Introduction
Oral squamous cell carcinoma (OSCC) is the sixth
most widespread cancer with an annual estimated incidence of
263,000 worldwide, two-thirds of these cases occurring in
developing countries (1,2). Although its incidence rate is not
increasing in the general population of Western countries, an
increase in the number of OSCC cases has been reported in young
adults in the United States and in some countries of Europe
(3–5). The 5-year survival rate remains
<60% of diagnosed cases, depending on the stage of the tumor at
presentation, in addition, OSCC has devastating consequences
negatively affecting the patients quality of life (5). Therefore, developments of new
approaches to prevent OSCC occurrence and growth are highly
desirable.
Epidemiological studies have identified several
nutrients derived from plants with anticancer properties and, among
these, flavonoids, a family of polyphenolic compounds, have proved
to be effective in cancer prevention (6). Apigenin (Api) is a flavonoid widely
found in fruits such as oranges and grapefruits, vegetables and in
plant-derived beverages such as chamomile, tea and wine (7). Due to its low toxicity and its
biological activities, i.e. anti-inflammatory and anti-oxidant
effects, there has been increasing interest in the potential use of
Api as an anticancer agent (8).
Several studies have proved Api to be effective in inhibiting tumor
growth in vitro in several types of cancer cells including
breast, cervical, prostate and colorectal ones (9–12).
The in vivo effectiveness of Api oral administration has
been demonstrated against prostate cancer (13,14).
Moreover, the topical application of Api was found to counteract
skin carcinoma growth in mice (15). Although Api has been widely studied
the cellular mechanisms responsible of its beneficial effects are
still far from being completely understood and scarce information
is available regarding the use of Api in oral cancer (16,17).
Api anticancer action depends on various mechanisms that can vary
according to cell type and it involves apoptosis, modulation of the
cell cycle and alteration of kinase pathways (8). Therefore, the aim of the present
study is to investigate the anti-proliferative effect of Api on
OSCC cells, elucidating the underlying molecular mechanisms. To
this end we evaluated Api’s effects on tongue derived OSCC cell
lines, SCC-25 and on the spontaneously immortalized keratinocytes,
HaCaT.
Materials and methods
Cell lines and materials
SCC-25, derived from a human oral squamous cell
carcinoma of the tongue (obtained from ATCC, USA) were maintained
in DMEM/F12 (Euroclone, Pero, Italy) supplemented with 10% fetal
bovine serum (FBS) (Euroclone), 400 ng/ml hydrocortisone
(Sigma-Aldrich, St. Louis, MO, USA) and 1% penicillin/streptomycin
(Euroclone). HaCaT cells (obtained from German Cancer Research
Center, Heidelberg, Germany) were cultured in high glucose DMEM
(Euroclone) supplemented with 10% FBS and 1%
penicillin/streptomycin. Cells were maintained at 37°C in a
humidified incubator with 5% CO2. Apigenin
(Sigma-Aldrich) was dissolved in pure DMSO and subsequently diluted
in culture medium. DMSO concentration did not exceed 0.1% during
treatments.
MTT assay
The effect of Api on cell vitality was evaluated by
thiazolyl blue tetrazolium bromide (MTT) assay. Cells were
transferred to 96-well plates (5,000 cells/cm2) and were
incubated with Api for 24 and 48 hours (h). Subsequently, the
medium was removed and the cells were incubated with MTT
(Sigma-Aldrich). Four hours later, formazan crystals were dissolved
with DMSO and the absorbance was recorded at 560 nm in a microplate
reader (Bio-Rad, Hercules, CA, USA). The effect of Api on cell
survival was calculated by normalizing the data to the absorbance
of vehicle-treated cells (considered as 100%). The IC50
value was estimated.
Annexin V-propidium iodide assay
In order to evaluate Api-induced apoptosis, the FITC
conjugated Annexin V-propidium iodide (PI) apoptosis detection kit
(BD Biosciences, Buccinasco, Italy) was used according to the
manufacturer’s instructions. Briefly, the cells were plated in
6-well plates (5,000 cells/cm2) and treated for 24 h
with Api 100 μM. Both floating and attached cells were
harvested, washed with PBS and, finally, suspended in binding
buffer containing Annexin V and PI. After 15-min incubation in the
dark, the presence of apoptotic cells was analysed by the
FACSCantoI flow cytometer (BD Biosciences).
Analysis of cell cycle distribution
After Api treatment, both floating and attached
cells were harvested, washed twice with cold PBS, spun in a
refrigerated centrifuge at 200 g for 10 min and resuspended in cold
70% ethanol overnight (ON) at 4°C. The following day the cells were
washed in cold PBS and then incubated in 5 μg/ml PI and 12.5
μg/ml RNAse (both from Sigma-Aldrich) for 48 h. When the
cell synchronization was required, SCC-25 and HaCaT cells were
maintained in serum-free medium for 30 and 48 h, respectively, and
subsequently treated with Api in complete medium. DNA content
analysis was subsequently performed by means of a FACSCantoI flow
cytometer (BD Biosciences, Buccinasco, Italy) and ≥20,000 events
were analyzed. Cell cycle distribution was then analyzed by ModFit
LT (Verity House Software, Topsham, ME, USA).
Western blotting
The Api-treated and control cells were washed twice
in cold PBS and lysed in 50 mM HEPES, pH 7.5, 150 mM NaCl, 10%
glycerol, 1% Triton X-100, 1.5 mM MgCl2, 5 mM EGTA, 4 mM
phenylmethylsulfonyl fluoride, 1% aprotinin, 10 mM sodium
orthovanadate, 20 mM sodium pyrophosphate (all from Sigma-Aldrich).
The lysates were clarified by centrifugation at 13,000 g for 15 min
at 4°C and the total protein content was evaluated with a Coomassie
Protein Assay Reagent kit (Pierce, Rockford, IL, USA). Cell lysates
(50 μg), mixed with Laemmli buffer (5% β-mercaptoethanol,
10% SDS, 50% glycerol, 400 mM Tris-HCl (pH 6.8) and 0.5%
bromophenol blue) (all from Sigma-Aldrich), were resolved overnight
in a 13% SDS-PAGE and then electroblotted onto nitrocellulose
membranes (GE Healthcare Life Science, Milan, Italy).
Non-specific binding sites were blocked by 1 h room
temperature (RT) incubation in 5% non-fat milk in TBS (10 mM
Tris-HCl pH 8, 150 mM NaCl, 0.05% Tween-20). Subsequently,
membranes were incubated overnight with the following primary
antibodies diluted in TBS plus 5% non-fat milk: cyclin
D1, cyclin E (Santa Cruz Biotechnology, Santa Cruz, CA,
USA), Actin (Santa Cruz Biotechnology), cyclin-dependent kinase 1
(CDK1), phospho-CDK1 (Cell Signaling Technology, Danvers, CA, USA).
Unbound antibodies were removed by TBS washing and the membranes
were subsequently incubated with peroxidase-conjugated specific
secondary antibodies for 1 h at RT. Finally, the immune complexes
were visualized using an enhanced chemiluminescence (ECL) system
(Genespin Srl., Milan, Italy). After blot scanning, the
densitometric analysis of the bands was performed using the Gel
Logic 100 Imaging System (Eastman Kodak, Rochester, NY, USA).
Results were expressed as a relative optical density (OD)
considering the ratio between the optical density of the protein
band of interest and that of the corresponding actin band.
Statistical analysis
Data are reported as means ± standard deviation from
at least three independent experiments. The statistical evaluation
of the results was performed using GraphPad Prism 3 Software
(GraphPad Softwar Inc., La Jolla, CA, USA). The differences between
controls and treated cell MTT results were analyzed by the one-way
ANOVA analysis of variance followed by Dunnett’s multiple
comparison test. Differences in the percentages of apoptotic cells
in the Annexin V-PI assay and differences in the cell cycle
distribution between controls and treated cells were analyzed by
Student’s t-test. Statistical significance was set at
p<0.05.
Results
Apigenin treatment inhibits cell
proliferation
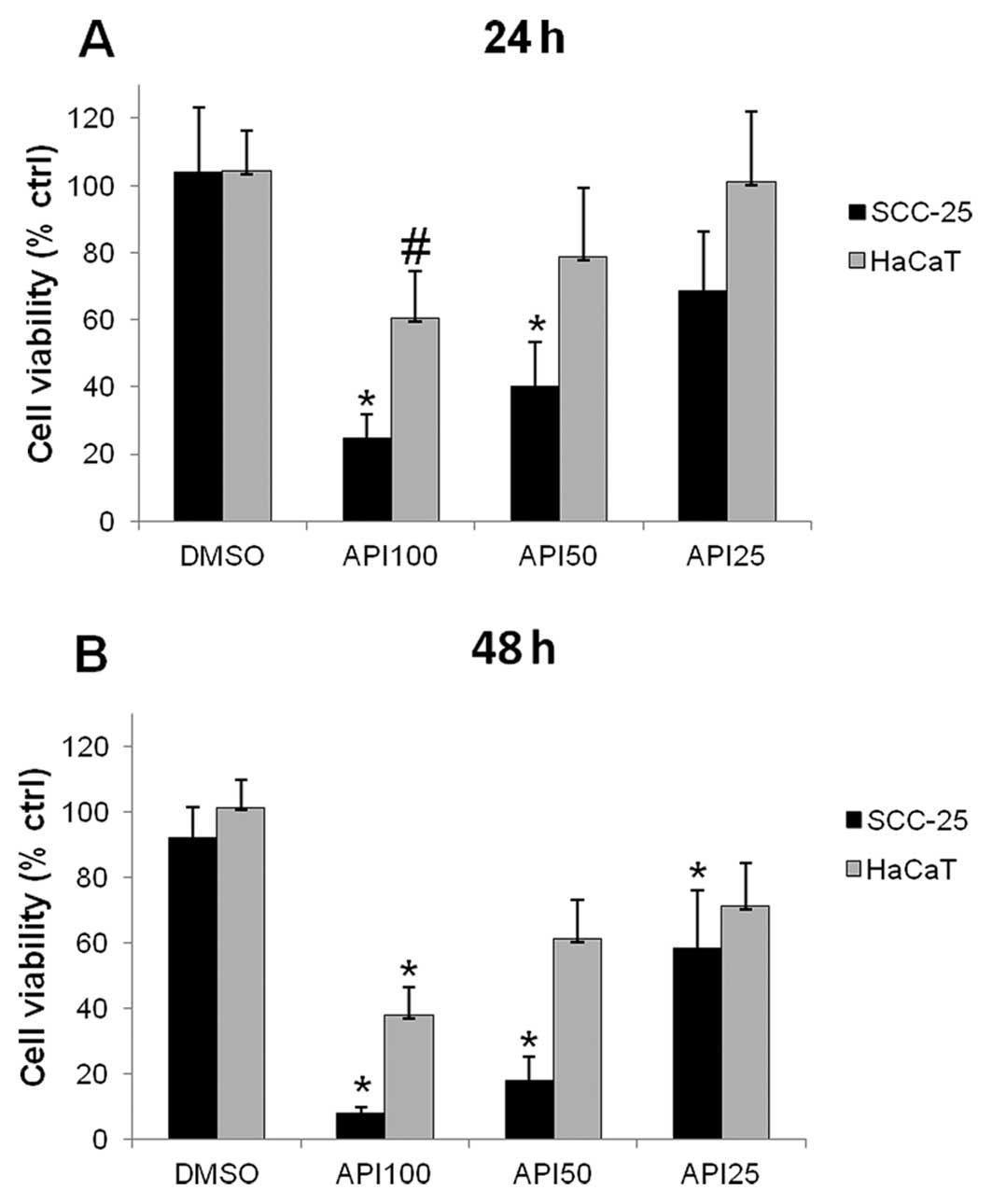
The effect of different Api concentrations on cell
survival was evaluated using MTT assay. The data obtained showed a
dose- and time-dependent decrease in cell vitality in both HaCaT
and SCC-25 cells (Fig. 1).
As can be observed in the graph of Fig. 1A, the highest doses of Api used
induced a marked, statistically significant, decrease in SCC-25
cells survival after 24-h treatment, in particular Api 50 and 100
μM reduced cell viability to values <50% with respect to
untreated controls. The Api-induced decrease in cell viability was
observed also in HaCaT cells; however, the effect of Api in these
cells was milder than in SCC-25 cells, in fact, only Api 100
μM significantly affected HaCaT cell viability.
After 48-h treatment all the tested doses
statistically significantly reduced SCC-25 cells viability, in
particular a dramatic drop in cell vitality to 18.1±7.% and
7.9±2.0% was observed after 48-h treatment with 50 and 100
μM Api, respectively. The Api-induced decrease in cell
viability with respect to vehicle-treated cells was observed also
in HaCaT cells; however, the effect of Api in these cells was
milder than in SCC-25 cells, in fact, only Api 100 μM
reduced HaCat cell viability below 50% (Fig. 1B).
Apigenin induces apoptosis in SCC-25
cells
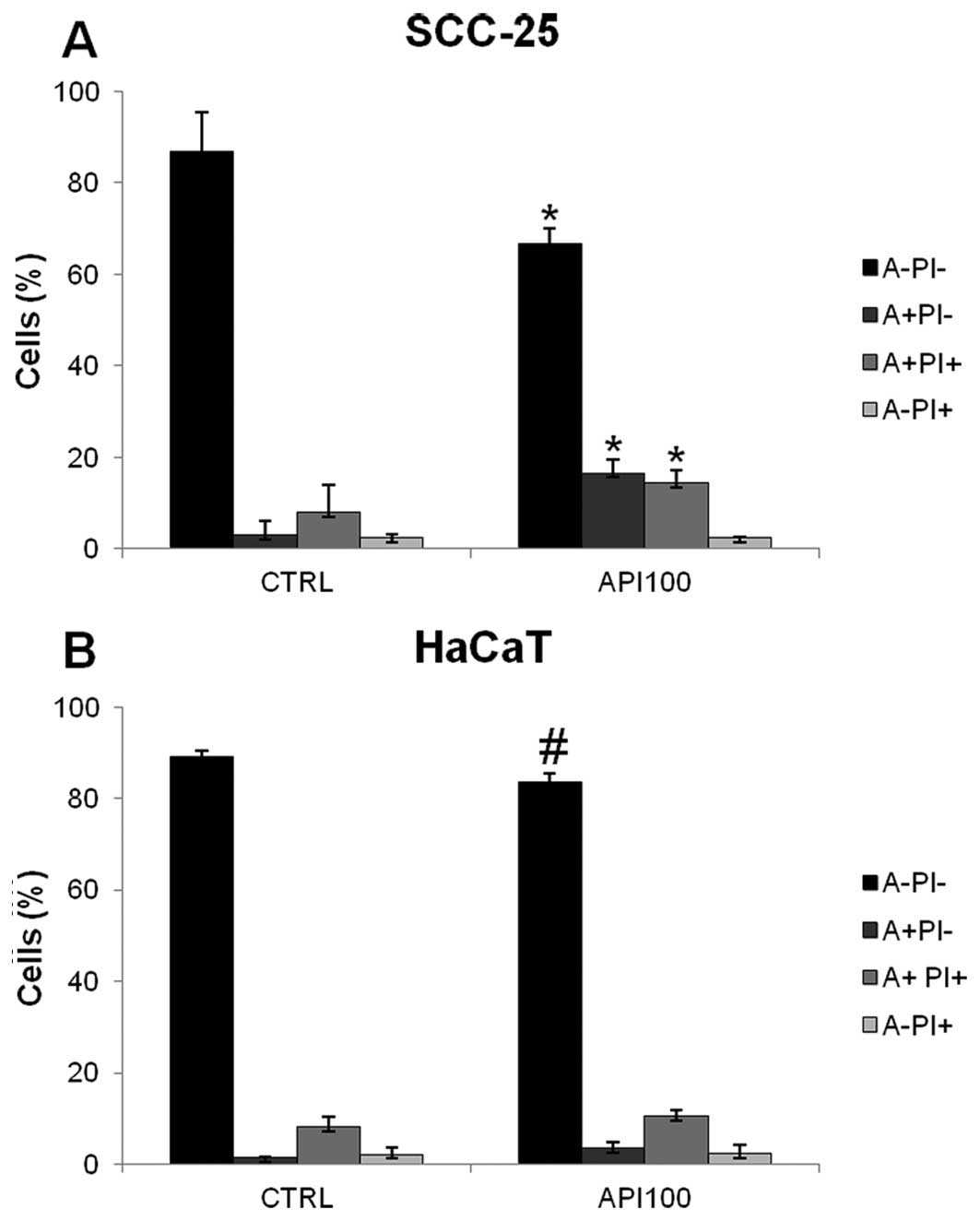
To assess if the Api-induced decrease in cell
survival was related to apoptosis, we performed a flow cytometric
analysis of the membrane translocation of phosphatidyl serine with
FITC-conjugated Annexin V (A), combined with PI DNA staining (PI).
As shown in Fig. 2A, 24 h exposure
to 100 μM Api significantly increased the percentage of
apoptotic SCC-25 cells (A+/PI−). In
particular, the population of cells showing
A+/PI− increased from <1.47±0.78% in the
controls to 19.8±4.1% in Api-treated cells (p<0.01), whereas the
percentage of cells in late apoptosis, being positive for both
Annexin V and PI staining (A+/PI+), increased
from 4.73±0.74 in the controls to 14.6±2.05% in Api-treated cells
(p<0.01) (Fig. 2A). On the
contrary, 100 μM Api treatment induced only a slight,
statistically insignificant, increase in the percentage of early
and late apoptotic cell populations in HaCaT cells (Fig. 2B).
Cell cycle alteration induced by
apigenin
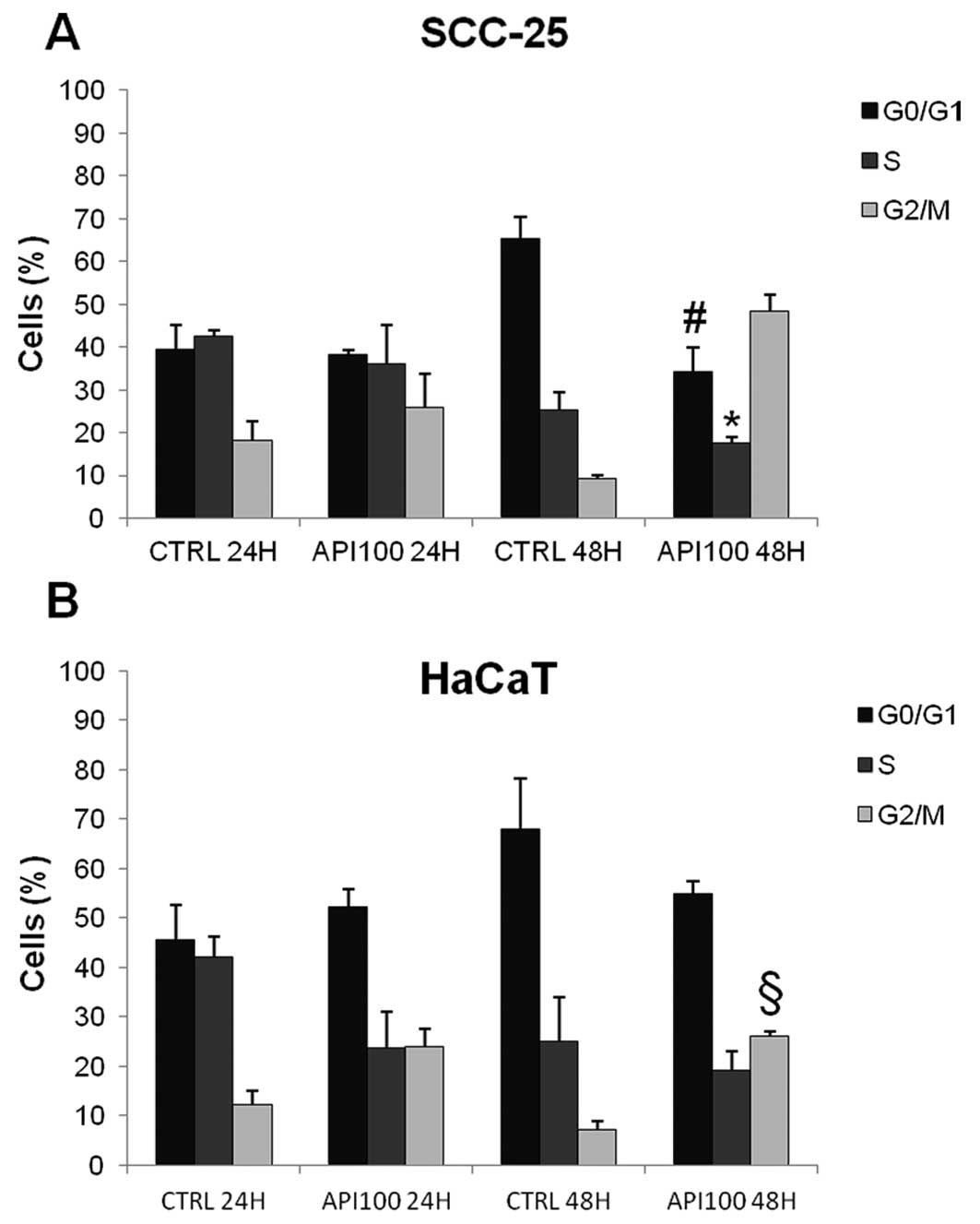
We aimed at correlating the Api-induced inhibition
of cell growth with an alteration of the cell cycle progression.
Therefore, we performed cell cycle analysis in cells treated with
the most effective dose of Api (100 μM) for 24 and 48 h.
An increase in the percentage of cells in the
G2/M phase in Api-treated cells was observed in both
HaCaT and SCC-25 cells compared with untreated controls. This
increase was associated with a slight, but statistically
significant, decrease in the percentage of cells in both
G0/G1 and S phase after 48 h exposure. The
modulation of SCC-25 cell cycle distribution due to Api, although
already notable after 24 h Api exposure, became statistically
significant after 48 h (Fig. 3A),
so that Api 100 μM treatment resulted in 48.2±4% of cells in
G2/M, versus the 9.3±0.8% observed in controls (Fig. 3A). In HaCaT cells, 48 h Api
treatment resulted in an increase in the G2/M population
from 7.1±1.7 in controls to 26.1±1 % in treated cells (p<0.001)
(Fig. 3B).
Despite the increase in the G2/M
population, there was a considerable number of cells still in the
G0/G1 phase, thus suggesting that another
checkpoint might be involved in Api-induced modulation of cell
cycle progression. In an attempt to better clarify the effect of
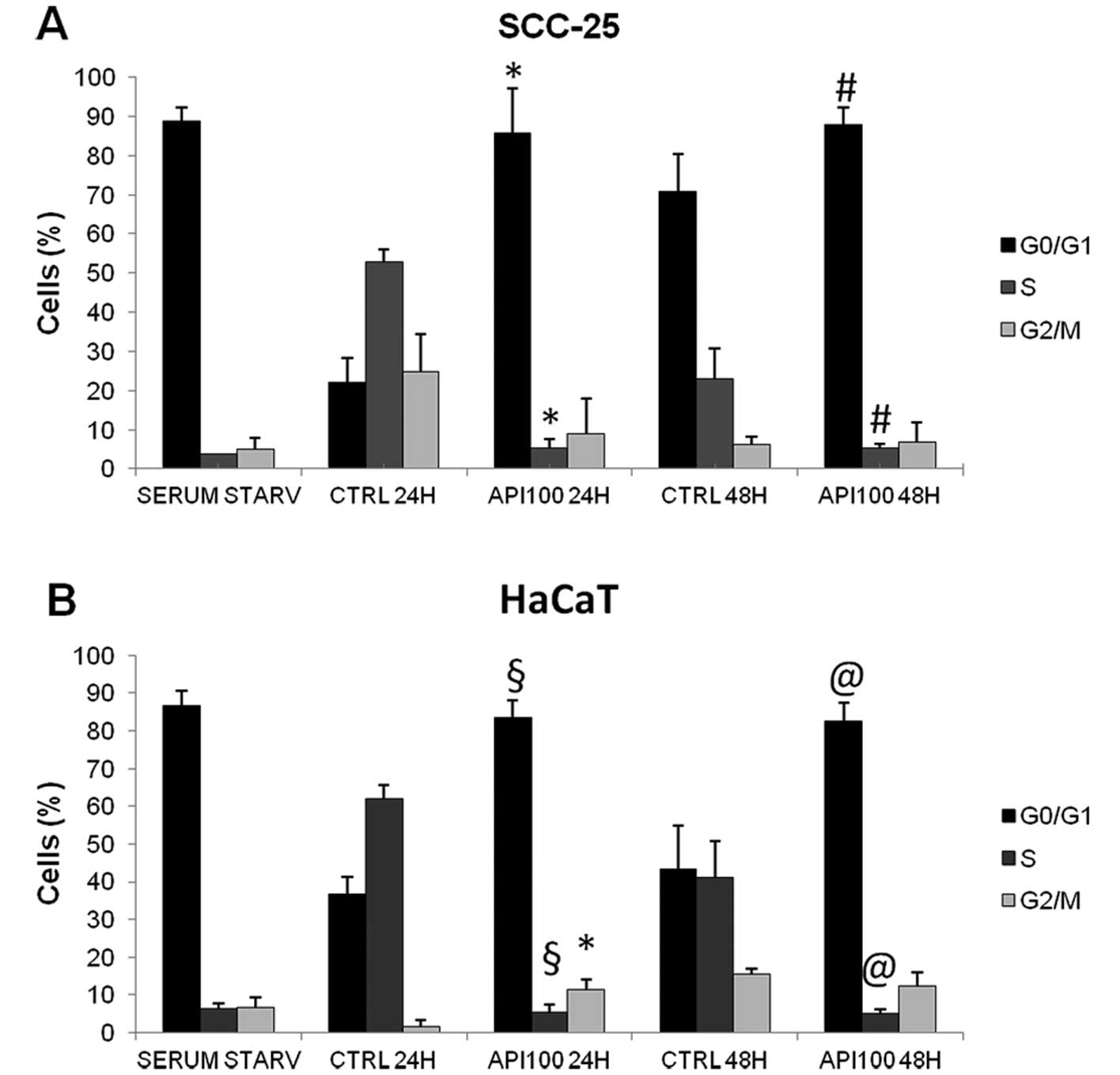
Api on cell cycle distribution, we analyzed the effect of 100
μM Api on synchronized cells. For this purpose, SCC-25 and
HaCaT cells were maintained in serum-free medium for 30 and 48 h,
respectively, then cells were allowed to re-enter the cell cycle by
culturing them in complete medium. As shown in Fig. 4, we obtained satisfactory
synchronization, with almost 90% of cells being in the
G0/G1 phase following serum starvation. Api
treatment on synchronized HaCaT and SCC-25 cells resulted in the
maintainance of cells into the G0/G1 phase,
in fact, while in untreated controls the majority of cells after 24
h were in the S phase as a result of cycle re-entering after serum
starvation, Api-treated cells maintained the same distribution of
serum starvation synchronized cells both after 24 and 48 h of
treatment (Fig. 4). In
synchronized SCC-25 cells, the exposure to 100 μM Api for 24
h resulted in 85.7±11.5% of cells being in
G0/G1 (p<0.01 versus controls) and a very
similar effect was observed in HaCaT cells. The blockage of cells
in G0/G1 persisted also after 48 h treatment.
Despite this, the blockage at G2/M transition was still
evident in HaCaT cells since a slight, but statistically
significant (p<0.01), increase in the percentage of cells in
G2/M was found in 24 h Api-treated cells when compared
to controls.
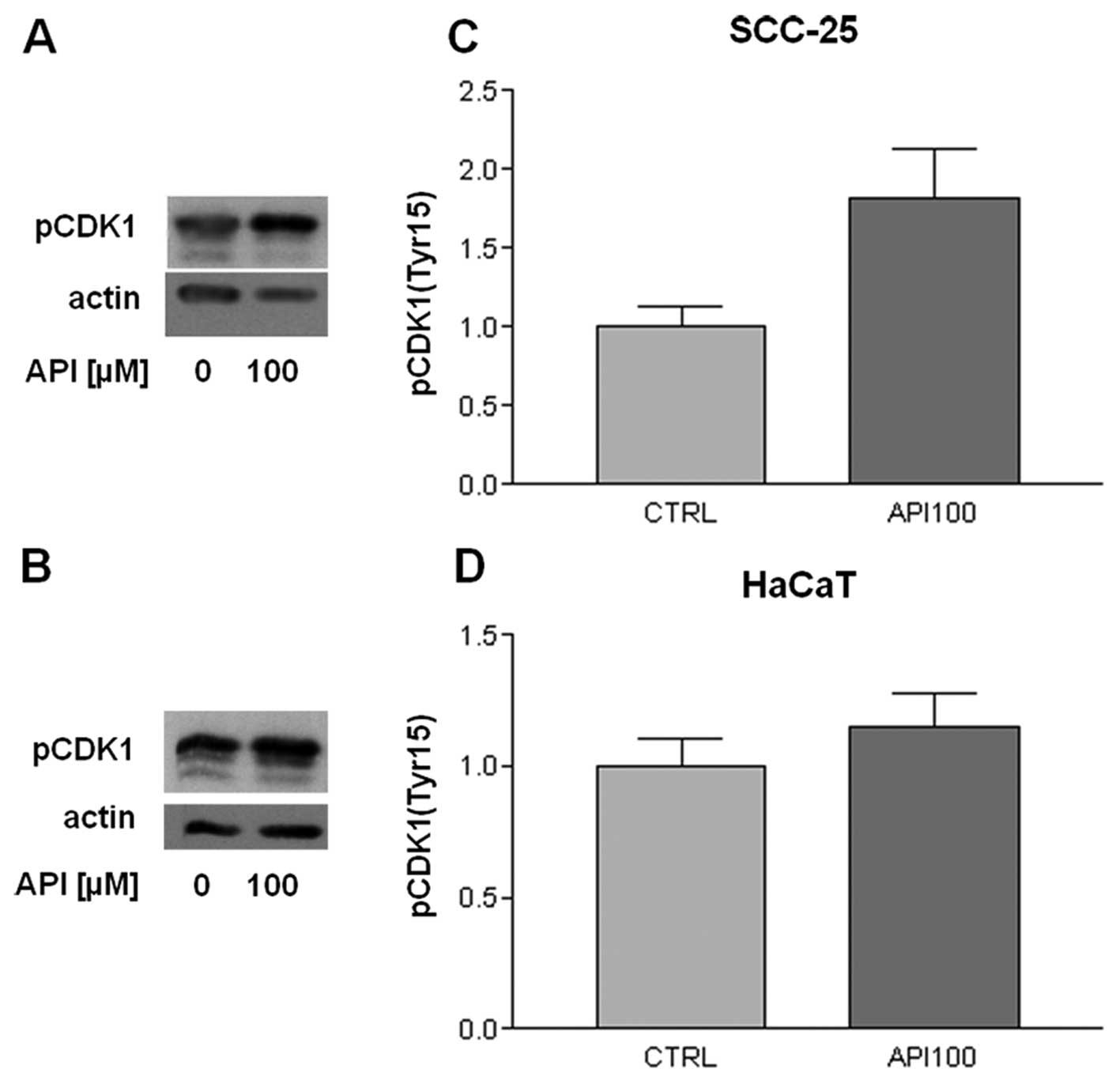
Apigenin inactivates CDK1 in SCC-25
cells
In order to address in more detail the effect of Api
on the cell cycle, we evaluated the expression of some cyclins and
the associated CDKs that regulate both G0/G1
and G2/M phase transition.
Therefore, we considered the protein level of cyclin
B1, which is involved in G2/M transition, in
unsynchronized cells. However, no significant modulations of its
expression by Api were evidenced either in SCC-25 or in HaCaT cell
lines (data not shown). We subsequently evaluated the level of the
CDK1, also known as p34cdc2, which is functionally
associated with cyclin B1. Even in this case we did not observe any
variation/modulation between treated and control cells.
Nevertheless the regulation of CDK1 is quite complex; in fact, it
is strictly regulated by phosphorylation so that its enzimatic
activity is inhibited by phosphorylation on the tyrosine in
position 15. Therefore, we evaluated the level of the tyrosine 15
phosphorylated form of CDK1. As shown in Fig. 5, a marked, although not
statistically significant, increase in tyrosine 15 phosphorylated
CDK1 was observed in SCC-25 cells following 100 μM Api
treatment for 24 h (Fig. 5C),
whereas Api treatment did not modulate the level of tyrosine 15
phosphorylation in HaCaT cells (Fig.
5D).
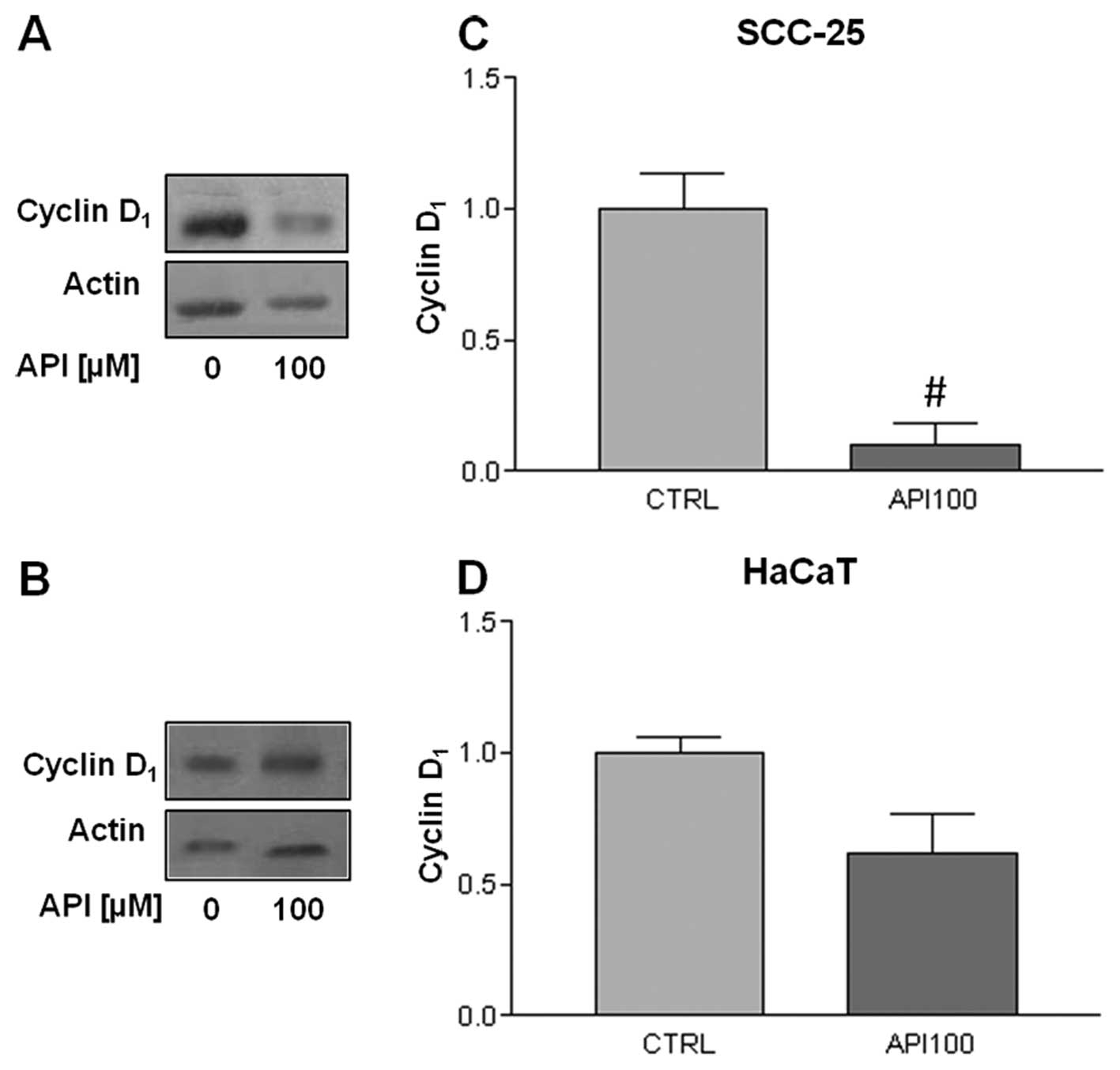
Apigenin modulates cyclin D1
and E expression
In view of the different Api action on the cell
cycle in synchronized versus asynchronous cells, we analyzed also
the cyclins and CDKs involved in the regulation of
G0/G1 transition in synchronized cells.
First, we evaluated the expression of cyclin D1, which
drives the progression through the G1 phase of cell
cycle, by exposing synchronized cells to 100 μM Api for 24
h. As can be seen in Fig. 6, Api
treatment modulated cyclin D1 expression in both cell
lines, with a statistically significant decrease being observed in
SCC-25 cells (Fig. 6C), while the
decrease in cyclin D1 was slight in HaCaT cells
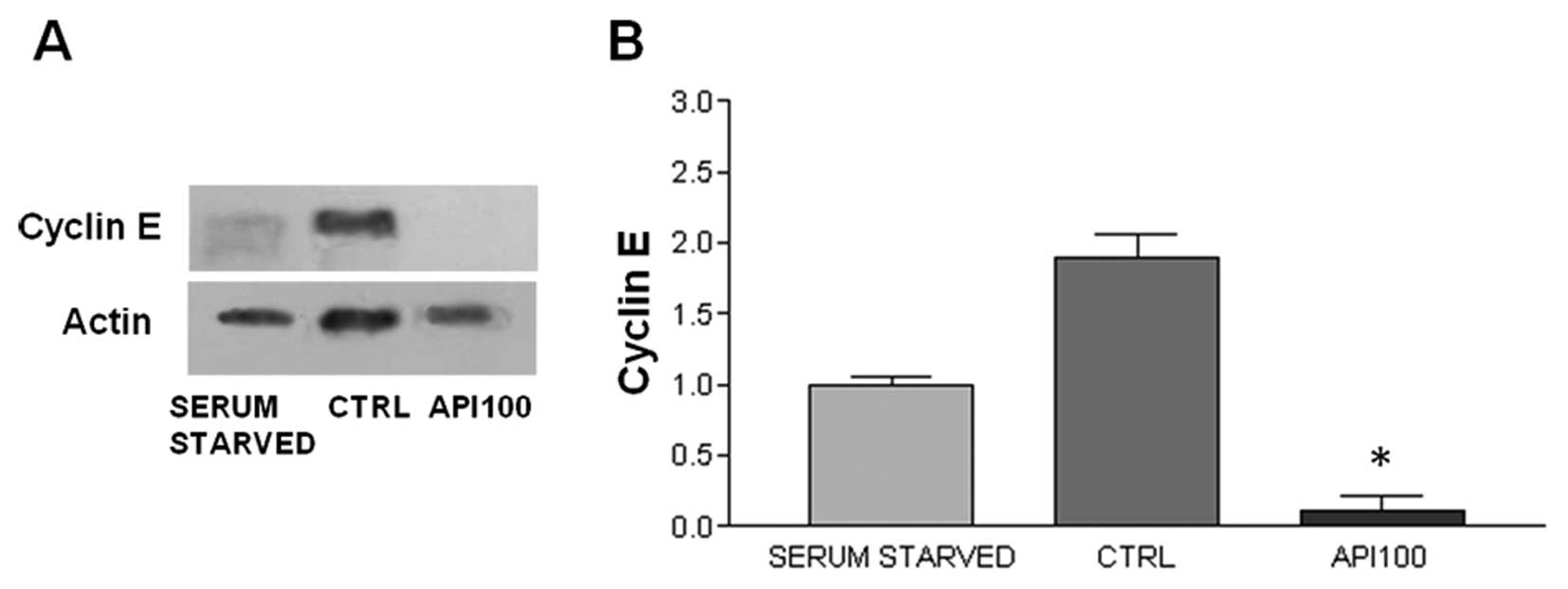
(Fig. 6B and D). Secondly, we
evaluated the Api effect on the expression of cyclin E, whose
levels are generally high during the mid G1 phase and
which is involved in the regulation of G1/S transition.
We did not find any cyclin E expression in SCC-25 cells (data not
shown). On the contrary, a statistically significant decrease in
HaCaT cells after Api treatment was found. The level of cyclin E
expression significantly dropped after 24 h Api treatment in HaCaT
cells (Fig. 7).
Discussion
Apigenin, a flavonoid belonging to the class of
flavones has proved its effectiveness against various cancer types
(8–13), but up to now scarce data are
available regarding the Api effects and molecular mechanisms on
OSCC.
In the present study, we aimed at evaluating Api’s
action on OSCC using the cell line SCC-25, derived from tongue
OSCC. In addition, Api’s effect was analyzed also on a
spontaneously immortalized, non-tumorigenic, keratinocyte cell
line: HaCaT cells. These cell lines have been widely used and they
represent a valid model for studying the action of potential
chemotherapeutic agents (18,19).
In addition, HaCaT cells may be considered a good model of the
initial transformation of squamous cell carcinoma. In fact,
although they can be referred to as normal keratinocytes, they show
some molecular alterations, found in the first steps of malignant
transformation of epithelial cells, such as the mutations in both
p53 alleles (20).
We demonstrated that Api inhibits cancer cell growth
in vitro; it was able to significantly decrease cell
survival in SCC-25 cells. In our paradigm, the SCC-25 cells were
much more sensitive to Api than immortalized keratinocytes (HaCaT).
In agreement with our findings a selective growth inhibitory effect
of Api in prostate cancer cells when compared with non-transformed
cells was reported (9). Masuelli
and colleagues (17) also reported
a comparable proliferation inhibition in SCC-15, OSCC derived
cells.
Consistent with data reported in breast (10) and in lung cancer (21), the analysis of the phosphatidyl
serine membrane translocation, an early apoptotic marker, showed
that Api induced apoptosis in SCC-25 cells. Apoptosis was not
detected in HaCaT cells, thus confirming a selective cytotoxic
action of Api towards tumoral SCC-25 cells. The absence of
apoptosis in HaCaT cells is not in agreement with previously
reported data from Abu-Yousif et al (22), where the exposure of HaCaT cells to
Api led to apoptosis in almost 15% of cells. One possible reason
for this difference might be that in the study of Abu-Yousif et
al Api treatment was conducted in serum-free medium, while in
the present study HaCaT cells were maintained in complete medium
during evaluation of Api cytotoxicity. However, the ratio of
apoptotic cells alone was not enough to explain the decrease in
cell viability observed in SCC-25 cells following Api treatment.
Furthermore, the absence of apoptosis in HaCaT cells, whose
proliferation was inhibited by Api, led us to hypothesize that
mechanisms other than apoptosis had a role in the cell number
decrease observed after Api treatment. We therefore investigated
whether the impairment of cell growth was associated with
alterations in the cell cycle progression. The analysis of cell
cycle distribution showed a marked increase in the percentage of
cells in the G2/M phase after Api treatment in both cell
lines. These results are in agreement with data reported by Ujiki
et al (23) and Choi and
Kim (10) that demonstrated an
Api-induced G2/M arrest in pancreatic and breast cancer
cells, respectively. In addition, an increase in the
G2/M population after Api exposure was observed also in
immortalized keratinocytes (24,25)
and in primary cultures of OSCC (16). On the contrary, our results differ
from the data reported by Masuelli and co-workers (17). Although they reported an
Api-induced decrease in cell survival similar to our findings, they
found a significant decrease in cells in both G1 and
G2/M phases after Api exposure in SCC-15. The main
regulator of G2/M progression is the CDK1/cyclin B
complex whose activity is required for cells to enter mitosis. In
our paradigm, Api induced only a slight modulation of the level of
cyclin B and CDK1 (data not shown). However, CDK1 activity is also
regulated by phosphorylation and dephosphorylation mediated by wee1
and cdc25 and, in particular dephosphorylation of CDK1 at the
tyrosine 15 site is required for its activity (26). Api treatment induced a significant
increase in the level of tyrosine 15 phosphorylated CDK1 in SCC-25
cells, thus indicating that Api can inactivate this CDK. Similar
results were obtained in human melanoma cells (27). Api’s ability to inactivate CDK1
through phosphorylation on tyrosine 15 sites was demonstrated also
by McVean et al (25) in
epidermal keratinocytes. On the contrary, HaCaT cells did not show
any modulation of CDK1 phosphorylation after Api treatment, thus
indicating that Api induces G2/M arrest through of
different mechanisms in different cell types.
Even after 48 h Api exposure, a relatively large
percentage of cells still remained in the
G0/G1 phase leading us to hypothesize that
Api could also impair G1 progression. Api treatment in
synchronized cells resulted in an almost complete
G0/G1 arrest in both the cell lines. These
data are consistent with results by Lepley and Pelling (28) who reported a G1
accumulation in synchronized skin fibroblasts exposed to Api, while
unsynchronized cells were arrested in G2/M. Western blot
analysis demonstrated that Api-induced G0/G1
arrest depended on different pathways in SCC-25 and HaCaT cells. A
statistically significant decrease in the expression of the cyclin
E cell was found in HaCaT, whereas we found no expression of this
cyclin in SCC-25 cells, although cyclin E overexpression is often
found in oral cancer (29).
However, the loss of cyclin E expression has been reported in two
other tongue-derived SCC cell lines (30). It is, therefore, conceivable that
cell growth deregulation in SCC-25 cells might depend on the loss
of cyclin E. These observations suggest that Api has a double
action on the cell cycle, inducing both a
G0/G1 and a G2/M arrest,
accompanied by a reduction in cyclin D1 and cyclin E
expression and inactivation of CDK1.
In conclusion, we demonstrated an inhibitory effect
on cell survival and apoptotic effect of Api in oral cancer cells.
The model of action we hypothesize implies the induction of
apoptosis in SCC-25 cells by Api, which also determines the cell
cycle arrest acting as a CDK1 inhibitor and inducing a decrease in
cyclin D1 expression.
Therefore, it appears that Api deserves to be
further studied as a potential agent for oral cancer prevention and
treatment since it is able to induce cell cycle arrest and
apoptosis in OSCC cells. In addition, Api seems to be a very
promising cell cycle regulating agent since it is able to induce a
double cell cycle arrest in different phases. It could, therefore,
be capable of arresting cell cycle progression also in cells, such
as SCC-25 where the cycle regulation machinery is deregulated but
not completely compromised.
References
|
1.
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics. CA Cancer J Clin. 60:277–300. 2010.
|
|
2.
|
Garavello W, Bertuccio P, Levi F, Lucchini
F, Bosetti C, Malvezzi M, Negri E and La Vecchia C: The oral cancer
epidemic in central and eastern Europe. Int J Cancer. 127:160–171.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Shiboski CH, Schmidt BL and Jordan RC:
Tongue and tonsilar carcinoma: increasing trends in the US
population ages 20–44 years. Cancer. 103:1843–1849. 2005.PubMed/NCBI
|
|
4.
|
Bonifazi M, Malvezzi M, Bertuccio P,
Edefonti V, Garavello W, Levi F, La Vecchia C and Negri E:
Age-period-cohort analysis of oral cancer mortality in Europe: the
end of an epidemic? Oral Oncol. 47:400–407. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Warnakulasuriya S: Living with oral
cancer: epidemiology with particular reference to prevalence and
life-style changes that influence survival. Oral Oncol. 46:407–410.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Rossi M, Garavello W, Talamini R, Negri E,
Bosetti C, Dal Maso L, Lagiou P, Tavani A, Polesel J, Barzan L,
Ramazzotti V, Franceschi S and La Vecchia C: Flavonoids and the
risk of oral and pharyngeal cancer: a case-control study from
Italy. Cancer Epidemiol Biomarkers Prev. 16:1621–1625. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Shukla S and Gupta S: Apigenin: a
promising molecule for cancer prevention. Pharm Res. 27:962–978.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Patel D, Shukla S and Gupta S: Apigenin
and cancer chemoprevention: progress, potential and promise
(Review). Int J Oncol. 30:233–245. 2007.PubMed/NCBI
|
|
9.
|
Gupta S, Afaq F and Mukhtar H: Selective
growth-inhibitory, cell-cycle deregulatory and apoptotic response
of apigenin in normal versus human prostate carcinoma cells.
Biochem Biophys Res Commun. 287:914–920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Choi EJ and Kim GH: Apigenin induces
apoptosis through a mtochondria/caspase-pathway in human breast
cancer MDA-MB-453 cells. J Clin Biochem Nutr. 44:260–265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhong Y, Krisanapun C, Lee SH, Nualsanit
T, Sams C, Peungvicha P and Baek SJ: Molecular targets of apigenin
in colorectal cancer cells: involvement of p21, NAG-1 and p53. Eur
J Cancer. 46:3365–3374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Xu Y, Xin Y, Diao Y, Lu C, Fu J, Luo L and
Yin Z: Synergistic effects of apigenin and paclitaxel on apoptosis
of cancer cells. PLoS One. 6:e291692011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kaur P, Shukla S and Gupta S: Plant
flavonoid apigenin inactivates Akt to trigger apoptosis in human
prostate cancer: an in vitro and in vivo study. Carcinogenesis.
29:2210–2217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Pandey M, Kaur P, Shukla S, Abbas A, Fu P
and Gupta S: Plant flavone apigenin inhibits HDAC and remodels
chromatin to induce growth arrest and apoptosis in human prostate
cancer cells: in vitro and in vivo study. Mol Carcinog. 51:952–962.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Wei H, Tye L, Bresnick E and Birt DF:
Inhibitory effect of apigenin, a plant flavonoid, on epidermal
ornithine decarboxylase and skin tumor promotion in mice. Cancer
Res. 50:499–502. 1990.PubMed/NCBI
|
|
16.
|
O’Prey J, Brown J, Fleming J and Harrison
PR: Effects of dietary flavonoids on major signal transduction
pathways in human epithelial cells. Biochem Pharmacol.
66:2075–2088. 2003.
|
|
17.
|
Masuelli L, Marzocchella L, Quaranta A,
Palumbo C, Pompa G, Izzi V, Canini A, Modesti A, Galvano F and Bei
R: Apigenin induces apoptosis and impairs head and neck carcinomas
EGFR/ErbB2 signaling. Front Biosci. 16:1060–1068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Scott RE, Wilke MS, Wille JJ Jr, Pittelkow
MR, Hsu BM and Kasperbauer JL: Human squamous carcinoma cells
express complex defects in the control of proliferation and
differentiation. Am J Pathol. 133:374–380. 1988.PubMed/NCBI
|
|
19.
|
Boukamp P, Petrussevska RT, Breitkreutz D,
Hornung J, Markham A and Fusenig NE: Normal keratinization in a
spontaneously immortalized aneuploid human keratinocyte cell line.
J Cell Biol. 106:761–771. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Komissarova EV and Rossman TG: Arsenite
induced poly(ADP-ribosyl)ation of tumor suppressor P53 in human
skin keratinocytes as a possible mechanism for carcinogenesis
associated with arsenic exposure. Toxicol Appl Pharmacol.
243:399–404. 2010. View Article : Google Scholar
|
|
21.
|
Lu HF, Chie YJ, Yang MS, Lee CS, Fu JJ,
Yang JS, Tan TW, Wu SH, Ma YS, Ip SW and Chung JG: Apigenin induces
caspase-dependent apoptosis in human lung cancer A549 cells through
Bax- and Bcl-2-triggered mitochondrial pathway. Int J Oncol.
36:1477–1484. 2010.PubMed/NCBI
|
|
22.
|
Abu-Yousif AO, Smith KA, Getsios S, Green
KJ, Van Dross RT and Pelling JC: Enhancement of UVB-induced
apoptosis by apigenin in human keratinocytes and organotypic
keratinocyte cultures. Cancer Res. 68:3057–3065. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ujiki MB, Ding XZ, Salabat MR, Bentrem DJ,
Golkar L, Milam B, Talamonti MS, Bell RH Jr, Iwamura T and Adrian
TE: Apigenin inhibits pancreatic cancer cell proliferation through
G2/M cell cycle arrest. Mol Cancer. 5:762006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lepley DM, Li B, Birt DF and Pelling JC:
The chemopreventive flavonoid apigenin induces G2/M arrest in
keratinocytes. Carcinogenesis. 17:2367–2375. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
McVean M, Weinberg WC and Pelling JC: A
p21(waf1)-independent pathway for inhibitory phosphorylation of
cyclin-dependent kinase p34(cdc2) and concomitant G(2)/M arrest by
the chemopreventive flavonoid apigenin. Mol Carcinog. 33:36–43.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Parker LL and Piwnica-Worms H:
Inactivation of the p34cdc2-cyclin B complex by the human WEE1
tyrosine kinase. Science. 257:1955–1957. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Casagrande F and Darbon JM: Effects of
structurally related flavonoids on cell cycle progression of human
melanoma cells: regulation of cyclin-dependent kinases CDK2 and
CDK1. Biochem Pharmacol. 61:1205–1215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lepley DM and Pelling JC: Induction of
p21/WAF1 and G1 cell-cycle arrest by the chemopreventive agent
apigenin. Mol Carcinog. 19:74–82. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Shintani S, Mihara M, Nakahara Y, Kiyota
A, Ueyama Y, Matsumura T and Wong DT: Expression of cell cycle
control proteins in normal epithelium, premalignant and malignant
lesions of oral cavity. Oral Oncol. 38:235–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Yamada S, Sumrejkanchanakij P, Amagasa T
and Ikeda MA: Loss of cyclin E requirement in cell growth of an
oral squamous cell carcinoma cell line implies deregulation of its
downstream pathway. Int J Cancer. 111:17–22. 2004. View Article : Google Scholar : PubMed/NCBI
|