Introduction
Glioblastoma multiforme, the most common primary
brain tumor in adults, is one of the deadliest of all human cancers
with a median survival of <2 years at best, even with the
current standard of care consisting of maximal surgical resection
followed by radiotherapy with concomitant and adjuvant temozolomide
(1,2). Early recurrence, which eventually
becomes fatal, is inevitable even after successful initial
treatment of glioblastoma, for which cancer stem cells are now
deemed responsible (3,4). Cancer stem cells are a small
subpopulation of cancer cells comprising a tumor that possess stem
cell-like properties as well as the capacity to initiate a tumor
and are often associated with resistance against conventional
chemo- and radiotherapies (5,6).
Elimination of the cancer stem cell population by overcoming their
therapy resistance, therefore, is considered a key to successful
management of glioblastoma as well as to realization of long-term
survival of patients with this devastating disease. Although it
still remains an open question whether or not cancer stem cells of
glioblastoma are invariably resistant to temozolomide, current
literature unanimously indicates that they often express
O6-methylguanine DNA methyltransferase (MGMT) and that
MGMT expression is associated with high resistance to temozolomide
(7). MGMT is a repair enzyme that
rapidly removes the methyl group attached by temozolomide at the
O6 position of the guanine residue. As such, MGMT
expression is considered to be a major mechanism for temozolomide
resistance of glioblastoma, which has indeed been supported by a
wealth of data from both clinical and basic studies (8–12).
Thus, the accumulating evidence supports the idea that elucidating
how MGMT expression is regulated in cancer stem cells of
glioblastoma would be a valuable strategy to control its expression
and, by so doing, render them susceptible to temozolomide
treatment.
In the present study, we dissected the molecular
mechanism of temozolomide resistance using glioblastoma cells
having cancer stem cell properties (stem-like glioblastoma cells)
that were derived directly from glioblastoma patients. We found
that the JNK pathway is critically involved in the temozolomide
resistance and MGMT expression of stem-like glioblastoma cells that
express MGMT. JNK inhibition failed to sensitize stem-like
glioblastoma cells without MGMT expression, suggesting that JNK may
contribute to temozolomide resistance of stem-like glioblastoma
cells in an MGMT expression-dependent manner.
Materials and methods
Reagents and antibodies
SP600125 and temozolomide were purchased from
Calbiochem (La Jolla, CA, USA) and LKT Laboratories, Inc. (St.
Paul, MN, USA), respectively, and were dissolved in
dimethylsulfoxide (DMSO) to prepare 50 mM stock solutions.
Anti-c-Jun (no. 9165) and anti-phospho-c-Jun (Ser 63) (no. 9261)
antibodies were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Anti-β-actin (A1978) was from Sigma (St. Louis,
MO, USA). Anti-JNK1 (sc-474) and anti-JNK2 (sc-7345) were from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-MGMT
(ab39253) was purchased from Abcam (Cambridge, UK).
Cell culture
Patient-derived stem-like glioblastoma cells used in
this study (GS-Y01, GS-Y03, GS-NCC01 and TGS01) were directly
established from glioblastoma tissues in accordance with protocols
approved by the Institutional Review Boards of the institutions
where they were established. The establishment and characterization
of the stem-like glioblastoma cells have been described (13–15).
The cells were maintained under the monolayer stem cell culture
condition reported previously (14,15).
Temozolomide treatment and live/dead cell
assays
Throughout this study, for temozolomide treatment,
cells were exposed to 50 μM temozolomide for 4 h, after
which they were cultured in the absence of temozolomide until being
subjected to analyses. In each set of treatment, cells were
examined for their viability at the end of pretreatment (i.e.,
SP600125 treatment and/or knockdown of JNK and/or MGMT), and an
equal number of viable cells were used for subsequent
temozolomide/control treatment. Live and dead cells were identified
by their ability and inability to exclude vital dyes [trypan blue
and propidium iodide (PI)], respectively. Unless otherwise
specified, both adherent and non-adherent cells in the dishes were
collected, and after centrifugation, re-suspended in
phosphate-buffered saline (PBS). The cell suspension was then mixed
with an equal volume of PBS containing trypan blue (0.4% w/v, final
concentration, 0.2%) and examined under a phase-contrast microscope
using a hemocytometer. The percentage of dead cells was defined as
100 × the number of dead cells / (the number of viable cells + the
number of dead cells). Alternatively, cells were incubated in
situ with PI (1 μg/ml) and Hoechst 33342 (10
μg/ml) for 10 min at 37°C in the CO2 incubator,
to stain dead cells and the cell nuclei, respectively. Then the
numbers of PI- and Hoechst-positive cells were scored under a
fluorescence microscope (CKX41; Olympus, Tokyo, Japan), and the
percentage of PI-positive cells (dead cells) against
Hoechst-positive cells (total cells) was determined.
Colony formation assay
For colony formation, cells were seeded at a low,
colony-forming density (5×103 cells per 60-mm dish) and
cultured for ∼4 weeks. The cells were then fixed with formaldehyde
(4% v/v) followed by staining with crystal violet (0.1% w/v).
Colonies [consisting of 8 or more cells (progenies) derived from a
single cell] were counted using a microscope.
Gene silencing by siRNA
siRNAs against human JNK1 (HSS108547), JNK2
(HSS108550), MGMT (HSS106519) and Stealth RNAi™ siRNA
negative control duplexes (Medium GC Duplexes no. 2) were purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA). Transfection
of siRNAs was performed using Lipofectamine RNAiMAX (Invitrogen
Life Technologies) according to the manufacturer’s
instructions.
Immunoblot analysis
Cells were washed with ice-cold PBS and lysed in the
RIPA buffer [10 mM Tris-HCl (pH 7.4), 0.1% SDS, 1% sodium
deoxycholate, 150 mM NaCl, 1 mM EDTA, 1.5 mM
Na3VO4, 10 mM NaF, 10 mM sodium
pyrophosphate, 10 mM sodium β-glycerophosphate and 1% protease
inhibitor cocktail set III (Calbiochem)]. After centrifugation for
10 min at 14,000 × g at 4°C, the supernatants were recovered as the
cell lysates, and the protein concentration of the cell lysates was
determined by the BCA protein assay kit (Pierce Biotechnology,
Inc., Rockford, IL, USA). Cell lysates containing equal amounts of
protein were separated by SDS-PAGE and transferred to a
polyvinylidene difluoride membrane. The membrane was probed with a
primary antibody and then with an appropriate HRP-conjugated
secondary antibody according to the protocol recommended by the
manufacturer of each antibody. Immunoreactive bands were visualized
using Immobilon Western Chemiluminescent HRP Substrate (Millipore,
Billerica, MA, USA).
RT-PCR
Total RNA was extracted with TRIzol (Invitrogen Life
Technologies). Total RNA was reverse-transcribed into cDNA using
PrimeScript™ 1st strand cDNA Synthesis kit (Takara,
Tokyo, Japan) according to the manufacturer’s instructions.
Amplification was performed with cycles of 97°C for 30 sec, 55°C
for 30 sec, and 72°C for 1 min in a thermal cycler (Takara PCR
Thermal Cycler Dice). PCR cycles were 30 for MGMT and 25 for
β-actin. RT-PCR analysis was performed with the following primers:
MGMT (forward: 5′-GCTGGAGCTGTCTGGTTGTGAG, reverse: 5′-GCGC
GGCTTTGGGGTTGC), β-actin (forward: 5′-CCCATGCCA TCCTGCGTCTG,
reverse: 5′-CGTCATACTCCTGCTTG CTG).
Statistical analysis
Results are expressed as the means ± SD, and
differences were compared using the 2-tailed Student’s t-test.
P-values <0.05 were considered statistically significant and
indicated with asterisks in the figures.
Results
SP600125, a JNK inhibitor, sensitizes
stem-like glioblastoma cells with MGMT expression to
temozolomide
We have previously demonstrated that the JNK pathway
is commonly activated in stem-like glioblastoma cells compared to
their differentiated counterparts and that the JNK activity is
required for the maintenance of the stemness/tumor-initiating
capacity of stem-like glioblastoma cells (14). Since cancer stem cells are in
general associated with chemo- and radio-resistance (6), we wondered if JNK also has a role in
determining their sensitivity/resistance against temozolomide, the
current chemo-therapeutic agent of choice in the treatment of
glioblastoma (16). To address
this question, we used stem-like glioblastoma cells derived
directly from glioblastoma patients and asked whether the cytotoxic
effect of temozolomide on these stem-like glioblastoma cells would
be modulated by treatment with SP600125, a chemical inhibitor of
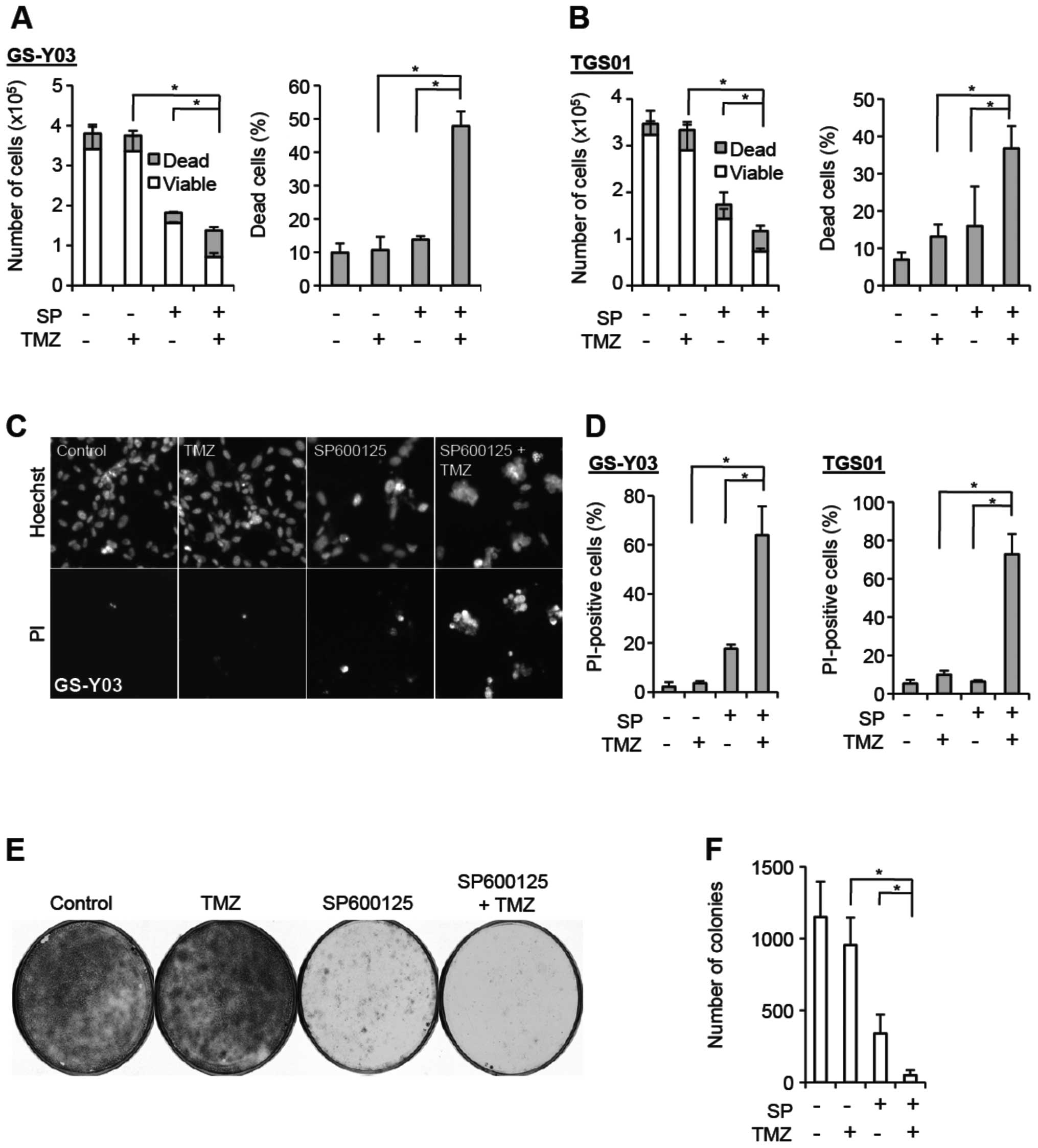
JNK. We first tested the idea using GS-Y03 stem-like glioblastoma
cells, which express MGMT and are virtually refractory to
clinically relevant concentrations (∼50 μM) of temozolomide
(Fig. 1A and C–F; see also
Fig. 3). When GS-Y03 cells
pretreated with SP600125 were exposed to temozolomide at 50
μM and collected 3 days after the temozolomide treatment to
determine their viability, we found that SP600125 pretreatment
significantly increased the proportion of dead cells after
temozolomide treatment, while SP600125 pretreatment alone caused
only marginal increase in the proportion of dead cells (Fig. 1A). Since fragile dead cells could
be lost during the cell collection procedure, we also stained dead
cells in situ with a fluorescent vital dye Hoechst 33342.
Although it turned out that SP600125 pretreatment alone somewhat
increased cell death when examined by this method, cell death was
further increased by exposing cells to temozolomide after SP600125
pretreatment, whereas temozolomide alone had no cell
death-increasing effect on GS-Y03 cells (Fig. 1C and D). Thus, the results of the
two cell death assays consistently suggested that SP600125 renders
GS-Y03 cells, which are otherwise highly resistant to temozolomide,
sensitive to the chemotherapeutic agent. We then wished to
determine whether SP600125 actually augments the suppressive effect
of temozolomide on clonogenic survival, instead of simply changing
the time kinetics of cell death, i.e., just letting cell death
occur at earlier time-points that would otherwise occur at later
time-points. To address this question, we conducted the colony
formation assay. SP600125 pretreatment of GS-Y03 cells alone
efficiently inhibited colony formation, most likely because
SP600125 caused a proliferation block associated with
differentiation. However, colony formation was further inhibited
when GS-Y03 cells were exposed to temozolomide after SP600125
pretreatment, whereas temozolomide alone had no discernible
inhibitory effect on colony formation (Fig. 1E and F). The results of the colony
formation assay thus indicated that SP600125 efficiently suppresses
clonogenic survival of stem-like glioblastoma cells in combination
with temozolomide. Intriguing enough, whereas we obtained
essentially similar results (i.e., sensitization to temozolomide by
SP600125 pretreatment) from TGS01 and GS-Y01 cells that express
MGMT (17,18) (Fig. 1B
and D; data not shown for GS-Y01), we observed no such effect
of SP600125 on another patient-derived stem-like glioblastoma cell
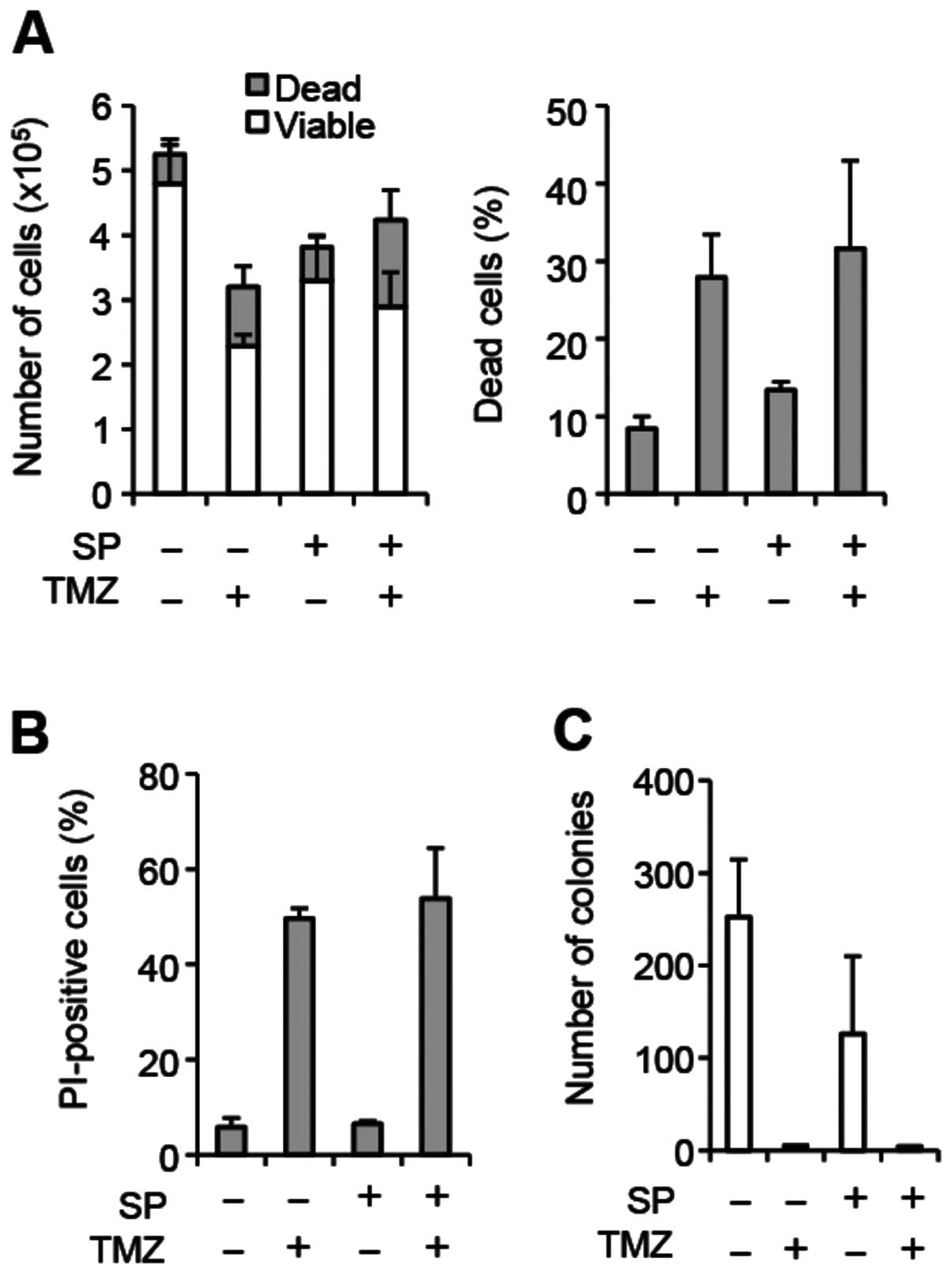
line GS-NCC01, which do not express MGMT (Fig. 2; Fig.
3A and F). In GS-NCC01 cells, temozolomide exposure alone
caused substantial cell death consistent with the lack of MGMT
expression, and SP600125 pretreatment failed to increase cell death
either alone or in combination with temozolomide (Fig. 2A and B). In line with the results
of the cell death assay, temozolomide exposure alone almost totally
eliminated colony formation, and we saw no further decrease in
colony formation by SP600125 pretreatment (Fig. 2C). Together, these results suggest
that SP600125 may specifically sensitize MGMT-expressing,
temozolomide-resistant stem-like glioblastoma cells to temozolomide
treatment.
Inhibition of MGMT expression in
stem-like glioblastoma cells by SP600125
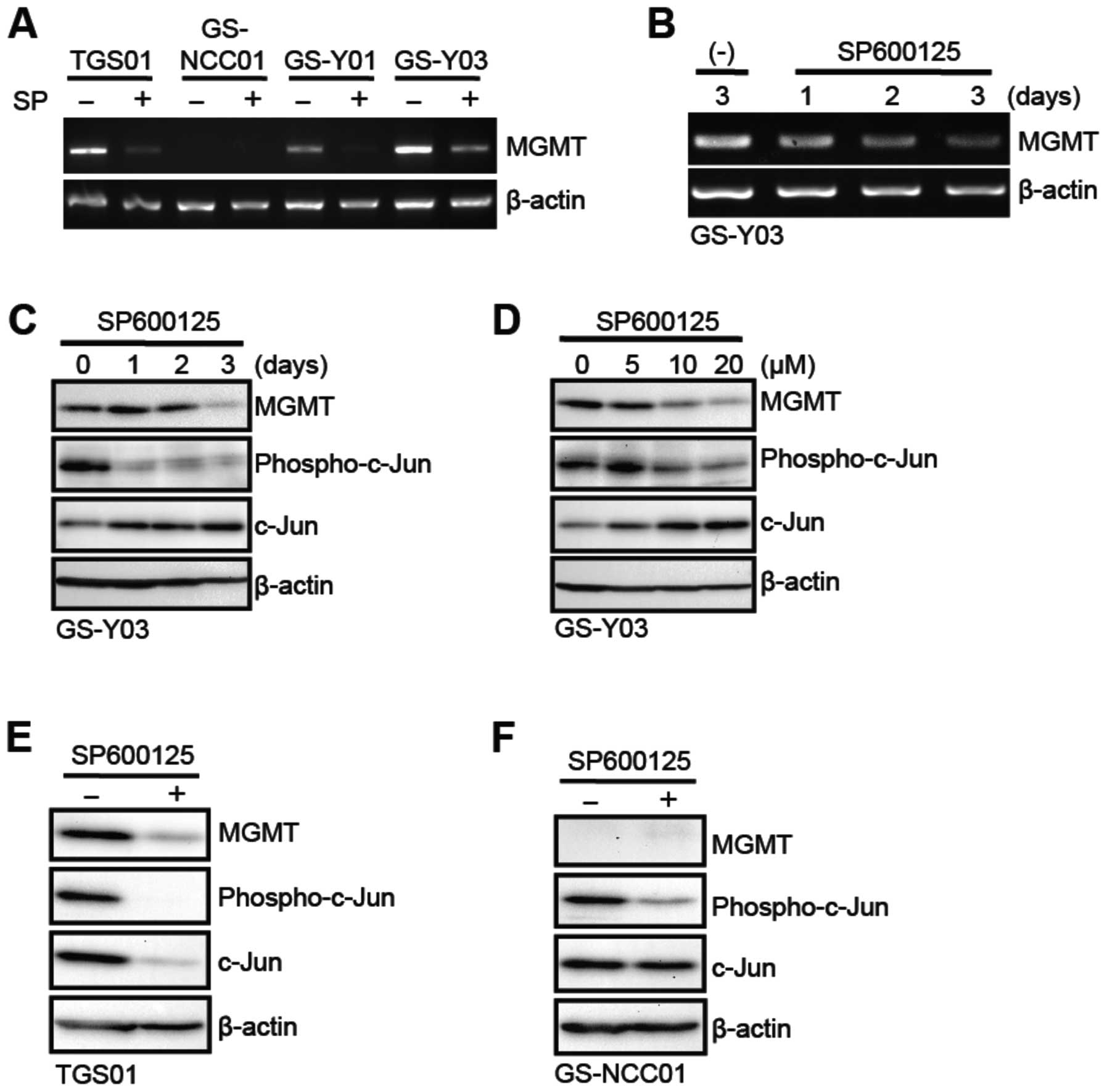
Since SP600125 specifically sensitized stem-like
glioblastoma cells with MGMT expression to temozolomide, we next
asked whether SP600125 did so through modulation of the expression
and/or activity of MGMT. To this end, we first examined the effect
of SP600125 on the mRNA expression of MGMT in stem-like
glioblastoma cells. The results indicated that GS-Y01, GS-Y03 and
TGS01 but not GS-NCC01 cells express detectable levels of MGMT
mRNA, which were significantly decreased after 3-day pretreatment
with SP600125 (Fig. 3A). The time
course analysis using GS-Y03 cells revealed that MGMT mRNA
gradually declined over 3 days during the SP600125 pretreatment
period (Fig. 3B). Consistent with
the change of the mRNA level, the SP600125 pretreatment also
decreased the expression of the MGMT protein, with some time lag
compared to that of mRNA (Fig.
3C). Further analysis showed that SP600125 inhibited both JNK
activity (as monitored by phospho-c-Jun expression) and MGMT
protein expression at 10 μM or higher concentrations
(Fig. 3D). Inhibition of MGMT
protein expression by SP600125 was similarly demonstrated in TGS01
and GS-Y01 cells (Fig. 3E; data
not shown for GS-Y01). We also confirmed that GS-NCC01 cells do not
express MGMT protein and that SP600125 does inhibit the JNK
activity in GS-NCC01 cells, excluding the possibility that the lack
of sensitization effect is due to failure of SP600125 to inhibit
JNK in GS-NCC01 cells (Fig.
3F).
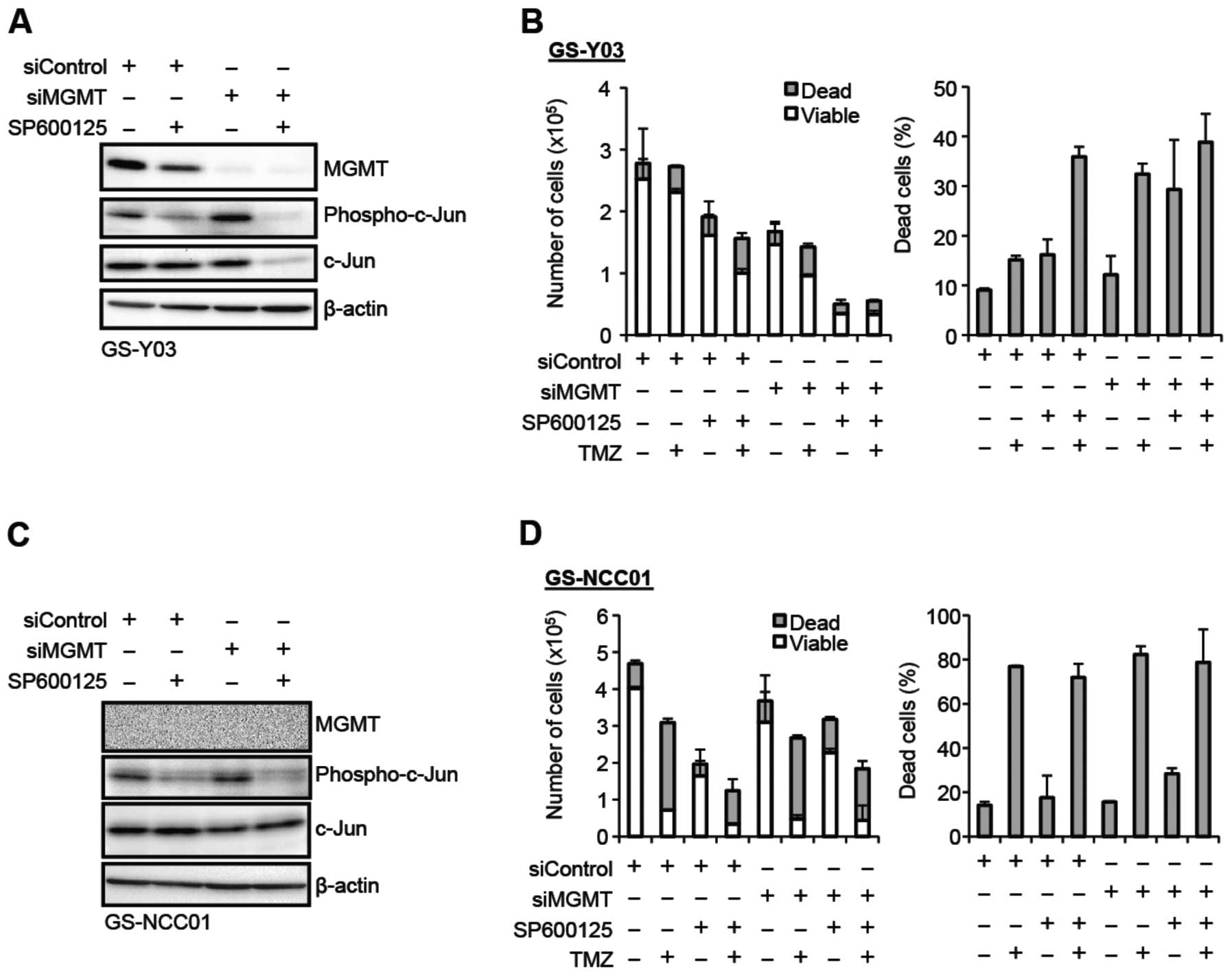
JNK knockdown inhibits MGMT expression
and sensitizes stem-like glioblastoma cells to temozolomide
To definitively establish that JNK is actually
involved in the regulation of the MGMT expression and temozolomide
resistance of stem-like glioblastoma cells, we knocked down JNK and
examined its effect on these parameters. To achieve effective
reduction in the JNK activity, we introduced into GS-Y03 and TGS01
cells a combination of siRNAs against JNK1 and JNK2. Successful
knockdown of JNK1 and JNK2 resulted in reduced expression of
phospho-c-Jun, and also caused decreased MGMT expression in
parallel with the reduced JNK activity (Fig. 4A). The stem-like glioblastoma cells
were then examined for their sensitivity to temozolomide in the
presence and absence of JNK knockdown. Whereas temozolomide
exposure alone in the absence of JNK knockdown did not have a
significant impact on cell viability, the proportion of dead cells
was substantially increased when cells were exposed to temozolomide
after JNK knockdown (Fig. 4B–E).
In line with the results of the cell death assay, JNK knockdown
cells formed apparently less colonies after temozolomide exposure
in the colony formation assay, in contrast to the control cells
which formed colonies quite efficiently irrespective of
temozolomide exposure (Fig. 4F and
G). In conjunction with the results of the analyses using
SP600125, these results demonstrate that JNK plays a critical role
in the maintenance of the MGMT expression and temozolomide
resistance of stem-like glioblastoma cells.
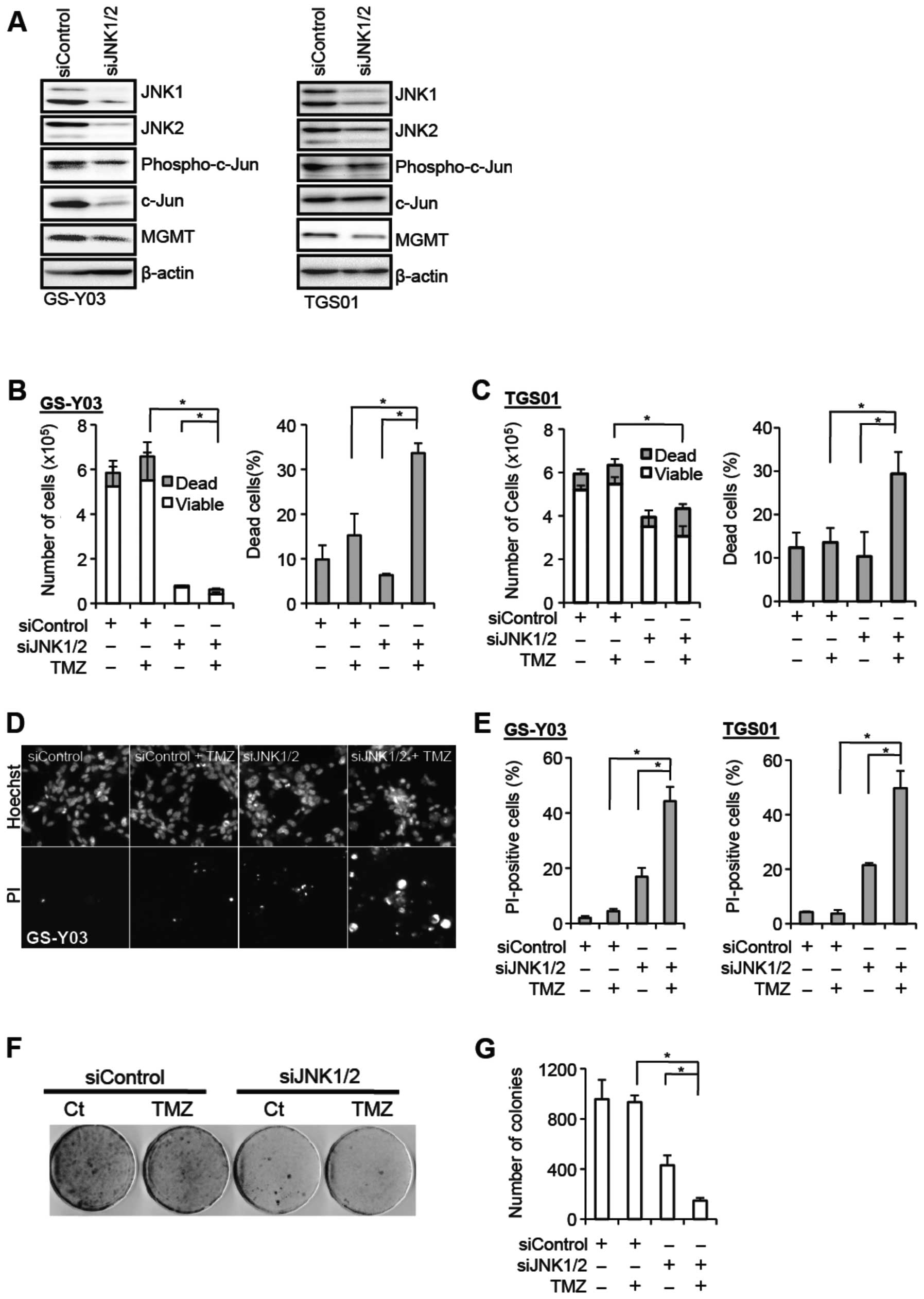 | Figure 4.JNK knockdown inhibits MGMT
expression and sensitizes stem-like glioblastoma cells to
temozolomide. (A) The indicated stem-like glioblastoma cells were
transiently transfected with a combination of siRNAs against JNK1
and JNK2 (siJNK1/2) or with a control siRNA (siControl). After 4
days, the transfected cells were subjected to immunoblot analysis
for the expression of the indicated proteins. (B–E) The indicated
stem-like glioblastoma cells transiently transfected with the
indicated siRNAs for 4 days were subsequently treated with or
without temozolomide (TMZ, 50 μM) for 4 h and cultured in
the absence of TMZ for 3 days thereafter. Then, the numbers of
viable and dead cells (left panel) as well as the percentage of
dead cells (right panel) were determined for GS-Y03 cells (B) and
TGS01 cells (C). Alternatively, the cells were subjected to cell
death analysis using propidium iodide (PI). Representative
fluorescence images of Hoechst- (upper rows) and PI- (lower rows)
positive cells (D), as well as the percentage of PI-positive cells
(dead cells) relative to Hoechst-positive cells (total cells) (E)
are shown. (F and G) GS-Y03 cells transiently transfected with a
combination of siRNAs against JNK1 and JNK2 (siJNK1/2) or with a
control siRNA (siControl) for 4 days were then subjected to colony
formation assay with or without temozolomide treatment (TMZ, 50
μM for 4 h). An image of a representative experiment (F) and
the number of colonies (G) are presented. In (B), (C), (E), and
(G), the values in the graphs represent means ± standard deviations
of three independent experiments. *P<0.05 [note that
the numbers of viable cells are compared in the left panels of (B)
and (C)]. |
MGMT-dependent contribution of JNK to
temozolomide resistance of stem-like glioblastoma cells
So far, we have demonstrated that JNK plays a role
in the maintenance of the MGMT expression and temozolomide
resistance of stem-like glioblastoma cells that express MGMT. We
then wished to ask whether downregulation of MGMT expression is the
mechanism for the JNK inhibition-mediated sensitization of
stem-like glioblastoma cells to temozolomide. To determine whether
the sensitization effect of JNK inhibition is dependent on MGMT
expression, we examined the impact of MGMT knockdown on the
temozolomide-sensitizing effect of JNK inhibition. In the control
experiment, GS-Y03 cells transfected with a control siRNA were
treated with or without temozolomide in the absence or presence of
SP600125 pretreatment. Consistent with the earlier results, under
this experimental condition, SP600125 inhibited MGMT expression and
sensitized the cells to temozolomide (Fig. 5A and B). In sharp contrast, when
the cells were transfected with a siRNA against MGMT to knockdown
MGMT expression, the cells became sensitive to temozolomide alone
(compare siMGMT and siMGMT+TMZ) and the temozolomide-sensitizing
effect of SP600125 pretreatment was lost under this condition
(compare siMGMT+TMZ and siMGMT+SP+TMZ) (Fig. 5A and B). Unexpectedly, SP600125
pretreatment promoted cell death when combined with MGMT knockdown,
the reason for which remains currently unknown. Nevertheless, the
observation that SP600125 pretreatment no longer promoted
temozolomide-induced cell death in MGMT knockdown cells supports
the idea that the temozolomide-sensitizing effect of JNK inhibition
is lost upon MGMT knockdown. Finally, to rule out the possibility
that the siRNA against MGMT cancelled the sensitizing effect of
SP600125 in an MGMT-independent manner, we conducted a similar
experiment using GS-NCC01 cells that do not express MGMT. The
results indicated GS-NCC01 cells remained equally sensitive to
temozolomide either in the absence or presence of SP600125
pretreatment irrespective of whether or not MGMT was knocked down
(Fig. 5C and D). Collectively,
these findings suggest that regulation of MGMT expression is a
mechanism by which JNK contributes to temozolomide resistance of
stem-like glioblastoma cells.
Discussion
Cancer stem cells are now increasingly recognized as
a critical therapeutic target in cancer treatment. Since cancer
stem cells are often associated with inherent resistance against
conventional chemo- and radio-therapies, overcoming their therapy
resistance is among the most important issues in cancer
therapeutics (3,4). In the case of glioblastoma, cancer
stem cells are frequently, albeit not always, resistant to
temozolomide, the standard chemotherapeutic agent for the treatment
of glioblastoma (7). Accumulating
evidence indicates that temozolomide-resistant glioblastoma cancer
stem cells are associated with increased MGMT expression (8–12),
and we and others have indeed demonstrated that MGMT expression is
responsible for the temozolomide resistance of glioblastoma cancer
stem cells (12,18). Thus, the evidence suggests that
therapies targeting MGMT expression/activity in combination with
temozolomide would be a rational strategy to treat
temozolomide-resistant glioblastoma cancer stem cells, and also
underscores the necessity of dissecting the mechanism of MGMT
regulation in glioblastoma cancer stem cells. In this regard, we
have previously demonstrated that the MAPK pathway regulates MGMT
expression in stem-like glioblastoma cells through the MDM2-p53
axis (18). Only recently, the
BMP2-HIF1-α, mGlu3-PI3K/NF-κB, and ZEB1-c-MYB pathways have been
implicated in the regulation of MGMT expression in glioma stem
cells (19–21). While these pathways are expected to
become attractive candidates for therapeutic targeting to overcome
temozolomide resistance of glioblastoma cancer stem cells,
apparently, the information is still limited and much remains
unknown regarding the MGMT regulation in glioblastoma cancer stem
cells.
Herein, we have provided lines of evidence that JNK
is involved in the maintenance of MGMT expression and temozolomide
resistance of stem-like glioblastoma cells. Both pharmacological
and genetic inhibition of JNK resulted in decreased MGMT expression
and reduced temozolomide resistance of otherwise
temozolomide-resistant stem-like glioblastoma cells with MGMT
expression. Significantly, JNK inhibition-mediated sensitization of
stem-like glioblastoma cells to temozolomide did not occur in cells
without MGMT expression (either originally lacking MGMT expression
or after MGMT knockdown). Thus, to our knowledge, this is the first
study to demonstrate the critical role of the JNK pathway in
MGMT-dependent temozolomide resistance of stem-like glioblastoma
cells. Intriguingly, an earlier report showed that SP600125
sensitized serum-cultured U87 cells to temozolomide (22). Given that U87 is a human
glioblastoma cell line widely known to be sensitive to temozolomide
due to lack of MGMT expression (23–25),
the mechanism for the temozolomide-sensitizing effect of SP600125
in that study was most likely MGMT-independent and therefore
distinct from the one proposed in the present study. Moreover,
since JNK inhibition in the study was done solely by use of
SP600125 known to have well-characterized off-target effects
(26–28), it could even be possible that the
sensitizing effect of SP600125 in U87 cells was JNK-independent. It
needs to be emphasized here, however, that the results of our study
do not necessarily exclude the possibility that JNK could also
contribute to temozolomide resistance independently of MGMT.
We have demonstrated in this study that JNK plays a
role in the control of MGMT expression in stem-like glioblastoma
cells, but the molecular pathway connecting JNK to MGMT expression
remains to be determined. Although MGMT could be regulated at the
protein level as exemplified by STAT3-mediated regulation of MGMT
(29), our data clearly indicate
that JNK regulation of MGMT expression occurs at the mRNA level,
suggesting that JNK likely contributes to MGMT expression through
promotion of its transcription. To date, a number of transcription
factors and complexes, such as Sp1, AP-1, c-Myc, HIF1-α, c-Myb, and
NF-κB, have been reported to directly bind and activate the MGMT
gene promoter (20,21,30–33).
Among these factors and complexes, Sp1 and c-Myc, let alone AP-1
which is a jun/fos heterodimer, may be of particular interest in
that they are direct targets of JNK-mediated phosphorylation and
activation. Earlier reports indicated that JNK maintains the
stability of the Sp1 protein through phosphorylation at Thr278/739
(34,35). Another report demonstrated that JNK
enhances the pro-apoptotic activity of c-Myc through
phosphorylation at Ser62/71 (36).
Whether and how JNK is connected with those and other transcription
factors/complexes, either directly or indirectly, is an intriguing
issue that still needs to be addressed.
While future studies are expected to shed light on
the detailed molecular mechanism underlying JNK-mediated control of
MGMT expression and temozolomide resistance in stem-like
glioblastoma cells, JNK would nonetheless remain superior as a
therapeutic target to any of the possible targets that would be
newly identified in future studies, because JNK is a key molecule
that stands at the ‘crossroads’ of the molecular pathways governing
stemness and chemoresistance. We have previously demonstrated that
JNK plays an essential role in the maintenance of the stem cell
properties and tumor-initiating capacity of stem-like glioblastoma
cells and that systemic administration of SP600125 as a monotherapy
effectively deprives their self-renewal and tumor-initiating
capacities in vivo (14).
As such, the data of our previous study alone would have suggested
use of JNK inhibitor as a maintenance monotherapy after the
completion of the initial chemoradiotherapy, for the purpose of
targeting glioblastoma cancer stem cells that have evaded
chemoradiotherapy. However, now that we have demonstrated that a
JNK inhibitor inhibited the temozolomide resistance of glioblastoma
cancer stem cells along with their stemness, there is a rationale
for concomitant use of a JNK inhibitor and temozolomide for the
treatment of glioblastoma. Such a combinational use of a JNK
inhibitor and temozolomide is also of value in another perspective,
because temozolomide would kill and irreversibly eliminate
glioblastoma cancer stem cells that have undergone differentiation
and lost the capacity to initiate tumors as a result of JNK
inhibitor treatment but may not necessarily be without the risk of
recovering their stem cell properties and tumor-initiating capacity
(37). Thus, JNK is a highly
beneficial molecular target to control the stemness and
chemoresistance of glioblastoma cancer stem cells at the same time
and therefore will remain an attractive therapeutic target to treat
glioblastoma cancer stem cells.
In conclusion, we have demonstrated in this study
that JNK contributes to the MGMT expression and temozolomide
resistance of stem-like glioblastoma cells. Together with the known
critical role of JNK in the maintenance of the stemness of
stem-like glioblastoma cells (14,38),
our current findings provide a molecular rationale for combining
JNK inhibitors with temozolomide to effectively target glioblastoma
stem cells. The therapeutic significance of such combination
treatment will be tested in future preclinical and clinical
studies.
Acknowledgements
We thank Drs Tomoki Todo and Nobuhito
Saito at the University of Tokyo for generously providing us with
the TGS01 cells and Dr Tomoko Kagawa for her continuous
support/encouragement. This study was supported by Grants-in-Aid
for Scientific Research, for Challenging Exploratory Research, and
for Young Scientists from the Ministry of Education, Culture,
Sports, Science and Technology of Japan, by a Grant-in-Aid from the
Global COE Program of the Japan Society for the Promotion of
Science, by the National Cancer Center Research and Development
Fund (23-A-20), and by a grant from the Japan Brain Foundation.
References
|
1.
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Khasraw M and Lassman AB: Advances in the
treatment of malignant gliomas. Curr Oncol Rep. 12:26–33. 2010.
View Article : Google Scholar
|
|
3.
|
Yu Y, Ramena G and Elble RC: The role of
cancer stem cells in relapse of solid tumors. Front Biosci (Elite
Ed). 4:1528–1541. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Binda E, Visioli A, Reynolds B and Vescovi
AL: Heterogeneity of cancer-initiating cells within glioblastoma.
Front Biosci (Schol Ed). 4:1235–1248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Malik B and Nie D: Cancer stem cells and
resistance to chemo and radio therapy. Front Biosci (Elite Ed).
4:2142–2149. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Beier D, Schulz JB and Beier CP:
Chemoresistance of glioblastoma cancer stem cells - much more
complex than expected. Mol Cancer. 10:1282011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Hermisson M, Klumpp A, Wick W, et al:
O6-methylguanine DNA methyltransferase and p53 status
predict temozolomide sensitivity in human malignant glioma cells. J
Neurochem. 96:766–776. 2006.
|
|
9.
|
Hegi ME, Liu L, Herman JG, et al:
Correlation of O6-methylguanine methyltransferase (MGMT)
promoter methylation with clinical outcomes in glioblastoma and
clinical strategies to modulate MGMT activity. J Clin Oncol.
26:4189–4199. 2008.
|
|
10.
|
Blough MD, Westgate MR, Beauchamp D, et
al: Sensitivity to temozolomide in brain tumor initiating cells.
Neurooncology. 12:756–760. 2010.PubMed/NCBI
|
|
11.
|
Spiegl-Kreinecker S, Pirker C, Filipits M,
et al: O6-methylguanine DNA methyltransferase protein
expression in tumor cells predicts outcome of temozolomide therapy
in glioblastoma patients. Neurooncology. 12:28–36. 2010.
|
|
12.
|
Kato T, Natsume A, Toda H, et al:
Efficient delivery of liposome-mediated MGMT-siRNA reinforces the
cytotoxity of temozolomide in GBM-initiating cells. Gene Ther.
17:1363–1371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ikushima H, Todo T, Ino Y, Takahashi M,
Miyazawa K and Miyazono K: Autocrine TGF-beta signaling maintains
tumorigenicity of glioma-initiating cells through Sry-related
HMG-box factors. Cell Stem Cell. 5:504–514. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Matsuda K, Sato A, Okada M, et al:
Targeting JNK for therapeutic depletion of stem-like glioblastoma
cells. Sci Rep. 2:5162012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Sato A, Sunayama J, Okada M, et al:
Glioma-initiating cell elimination by metformin activation of FOXO3
via AMPK. Stem Cells Transl Med. 1:811–824. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hart MG, Garside R, Rogers G, Stein K and
Grant R: Temozolomide for high grade glioma. Cochrane Database Syst
Rev. 4:CD0074152013.
|
|
17.
|
Hegi ME, Diserens AC, Godard S, et al:
Clinical trial substantiates the predictive value of
O-6-methylguanine-DNA methyltransferase promoter methylation in
glioblastoma patients treated with temozolomide. Clin Cancer Res.
10:1871–1874. 2004. View Article : Google Scholar
|
|
18.
|
Sato A, Sunayama J, Matsuda K, et al:
MEK-ERK signaling dictates DNA-repair gene MGMT expression and
temozolomide resistance of stem-like glioblastoma cells via the
MDM2-p53 axis. Stem Cells. 29:1942–1951. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ciceroni C, Bonelli M, Mastrantoni E, et
al: Type-3 metabotropic glutamate receptors regulate
chemoresistance in glioma stem cells, and their levels are
inversely related to survival in patients with malignant gliomas.
Cell Death Differ. 20:396–407. 2013. View Article : Google Scholar
|
|
20.
|
Persano L, Pistollato F, Rampazzo E, et
al: BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by
affecting HIF-1alpha stability and MGMT expression. Cell Death Dis.
3:e4122012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Siebzehnrubl FA, Silver DJ, Tugertimur B,
et al: The ZEB1 pathway links glioblastoma initiation, invasion and
chemoresistance. EMBO Mol Med. 5:1196–1212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Ohba S, Hirose Y, Kawase T and Sano H:
Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced
cytotoxicity in human glioma cells. J Neurooncol. 95:307–316. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kanzawa T, Germano IM, Kondo Y, Ito H, Kyo
S and Kondo S: Inhibition of telomerase activity in malignant
glioma cells correlates with their sensitivity to temozolomide. Br
J Cancer. 89:922–929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Aghi M, Rabkin S and Martuza RL: Effect of
chemotherapy-induced DNA repair on oncolytic herpes simplex viral
replication. J Natl Cancer Inst. 98:38–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Nadkarni A, Shrivastav M, Mladek AC, et
al: ATM inhibitor KU-55933 increases the TMZ responsiveness of only
inherently TMZ sensitive GBM cells. J Neurooncol. 110:349–357.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Miyazawa K: A negative regulator or just
an unconcerned passerby: phosphoinositide 3-kinase signalling in
IL-12 production. J Biochem. 152:497–499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Jemaa M, Vitale I, Kepp O, et al:
Selective killing of p53-deficient cancer cells by SP600125. EMBO
Mol Med. 4:500–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Schmidt M, Budirahardja Y, Klompmaker R
and Medema RH: Ablation of the spindle assembly checkpoint by a
compound targeting Mps1. EMBO Rep. 6:866–872. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kohsaka S, Wang L, Yachi K, et al: STAT3
inhibition overcomes temozolomide resistance in glioblastoma by
downregulating MGMT expression. Mol Cancer Ther. 11:1289–1299.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Bocangel D, Sengupta S, Mitra S and Bhakat
KK: p53-mediated down-regulation of the human DNA repair gene
O6-methylguanine-DNA methyltransferase (MGMT) via
interaction with Sp1 transcription factor. Anticancer Res.
29:3741–3750. 2009.PubMed/NCBI
|
|
31.
|
Boldogh I, Ramana CV, Chen Z, et al:
Regulation of expression of the DNA repair gene
O6-methylguanine-DNA methyltransferase via protein
kinase C-mediated signaling. Cancer Res. 58:3950–3956.
1998.PubMed/NCBI
|
|
32.
|
Pyko IV, Nakada M, Sabit H, et al:
Glycogen synthase kinase 3beta inhibition sensitizes human
glioblastoma cells to temozolomide by affecting
O6-methylguanine DNA methyltransferase promoter
methylation via c-Myc signaling. Carcinogenesis. 34:2206–2217.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lavon I, Fuchs D, Zrihan D, et al: Novel
mechanism whereby nuclear factor kappaB mediates DNA damage repair
through regulation of O(6)-methylguanine-DNA-methyltransferase.
Cancer Res. 67:8952–8959. 2007. View Article : Google Scholar
|
|
34.
|
Chuang JY, Wang YT, Yeh SH, Liu YW, Chang
WC and Hung JJ: Phosphorylation by c-Jun NH2-terminal kinase 1
regulates the stability of transcription factor Sp1 during mitosis.
Mol Biol Cell. 19:1139–1151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Wang SA, Chuang JY, Yeh SH, et al: Heat
shock protein 90 is important for Sp1 stability during mitosis. J
Mol Biol. 387:1106–1119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Noguchi K, Kitanaka C, Yamana H, Kokubu A,
Mochizuki T and Kuchino Y: Regulation of c-Myc through
phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J
Biol Chem. 274:32580–32587. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Kitanaka C, Sato A and Okada M: JNK
signaling in the control of the tumor-initiating capacity
associated with cancer stem cells. Genes Cancer. Jan 22–2013.(Epub
ahead of print). View Article : Google Scholar
|
|
38.
|
Yoon CH, Kim MJ, Kim RK, et al: c-Jun
N-terminal kinase has a pivotal role in the maintenance of
self-renewal and tumorigenicity in glioma stem-like cells.
Oncogene. 31:4655–4666. 2012. View Article : Google Scholar : PubMed/NCBI
|