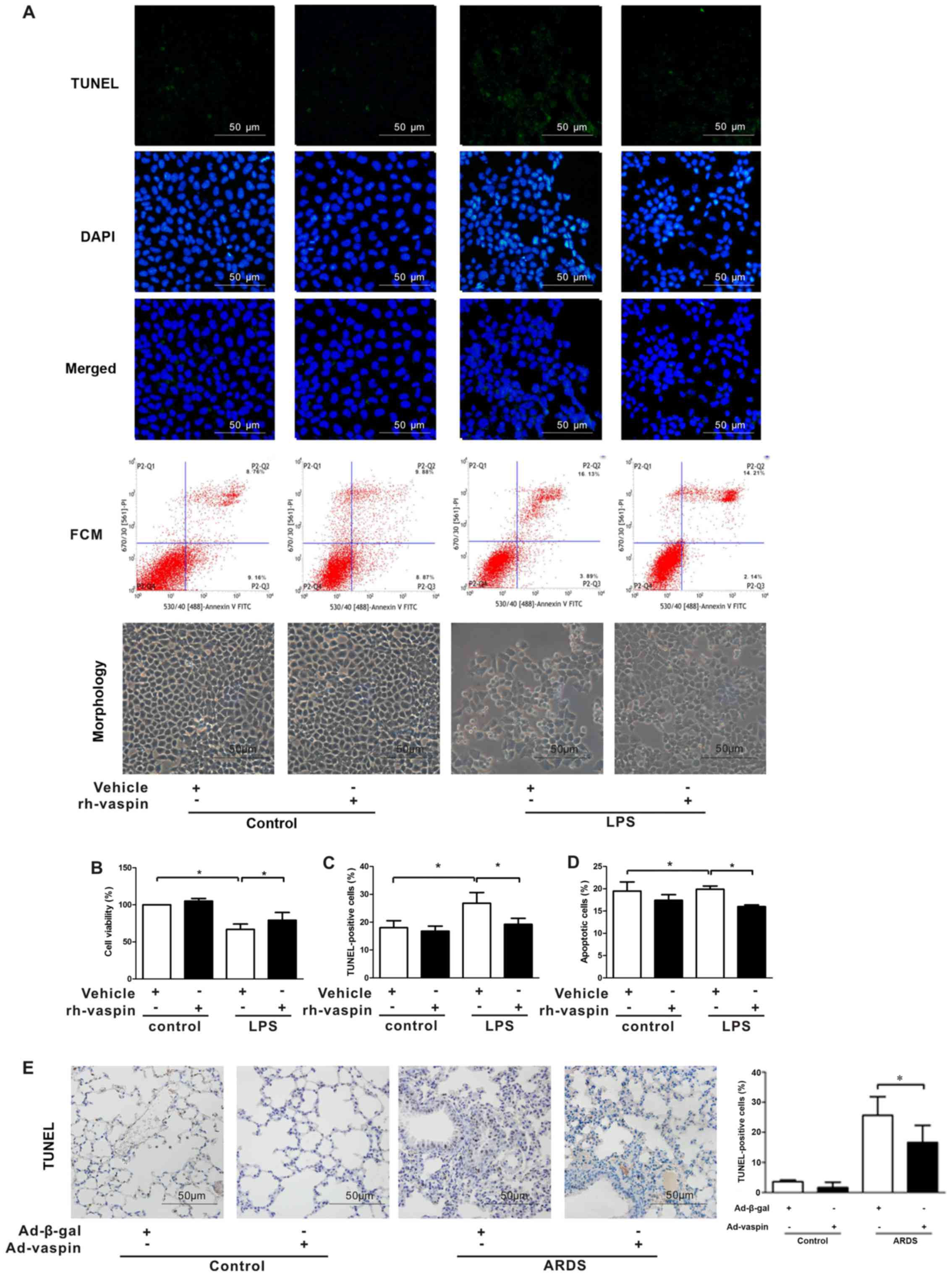 | Figure 4Vaspin improves survival and
suppresses apoptosis following LPS insult in vitro and in
vivo. HPMECs were pretreated with rh-vaspin (10 ng/ml) or PBS
as a control for 24 h, and were then exposed to PBS or LPS (100
ng/ml) for 2 h. (A) Representative images of TUNEL staining and FCM
(n=3 cultures from each group assessed in triplicate). (B) HPMEC
survival was measured by Cell Counting kit-8 analyses (n=5 cultures
from each group assessed in triplicate). (C) Quantitative analysis
of TUNEL-positive HPMECs. (D) Quantitative analysis of apoptotic
HPMECs, as determined by FCM. (E) Left panel, representative images
of TUNEL staining of lung tissues; right panel, quantitative
analysis of the number of TUNEL-positive cells. Data are presented
as the mean ± standard deviation. *P<0.05. Ad-β-gal,
adenoviral vector expressing β-galactosidase; Ad-vaspin, adenoviral
vector expressing vaspin; FCM, flow cytometry; HPMECs, human
pulmonary microvascular endothelial cells; LPS, lipopolysaccharide;
rh-vaspin, recombinant human vaspin; TUNEL, TdT-mediated dUTP nick
end labeling. |
Vaspin inhibits LPS-induced ROS
generation and NOX activation following LPS insult in vitro
A previous study demonstrated that ROS and NOX
mediated EC apoptosis under various insults. Furthermore, vaspin
has been demonstrated to inhibit ROS generation by suppressing NOX
activation in ECs, including HUVECs. Therefore, the present study
further evaluated the effects of vaspin on intracellular ROS
production by measuring the fluorescence intensity of the
intracellular fluorescent probe, DHE. Compared with HPMECs exposed
to LPS alone, pretreatment with rh-vaspin significantly
counteracted LPS-induced ROS production in HPMECs (Fig. 5A and B). Constitutive NOX
functions as an oxygen sensor that regulates intracellular
superoxide organization. Therefore, to elucidate the upstream
mechanisms, the effects of vaspin on NOX were further assessed by
measuring NOX activity and expression. The results indicated that
LPS induced an increase in NOX activity (Fig. 5C) and expression (Fig. 5D and E); however, this was
markedly abrogated by pretreatment of HPMECs with vaspin. These
findings indicated that the protective effects of vaspin against
LPS insults in ECs may be associated with antioxidative
properties.
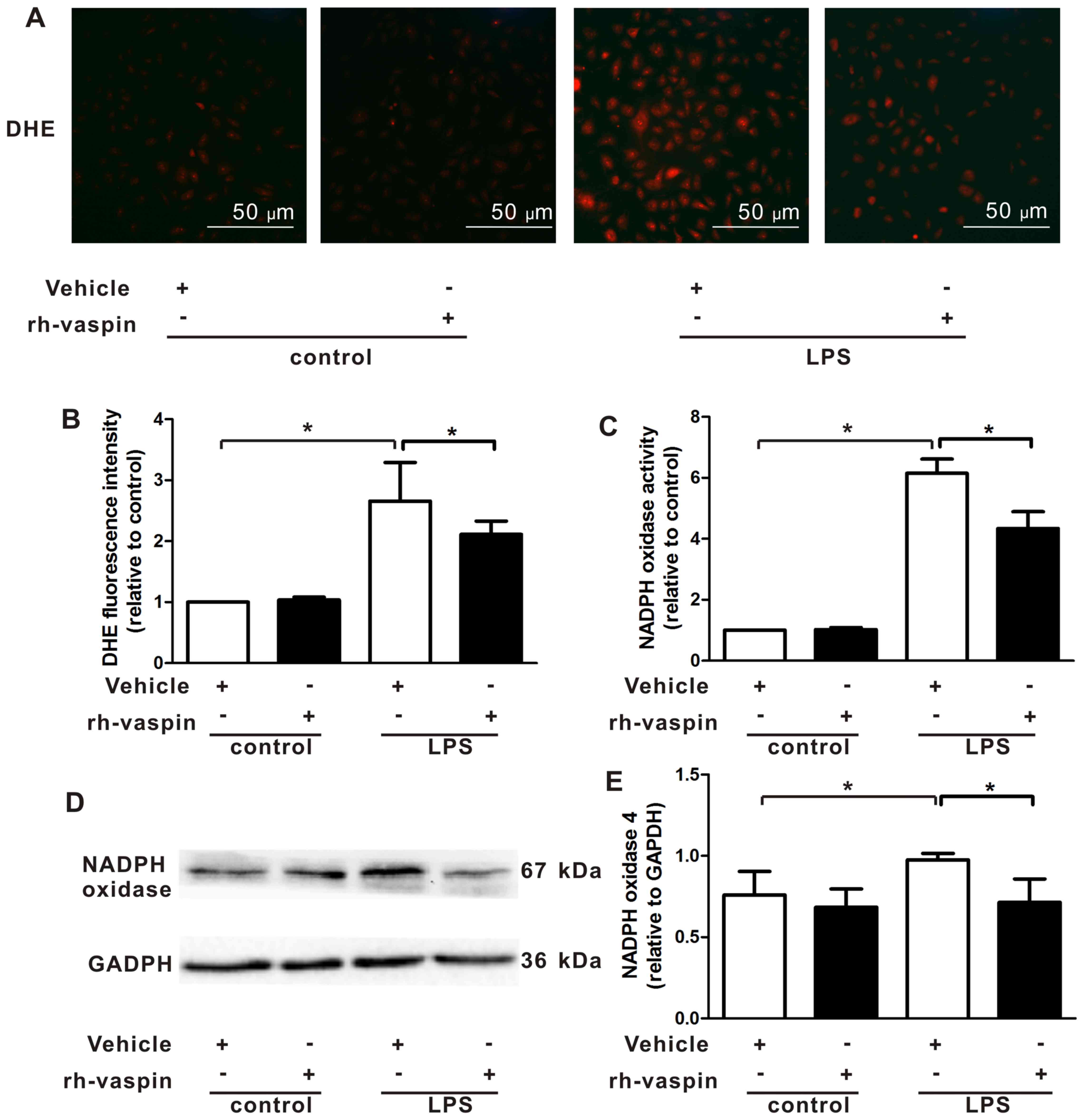 | Figure 5Vaspin inhibits LPS-induced ROS
generation and NADPH oxidase activation following LPS insults in
vitro. HPMECs were pretreated with rh-vaspin (10 ng/ml) or PBS
as a control for 24 h, and were then exposed to PBS or LPS (100
ng/ml) for 2 h. (A) Representative images of DHE staining of HPMECs
(n=3 cultures from each group assessed in triplicate). (B)
Quantitative analysis of ROS production in HPMECs, as determined by
fluorescence staining using DHE. (C) Quantitative analysis of NADPH
oxidase activity in HPMECs, as determined by lucigenin assay. (D)
Expression of NADPH oxidase was determined by western blot
analysis. Relative protein expression levels were semi-quantified
by measuring band intensities, and values were expressed relative
to GADPH (n=5 cultures from each group assessed in triplicate). (E)
Semi-quantitative analysis of NADPH oxidase expression in HPMECs,
as determined by western blot analysis. Data are presented as the
mean ± standard deviation. *P<0.05. DHE,
dihydroethidium; HPMECs, human pulmonary microvascular endothelial
cells; LPS, lipopolysaccharide; NADPH, nicotinamide adenine
dinucleotide phosphate; rh-vaspin, recombinant human vaspin; ROS,
reactive oxygen species. |
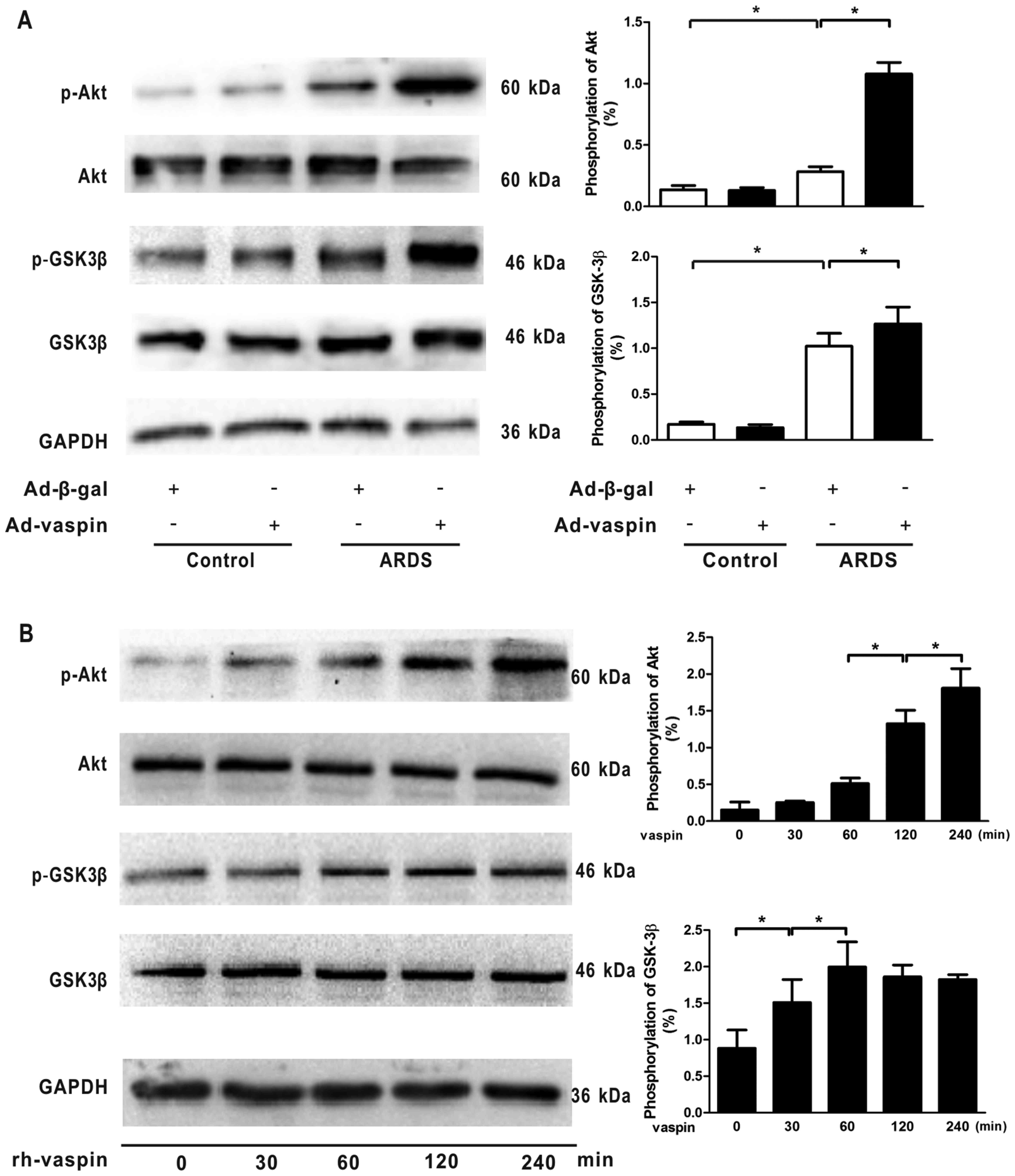
Vaspin activates the Akt-GSK3β signaling
pathway in vivo and in vitro
The PI3K/Akt signaling pathway acts as a
compensatory regulator of ARDS through its anti-inflammatory,
anti-apoptotic and antioxidant effects in response to numerous
growth factors. Therefore, to assess the effects of vaspin on the
activation of Akt-related signaling in vivo and in
vitro, the phosphorylation of Akt and its downstream target
GSK3β were assessed by western blotting. The results indicated that
p-Akt and p-GSK3β levels were low under non-stressed conditions but
were increased in a mouse model of LPS-induced ARDS; however, the
differences between these groups were not statistically
significant, thus suggesting an endogenous negative feedback
mechanism underlying the effects of LPS on the PI3K/Akt pathway.
Notably, administration of vaspin enhanced the phosphorylation of
Akt and GSK3β in mouse lungs subjected to LPS (Fig. 6A). At the cellular level,
rh-vaspin stimulated the phosphorylation of Akt and GSK3β (Fig. 6B). Collectively, these data
suggested that vaspin acts as a stimulatory factor for the
Akt-GSK3β signaling pathway, and vaspin-induced protection of
pulmonary ECs during ARDS may be mediated, at least partially, by
activating the Akt-GSK3β signaling pathway.
Discussion
Pulmonary vascular hyperpermeability has long been
considered a principal pathological hallmark of ARDS, which is
largely responsible for pulmonary edema formation (3,4,32).
Therefore, stability and integrity of the pulmonary endothelial
barrier are important in the pathogenesis of ARDS. To the best of
our knowledge, the present study is the first to provide evidence
to suggest that vaspin protects the pulmonary endothelial barrier
during LPS-induced ARDS by limiting inflammation, apoptosis and ROS
generation in pulmonary ECs. In a murine model of ARDS, systemic
administration of Ad-vaspin attenuated lung injuries by suppressing
the pulmonary inflammatory response and restoring pulmonary
endothelial barrier functions. In HPMECs, treatment with a
physiological concentration of rh-vaspin led to attenuation of the
inflammatory response, cell apoptosis and NOX-dependent ROS
generation, as well as enhancement of EC survival, with no
alterations in cell AJs expression or cytoskeletal rearrangement. A
further mechanistic study suggested that the beneficial effects of
vaspin are, at least partially, mediated by activation of the
Akt-GSK3β signaling pathway. Taken together, these data indicated
that vaspin may promote the function and refine stability of the
pulmonary endothelial barrier via its anti-inflammatory,
anti-apoptotic and antioxidative properties in ECs, thus protecting
against LPS-induced endothelial barrier hyperpermeability and
disruption in the experimental systems of ARDS. Therapeutic
interventions that aim to restore vaspin levels may be favorable
for the prevention or treatment of ARDS.
At present, adipose tissue is considered a
significant endocrine organ that is capable of crosstalk with
peripheral organs through various multifunctional adipokines
(11). Compared with the
well-elucidated effects of adipokines on cardiovascular disease,
the association between adipokines and the pathogenesis of ARDS has
only recently begun to be elucidated (12,33). Given the systemic abnormalities
associated with obesity and the accompanying metabolic syndrome,
the effects of obesity on the pathogenesis of ARDS may be
associated with numerous facets of the obese state, including
dysregulation of adipokine release, which are known to contribute
to the development of obesity-associated vascular disorders. The
majority of adipokines are proinflammatory, whereas a small number
of anti-inflammatory adipokines exert beneficial effects on
obesity-associated complications (13). Vaspin functions as a novel
predictor of obesity-associated vascular disorders and endothelial
damage in human studies (21,23). Furthermore, it has been
demonstrated to be involved in the modulation of diverse biological
process, including inflammation, apoptosis and oxidative stress in
experimental studies (24–28,34),
which are potentially implicated in the pathogenesis of ARDS. As
the homologous gene of vaspin, α1-antitrypsin is well known for its
function in inhibiting serum serine proteases, including neutrophil
elastase, trypsin, thrombin and proteinase-3 (35). In addition, α1-antitrypsin has
been reported to exert pleiotropic effects in the airways via its
antioxidant, antiprotease, anti-inflammatory and anti-apoptotic
properties (36–40). From a clinical perspective, it has
previously been indicated that there is no therapeutic advantages
to enhancing anti-neutrophil elastase activity in ARDS (41,42). Nevertheless, to the best of our
knowledge, no study has assessed the effects of vaspin on pulmonary
ECs, particularly in the setting of ARDS. Therefore, the effects of
vaspin on pulmonary ECs were explored in the present study.
Inflammation serves a vital role in the initiation
and progression of ARDS (43,44), and vaspin has been reported to
exert anti-inflammatory effects against various insults (26,34). The present study demonstrated that
vaspin suppressed the inflammatory response to LPS in lung tissues
and HPMECs, as evidenced by a decrease in the levels of
proinflammatory cytokines (IL-6 and TNF-α) and ICAM-1 in lung
tissues, and a reduction in the activation of NF-κB, which is a
pivotal inflammatory mediator, post-LPS stimulation. Consistent
with these findings, previous studies have indicated that vaspin
may protect ECs from proinflammatory cytokines-induced inflammation
by inhibiting activation of NF-κB and the production of its
downstream molecules, including ICAM-1, VCAM-1 and MCP-1 (34). However, EC barrier
hyperpermeability in ARDS involves complex interactions between ECs
and numerous cell types. Furthermore, vaspin is a pleiotropic
adipokine, the effects of which in obesity-associated vascular
disease are not exclusive to a specific cell type via a single
mechanism. Therefore, it cannot be ruled out that vaspin exerts its
potential effects on other cell types, including vascular SMCs
(VSMCs), polymorphonuclear leukocytes (PMN) or macrophages, all of
which serve important but distinct roles in ARDS progression.
Results from previous studies have indicated that vaspin has
anti-inflammatory and antimigratory roles in VSMCs, and may prevent
the expression of ICAM-1 in cultured VSMCs (45,46). Further investigation is still
required to fully elucidate the comprehensive effects of vaspin on
ARDS. Taken together, although the present study did not directly
detect or rule out the effects of vaspin on numerous cell types in
ARDS, the present findings suggested that vaspin, as a
multifunctional adipokine, exerts beneficial effects on ARDS via
its anti-inflammatory function in lung endothelium.
It is widely accepted that compromised EC integrity
and subsequent dysfunction of the pulmonary EC barrier contribute
to pulmonary endothelial barrier hyperpermeability, thus leading to
protein-rich leakage and PMN infiltration under ARDS conditions
(4,32). Therefore, therapeutic approaches
that restore pulmonary endothelial barrier integrity and function
are of vital importance in the progression of ARDS (3,4).
In the present study, aggravation of pulmonary microvascular
hyperpermeability in a mouse model of ARDS was significantly
suppressed following systemic administration of Ad-vaspin, thus
suggesting that vaspin promotes endothelial barrier function in
vivo, as evidenced by a significant reduction in capillary
leakage (measured by BALF protein concentrations, EBDA
extravasation and W/D ratios). Further in vitro
investigations supported this beneficial property at the cellular
level; the in vitro results demonstrated that vaspin
reversed LPS-induced increases in the influx of FITC-dextran in
HPMECs. The EC barrier consists of the cytoskeleton, AJs and tight
junctions. Stabilization of endothelial AJs and the actin
cytoskeleton is essential for a restrictive pulmonary EC barrier
(47,48). Notably, in the present study, no
difference in AJ protein expression and cytoskeletal organization
was detected in HPMECs treated with LPS. Therefore, it may be
hypothesized that vaspin-induced promotion of EC barrier function
is mediated by other mechanisms associated with cell integrity,
rather than regulation of cell structure.
It has previously been hypothesized that a reduction
in pulmonary endothelial cell apoptosis represents an important
therapeutic target for ARDS (49). Vaspin has been reported to
ameliorate vascular injuries under the diabetic milieu, thus
exerting beneficial effects on diabetic vascular complications by
promoting the proliferation and inhibiting the apoptosis of ECs
(24). In addition, vaspin has
been demonstrated to attenuate methylglyoxal-induced cell death in
HUVECs (25). To confirm whether
vaspin-induced suppression of pulmonary hyperpermeability in
response to LPS is due to its protection of pulmonary ECs, the
present study examined the direct effects of vaspin on pulmonary
endothelium at the cellular level. Vaspin-mediated attenuation of
EC barrier dysfunction was associated with a significant decrease
in HPMECs apoptosis under LPS stimulation. These findings suggested
that vaspin exerts its protective effects on pulmonary EC barrier
during the LPS-induced ARDS milieu is dependent, at least
partially, on its ability to attenuate apoptosis of ECs, thus
reinforcing barrier integrity and restoring EC barrier
function.
Endothelial dysfunction can be caused by various
insults, among which ROS production is markedly involved in ARDS
pathogenesis (50,51). ROS are produced by NOX and
function as second messengers of cytokines that are formed in
response to various stimuli. It has previously been demonstrated
that the primary downstream target of ROS is apoptosis
signal-regulating kinase 1, which regulates the balance between
cell death and survival (52,53). In the present study, the results
demonstrated that vaspin may inhibit ROS production and NOX
activation, thus alleviating damage to the pulmonary ECs barrier
caused by LPS-induced oxidative burst. Consistent with these
findings, vaspin has been reported to exert an antioxidative effect
on various types of cells, including ECs (25,46).
The present study also explored the underlying
mechanism mediating the favorable effects of vaspin. The
Akt-associated pathway has a central role in cell signaling with
diverse cellular functions, including cell viability,
proliferation, angiogenesis and migration. The Akt signaling
pathway acts as an endogenous negative feedback or compensatory
mechanism that serves to suppress proinflammatory and apoptotic
events in response to injurious stimuli (54). According to the findings of a
previous study, vaspin can activate Akt-associated signaling
(27); therefore, the present
study detected the phosphorylation levels of Akt and its substrate
GSK3β; the results confirmed that the beneficial effects mediated
by vaspin in ARDS were associated with activation of the Akt/GSK3β
signaling pathway. However, it is well known that a high degree of
functional versatility and overlap exists among the Akt-associated
signaling pathways, and the inflammatory response can be regulated
by numerous intercellular signals that overlap with each other to
modulate cell biology in an integrated manner. With regards to
inflammation-associated signaling pathways, we aim to investigate
those that induce activation of NF-κB, including stress-activated
protein kinases/c-Jun N-terminal kinases, p38 mitogen-activated
protein kinases and extracellular signal-regulated kinases 1/2
pathways, in our subsequent studies. Furthermore, although GRP78 is
identified as one of the interacting molecules of vaspin on the
surface of aortic ECs, further studies are required to confirm the
specific mechanism that mediates the beneficial effects of vaspin
on pulmonary ECs, more specifically in the pathogenesis of
ARDS.
In conclusion, the present study demonstrated that
vaspin protects against LPS-induced ARDS through reversing EC
barrier dysfunction via the suppression of inflammation, apoptosis
and ROS generation in pulmonary ECs, at least partially via
activation of the Akt/GSK3β signaling pathway. These findings
provide evidence of a causal link between vaspin and EC barrier
dysfunction during ARDS, and suggest a potential therapeutic
intervention for patients with ARDS in clinical practice.
Abbreviations:
|
ARDS
|
acute respiratory distress
syndrome
|
|
CAD
|
coronary artery disease
|
|
IJV
|
internal jugular vein
|
|
LPS
|
lipopolysaccharide
|
|
Ad-vaspin
|
adenoviral vector expressing
vaspin
|
|
Ad-β-gal
|
adenoviral vector expressing
β-galactosidase
|
|
rh-vaspin
|
recombinant human vaspin
|
|
ECs
|
endothelial cells
|
|
EBDA
|
Evans blue dyed-albumin
|
|
W/D
|
wet/dry
|
|
HPMECs
|
human pulmonary microvascular
endothelial cells
|
|
SMCs
|
smooth muscle cells
|
|
CCK-8
|
Cell Counting kit-8
|
|
TUNEL
|
TdT-mediated dUTP nick end
labeling
|
|
FCM
|
flow cytometry
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
ICAM
|
intercellular cell adhesion
molecule
|
|
VCAM
|
vascular cell adhesion molecule
|
|
MCP-1
|
monocyte chemotactic protein-1
|
|
AJ
|
adherens junction
|
|
DHE
|
dihydroethidium
|
|
ROS
|
reactive oxygen species
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81670071).
References
|
1
|
Ranieri VM, Rubenfeld GD, Thompson BT,
Ferguson ND, Caldwell E, Fan E and Camporota L: Acute respiratory
distress syndrome: The Berlin Definition. JAMA. 307:2526–2533.
2012.PubMed/NCBI
|
|
2
|
Phua J, Badia JR, Adhikari NK, Friedrich
JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, et
al: Has mortality from acute respiratory distress syndrome
decreased over time?: A systematic review. Am J Respir Crit Care
Med. 179:220–227. 2009. View Article : Google Scholar
|
|
3
|
Müller-Redetzky HC, Suttorp N and
Witzenrath M: Dynamics of pulmonary endothelial barrier function in
acute inflammation: Mechanisms and therapeutic perspectives. Cell
Tissue Res. 355:657–673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lucas R, Verin AD, Black SM and Catravas
JD: Regulators of endothelial and epithelial barrier integrity and
function in acute lung injury. Biochem Pharmacol. 77:1763–1772.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gong MN, Bajwa EK, Thompson BT and
Christiani DC: Body mass index is associated with the development
of acute respiratory distress syndrome. Thorax. 65:44–50. 2010.
View Article : Google Scholar
|
|
6
|
Anzueto A, Frutos-Vivar F, Esteban A,
Bensalami N, Marks D, Raymondos K, Apezteguía C, Arabi Y, Hurtado
J, González M, et al Ventila group: Influence of body mass index on
outcome of the mechanically ventilated patients. Thorax. 66:66–73.
2011. View Article : Google Scholar
|
|
7
|
Stapleton RD and Suratt BT: Obesity and
nutrition in acute respiratory distress syndrome. Clin Chest Med.
35:655–671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Konter J, Baez E and Summer RS: Obesity:
'priming' the lung for injury. Pulm Pharmacol Ther. 26:427–429.
2013. View Article : Google Scholar
|
|
9
|
Matsuzawa Y, Funahashi T and Nakamura T:
The concept of metabolic syndrome: Contribution of visceral fat
accumulation and its molecular mechanism. J Atheroscler Thromb.
18:629–639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Van Gaal LF, Mertens IL and De Block CE:
Mechanisms linking obesity with cardiovascular disease. Nature.
444:875–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamawaki H: Mechanisms of action of novel
adipocytokines on the vascular system. Nihon Yakurigaku Zasshi.
137:131–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mattu HS and Randeva HS: Role of
adipokines in cardiovascular disease. J Endocrinol. 216:T17–T36.
2013. View Article : Google Scholar
|
|
13
|
Ohashi K, Shibata R, Murohara T and Ouchi
N: Role of anti-inflammatory adipokines in obesity-related
diseases. Trends Endocrinol Metab. 25:348–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang C: Obesity, inflammation, and lung
injury (OILI): The good. Mediators Inflamm. 2014:9784632014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shah D, Romero F, Duong M, Wang N, Paudyal
B, Suratt BT, Kallen CB, Sun J, Zhu Y, Walsh K, et al:
Obesity-induced adipokine imbalance impairs mouse pulmonary
vascular endothelial function and primes the lung for injury. Sci
Rep. 5:113622015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Konter JM, Parker JL, Baez E, Li SZ,
Ranscht B, Denzel M, Little FF, Nakamura K, Ouchi N, Fine A, et al:
Adiponectin attenuates lipopolysaccharide-induced acute lung injury
through suppression of endothelial cell activation. J Immunol.
188:854–863. 2012. View Article : Google Scholar
|
|
17
|
Fan XF, Xue F, Zhang YQ, Xing XP, Liu H,
Mao SZ, Kong XX, Gao YQ, Liu SF and Gong YS: The Apelin-APJ axis is
an endogenous counterinjury mechanism in experimental acute lung
injury. Chest. 147:969–978. 2015. View Article : Google Scholar
|
|
18
|
Qi D, Tang X, He J, Wang D, Zhao Y, Deng
W, Deng X, Zhou G, Xia J, Zhong X, et al: Omentin protects against
LPS-induced ARDS through suppressing pulmonary inflammation and
promoting endothelial barrier via an Akt/eNOS-dependent mechanism.
Cell Death Dis. 7:e23602016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hida K, Wada J, Eguchi J, Zhang H, Baba M,
Seida A, Hashimoto I, Okada T, Yasuhara A, Nakatsuka A, et al:
Visceral adipose tissue-derived serine protease inhibitor: A unique
insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci
USA. 102:10610–10615. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dimova R and Tankova T: The role of vaspin
in the development of metabolic and glucose tolerance disorders and
atherosclerosis. BioMed Res Int. 2015:8234812015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kobat MA, Celik A, Balin M, Altas Y,
Baydas A, Bulut M, Aydin S, Dagli N, Yavuzkir MF and Ilhan S: The
investigation of serum vaspin level in atherosclerotic coronary
artery disease. J Clin Med Res. 4:110–113. 2012.PubMed/NCBI
|
|
22
|
Aust G, Richter O, Rohm S, Kerner C, Hauss
J, Klöting N, Ruschke K, Kovacs P, Youn BS and Blüher M: Vaspin
serum concentrations in patients with carotid stenosis.
Atherosclerosis. 204:262–266. 2009. View Article : Google Scholar
|
|
23
|
Wang HH and Wang QF: Low vaspin levels are
related to endothelial dysfunction in patients with ankylosing
spondylitis. Braz J Med Biol Res. 49:e52312016. View Article : Google Scholar :
|
|
24
|
Nakatsuka A, Wada J, Iseda I, Teshigawara
S, Higashio K, Murakami K, Kanzaki M, Inoue K, Terami T, Katayama
A, et al: Visceral adipose tissue-derived serine proteinase
inhibitor inhibits apoptosis of endothelial cells as a ligand for
the cell-surface GRP78/voltage-dependent anion channel complex.
Circ Res. 112:771–780. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Phalitakul S, Okada M, Hara Y and Yamawaki
H: Vaspin prevents methylglyoxal-induced apoptosis in human
vascular endothelial cells by inhibiting reactive oxygen species
generation. Acta Physiol (Oxf). 209:212–219. 2013.
|
|
26
|
Jung CH, Lee MJ, Kang YM, Lee YL, Yoon HK,
Kang SW, Lee WJ and Park JY: Vaspin inhibits cytokine-induced
nuclear factor-kappa B activation and adhesion molecule expression
via AMP-activated protein kinase activation in vascular endothelial
cells. Cardiovasc Diabetol. 13:412014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung CH, Lee WJ, Hwang JY, Seol SM, Kim
YM, Lee YL and Park JY: Vaspin protects vascular endothelial cells
against free fatty acid-induced apoptosis through a
phosphatidylinositol 3-kinase/Akt pathway. Biochem Biophys Res
Commun. 413:264–269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin Y, Zhuang J, Li H, Zhu G, Zhou S, Li
W, Peng W and Xu Y: Vaspin attenuates the progression of
atherosclerosis by inhibiting ER stress-induced macrophage
apoptosis in apoE/mice. Mol Med Rep. 13:1509–1516. 2016. View Article : Google Scholar
|
|
29
|
National Research Council (US) Institute
for Laboratory Animal Research: Guidance for the description of
animal research in scientific publications. Washington (DC):
National Academies Press (US); 2011
|
|
30
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute Lung
Injury in Animals Study Group: An official American Thoracic
Society workshop report: features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Ryan D, Frohlich S and McLoughlin P:
Pulmonary vascular dysfunction in ARDS. Ann Intensive Care.
4:282014. View Article : Google Scholar
|
|
33
|
Shibata R, Ohashi K, Murohara T and Ouchi
N: The potential of adipokines as therapeutic agents for
cardiovascular disease. Cytokine Growth Factor Rev. 25:483–487.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu S, Dong Y, Wang T, Zhao S, Yang K,
Chen X and Zheng C: Vaspin inhibited proinflammatory cytokine
induced activation of nuclear factor-kappa B and its downstream
molecules in human endothelial EA.hy926 cells. Diabetes Res Clin
Pract. 103:482–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Taylor NJ and Shawcross DL:
Alpha1-antitrypsin deficiency. N Engl J Med. 361:2102author reply
2102. 2009.PubMed/NCBI
|
|
36
|
Tumen J, Meyrick B, Berry L Jr and Brigham
KL: Antiproteinases protect cultured lung endothelial cells from
endotoxin injury. J Appl Physiol (1985). 65:835–843. 1988.
|
|
37
|
Feng Y, Hu L, Xu Q, Yuan H, Ba L, He Y and
Che H: Cytoprotective role of alpha-1 antitrypsin in vascular
endothelial cell under hypoxia/reoxygenation condition. J
Cardiovasc Pharmacol. 66:96–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stockley RA: The multiple facets of
alpha-1-antitrypsin. Ann Transl Med. 3:1302015.PubMed/NCBI
|
|
39
|
Jie Z, Cai Y, Yang W, Jin M, Zhu W and Zhu
C: Protective effects of alpha 1-antitrypsin on acute lung injury
in rabbits induced by endotoxin. Chin Med J (Engl). 116:1678–1682.
2003.
|
|
40
|
Petrache I, Fijalkowska I, Medler TR,
Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR and Tuder RM:
α-1 antitrypsin inhibits caspase-3 activity, preventing lung
endothelial cell apoptosis. Am J Pathol. 169:1155–1166. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wewers MD, Herzyk DJ and Gadek JE:
Alveolar fluid neutrophil elastase activity in the adult
respiratory distress syndrome is complexed to
alpha-2-macroglobulin. J Clin Invest. 82:1260–1267. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gadek JE and Pacht ER: The interdependence
of lung antioxidants and antiprotease defense in ARDS. Chest.
110(Suppl 6): 273S–277S. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gando S, Kameue T, Matsuda N, Sawamura A,
Hayakawa M and Kato H: Systemic inflammation and disseminated
intravascular coagulation in early stage of ALI and ARDS: Role of
neutrophil and endothelial activation. Inflammation. 28:237–244.
2004. View Article : Google Scholar
|
|
44
|
Meduri GU, Annane D, Chrousos GP, Marik PE
and Sinclair SE: Activation and regulation of systemic inflammation
in ARDS: Rationale for prolonged glucocorticoid therapy. Chest.
136:1631–1643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Phalitakul S, Okada M, Hara Y and Yamawaki
H: A novel adipocytokine, vaspin inhibits platelet-derived growth
factor-BB-induced migration of vascular smooth muscle cells.
Biochem Biophys Res Commun. 423:844–849. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Phalitakul S, Okada M, Hara Y and Yamawaki
H: Vaspin prevents TNF-α-induced intracellular adhesion molecule-1
via inhibiting reactive oxygen species-dependent NF-κB and PKCθ
activation in cultured rat vascular smooth muscle cells. Pharmacol
Res. 64:493–500. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kása A, Csortos C and Verin AD:
Cytoskeletal mechanisms regulating vascular endothelial barrier
function in response to acute lung injury. Tissue Barriers.
3:e9744482015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367. 2006.
View Article : Google Scholar
|
|
49
|
Galani V, Tatsaki E, Bai M, Kitsoulis P,
Lekka M, Nakos G and Kanavaros P: The role of apoptosis in the
pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An
up-to-date cell-specific review. Pathol Res Pract. 206:145–150.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lang JD, McArdle PJ, O'Reilly PJ and
Matalon S: Oxidant-antioxidant balance in acute lung injury. Chest.
122(Suppl 6): 314S–320S. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tasaka S, Amaya F, Hashimoto S and
Ishizaka A: Roles of oxidants and redox signaling in the
pathogenesis of acute respiratory distress syndrome. Antioxid Redox
Signal. 10:739–753. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shiizaki S, Naguro I and Ichijo H:
Activation mechanisms of ASK1 in response to various stresses and
its significance in intracellular signaling. Adv Biol Regul.
53:135–144. 2013. View Article : Google Scholar
|
|
53
|
Matsuzawa A, Nishitoh H, Tobiume K, Takeda
K and Ichijo H: Physiological roles of ASK1-mediated signal
transduction in oxidative stress- and endoplasmic reticulum
stress-induced apoptosis: advanced findings from ASK1 knockout
mice. Antioxid Redox Signal. 4:415–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Manukyan MC, Weil BR, Wang Y, Abarbanell
AM, Herrmann JL, Poynter JA and Meldrum DR: The phosphoinositide-3
kinase survival signaling mechanism in sepsis. Shock. 34:442–449.
2010. View Article : Google Scholar : PubMed/NCBI
|