Introduction
The oncosuppressor p53 plays a critical role in
cancer cell growth inhibition and apoptosis in response to a
variety of stress signals, including DNA damage, hypoxia and
aberrant proliferation signals such as oncogene activation
(1). Most of the p53
oncosuppressor activities, such as cell cycle arrest, apoptosis, or
senescence, are achieved through the control of p53-mediated gene
transcription (2). The activation
of the p53 pathway has been implicated in an individual’s ability
to suppress tumor formation and to respond to many types of cancer
therapies (3). Therefore, p53 is
the most inactivated oncosuppressor in human tumors mainly by gene
mutations or deregulation of protein activity (4). In this regard, we previously showed
that p53 apoptotic activity may be inhibited by overexpression of
NADPH oxidase 1 (Nox1) that interferes with p53 binding to
apoptotic gene promoters by affecting p53 post-translational
modifications (5,6). NADPH oxidases are a family of enzymes
that regulate redox-sensitive signalling pathways involved in
cancer development and progression and are often overexpressed in
different types of human cancers favouring tumor progression and
angiogenesis (7,8).
Cationic triphenylmethane pharmacophore (TPM)
gentian violet (GV) has a long history of use as antifungal and
antibacterial agent (9) and is
also exploited in other research fields (10). Of note, GV has been shown to have
potent anticancer and anti-angiogenic activities in mice and human
(11,12) and to have an effect against
melanoma metastases (13),
although the anticancer molecular mechanisms are not fully
understood. GV was originally shown to have oxidation-reduction
potential (14) with likely free
radical formation. Recent studies showed that GV may have potent
inhibitory activity against NADPH oxidase Nox2 and Nox4 (15) and to induce cell death by
disruption of the mitochondrial system (16). GV enhances the cytotoxic activity
of thiostrepton (TS), a thiazole antibiotic that inhibits the
expression of oncogenic transcription factor FOXM1, required for
cell cycle progression and resistance to oncogene-induced oxidative
stress (17). The GV and TS
combinatorial approach may thus be particularly useful in treating
tumors with aberrant mitochondrial oxidant production.
The success of GV in inhibiting Nox activity and to
induce anticancer effects prompted us to examine the potential
effect of this agent on wild-type (wt) p53 activity, which has not
been addressed yet. We found that GV counteracted the Nox1
inhibitory effect toward p53 transcriptional activity, restoring
p53 transactivation. Noteworthy, GV was able to directly induce p53
oncosuppressor activity by stimulating p53/DNA binding and
transactivation activities. Therefore, GV anticancer activity may
depend also on p53 activation.
Materials and methods
Cell culture and reagents
Human lung cancer H1299 (p53 null), colon cancer RKO
(carrying wt-p53), RKO stable interfered for p53 function
(RKO-sip53) (18) (a kind gift
from S. Soddu, Regina Elena National Cancer Institute, Rome,
Italy), colon cancer HCT116 (wtp53), HCT116-p53−/− (a
kind gift from B. Vogelstein, Johns Hopkins University, Baltimore,
MD), glioblastoma ADF (wtp53) (19,20)
cell lines were routinely maintained in RPMI-1640
(Life-Technology-Invitrogen) medium containing 10% heat-inactivated
fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin, and
glutamine, in 5% CO2 humidified incubator at 37°C.
The following reagents were used: gentian violet
(GV) stored at 1 mM in DMSO was used at 0.2, 0.5, 1, and 2
μM; p53 inhibitor pifithryn-α (PFT) (21) (Enzo Life Sciences, Lausen,
Switzerland) was used at 30 μM; chemotherapeutic agent
adriamycin (ADR) stored at 2 μg/μl in PBS was used at
2 μg/ml. PBS and DMSO solvents were used as control.
RNA isolation and reverse transcription
(RT)-PCR analysis
Cells were harvested in TRIzol Reagent and total RNA
was isolated following the manufacturer’s instructions
(Invitrogen). The first-strand cDNA was synthesized from 2
μg of total RNA with MuLV reverse transcriptase kit (Applied
Biosystems). Semi-quantitative reverse-transcribed (RT)-PCR was
carried out by using Hot-Master Taq polymerase (Eppendorf) with 2
μl cDNA reaction and genes specific oligonucleotides under
conditions of linear amplification. PCR products were run on a 2%
agarose gel and visualized with ethidium bromide. The housekeeping
β-actin gene, used as internal standard, was amplified from the
same cDNA reaction mixture. Densitometric analysis was applied to
quantify mRNA levels compared to control gene expression.
Viability assay
Exponentially proliferating cells were exposed to
different concentrations of GV for 24 h. Both floating and adherent
cells were collected and counted in hemocytometer after addition of
trypan blue. The percentage of dead cells (i.e. blue/total cells)
was determined by scoring 100 cells per chamber for three times. At
least three independent experiments were performed and cell numbers
were determined in triplicates.
Western blotting
Total cell extracts were prepared by incubation in
lysis buffer containing 50 mmol/l Tris-HCl, pH 7.5, 150 mmol/l
NaCl, 150 mmol/l KCl, 1 mmol/l dithiothreitol, 5 mmol/l EDTA, pH
8.0, 1% Nonidet P-40 plus a mix of protease and phosphatase
inhibitors (Roche Diagnostic) and resolved by SDS-polyacrylamide
gel electrophoresis. Proteins were transferred to a polyvinylidene
difluoride membrane (PVDF, Millipore) and incubated with the
primary antibodies followed by an anti-immunoglobulin-G-horseradish
peroxidase antibody (Bio-Rad). Specific proteins were detected by
enhanced chemiluminescence (ECL) (Amersham). Immunoblotting was
performed with: mouse monoclonal anti-Poly (ADP-ribose) polymerase
(PARP) (cleaved form, BD Pharmingen), rabbit polyclonal anti-p53
(FL393) mouse monoclonal anti-p53 (DO1) (both from Santa Cruz
Biotechnology), monoclonal anti-GFP (Roche Diagnostic), polyclonal
anti-Bax (N20, Santa Cruz Biotechnology), monoclonal
anti-phospho-Histone H2A.X (Ser139) (Millipore) (a kind gift from
S. Soddu) and monoclonal anti-β-actin (Calbiochem) antibodies.
Transfection and transactivation
assay
For the assay, H1299 cells, plated at subconfluence
in 60-mm Petri dishes, were transiently co-transfected using the
N,N-bis-(2- hydroxyethyl)-2-amino-ethanesulphonic acid-buffered
saline (BBS) version of the calcium phosphate procedure (22) with the luciferase reporter gene
driven by the p53-dependent natural Noxa-luc (kindly provided by T.
Taniguchi, University of Tokyo, Japan) promoter, wtp53 (0.5
μg/sample) and Nox1-GFP (4 μg/sample) (kindly
provided by M. Bignami, National Institute of Health, ISS, Rome,
Italy) expression vectors. Twenty-four hours after transfection
cells were treated with GV (1 μM) for further 24 h.
Transfection efficiency was normalized with the use of a
co-transfected β-galactosidase plasmid. Luciferase activity was
assayed on whole cell extract and the luciferase values were
normalized to β-galactosidase activity and protein content.
Chromatin immunoprecipitation (ChIP)
assay
ChIP analysis was carried out essentially as
previously described (23).
Briefly, cells were crosslinked with 1% formaldehyde for 10 min at
room temperature and then inactivated by the addition of 125 mM
glycine. Chromatin extracts containing DNA fragments with an
average size of 500 bp were incubated overnight at 4°C with milk,
shaking using polyclonal anti-p53 antibody (FL393, Santa Cruz
Biotechnology). Before use, protein G (Pierce) was blocked with 1
μg/μl sheared herring sperm DNA and 1
μg/μl BSA for 3 h at 4°C and then incubated with
chromatin and antibody for 2 h at 4°C. PCR was performed with
Hot-Master Taq (Eppendorf) using 2 μl of immunoprecipitated
DNA and promoter-specific primers spanning p53 binding sites.
Immunoprecipitation with non-specific immunoglobulins (IgG, Santa
Cruz Biotechnology) was performed as negative controls. PCR
products were run on a 2% agarose gel and visualized with ethidium
bromide.
Statistical analysis
Each experiment, unless otherwise specified, was
performed at least three times, and data are presented as the mean
± SD. Statistical significance was determined using Student’s
t-test. A P-value of ≤0.05 was considered statistically
significant.
Results
GV counteracts the Nox1 inhibitory effect
on p53 transcriptional activity
Nox1 overexpression in cancer cells has been
recently shown to inhibit p53 apoptotic transcriptional activity
either after p53 overexpression or after drug-induced p53
activation (6). On the other hand,
GV has been reported to efficiently inhibit Nox activity and block
tumor growth in mice (15).
Therefore, we evaluated whether GV could re-establish wtp53
transcriptional activity in the presence of Nox1 overexpression. To
test this hypothesis, luciferase assay was performed in p53-null
H1299 cells co-transfected with wtp53 and Nox1-GFP expression
vectors and the report vector containing the luciferase gene under
the control of p53 target Noxa gene promoter (Noxa-luc). As shown
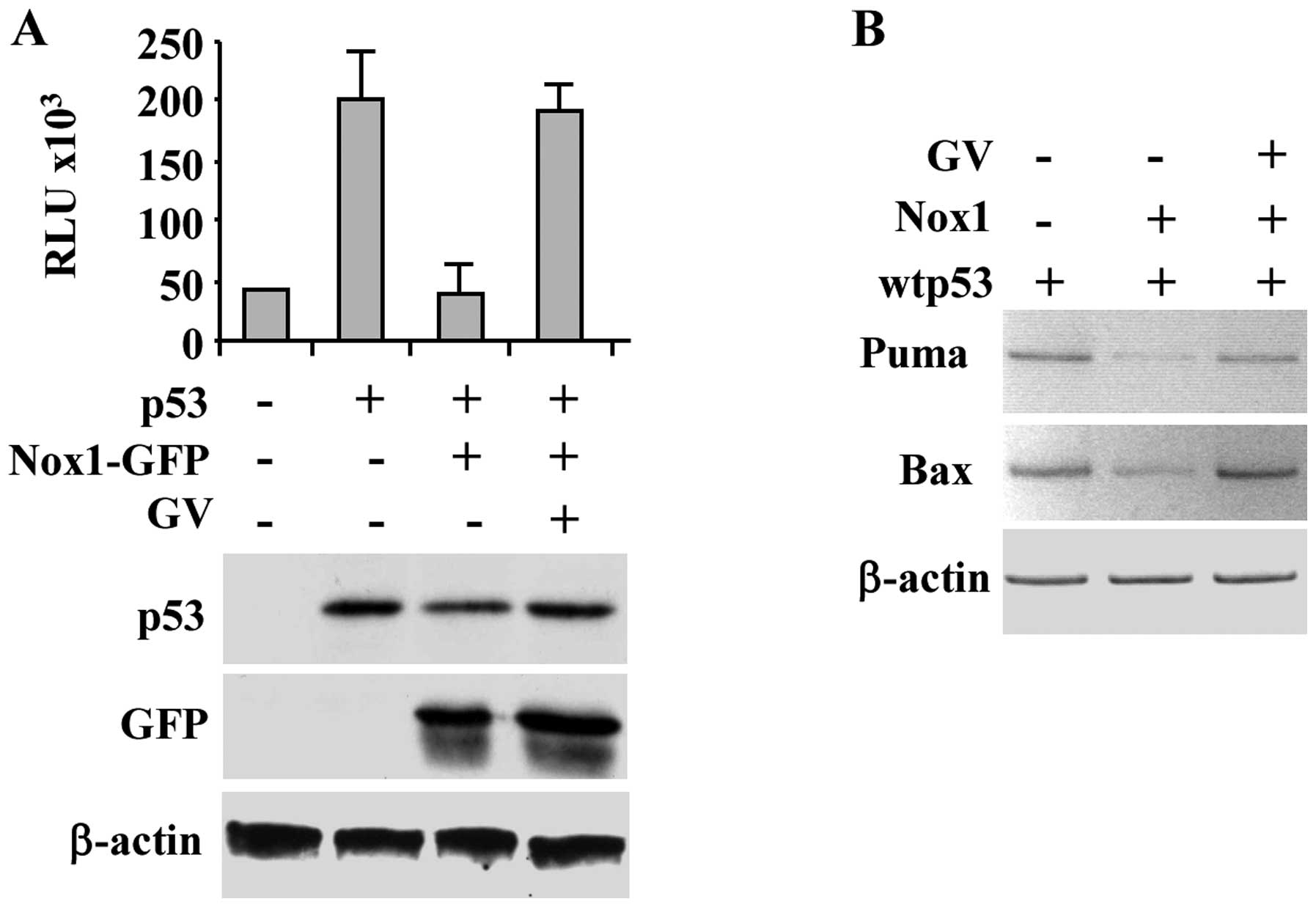
in Fig. 1A, Nox1 repressed the
p53-induced Noxa-luciferase reporter activity, as previously
reported (6), while GV treatment
efficiently restored wtp53 transcriptional activity. Western
immunoblotting of the overexpressed proteins is shown (Fig. 1A, lower panel). The positive effect
of GV on rescue of p53 transcriptional activity despite Nox1
overexpression was confirmed by in vivo RT-PCR analysis
whereas the p53-induced target gene transcription (i.e., Puma and
Bax) was inhibited by Nox1 co-transfection and restored by
concomitant GV treatment (Fig.
1B). These results support the hypothesis that GV inhibits Nox1
activity restoring wtp53 transcriptional activity.
GV induces γH2AX phosphorylation and p53
protein stabilization
Next, we evaluated whether GV treatment might induce
histone H2AX phosphorylation. The phosphorylation of the subtype of
histone H2A, called H2AX, in the position of Ser139 producing
γH2AX, occurs in response to formation of double strand brakes
(DSB) and is an early sign of replication stalling. In general,
analysis of γH2AX expression can be used to detect the genotoxic
effect of different anticancer agents (24). Herein we found that GV treatment
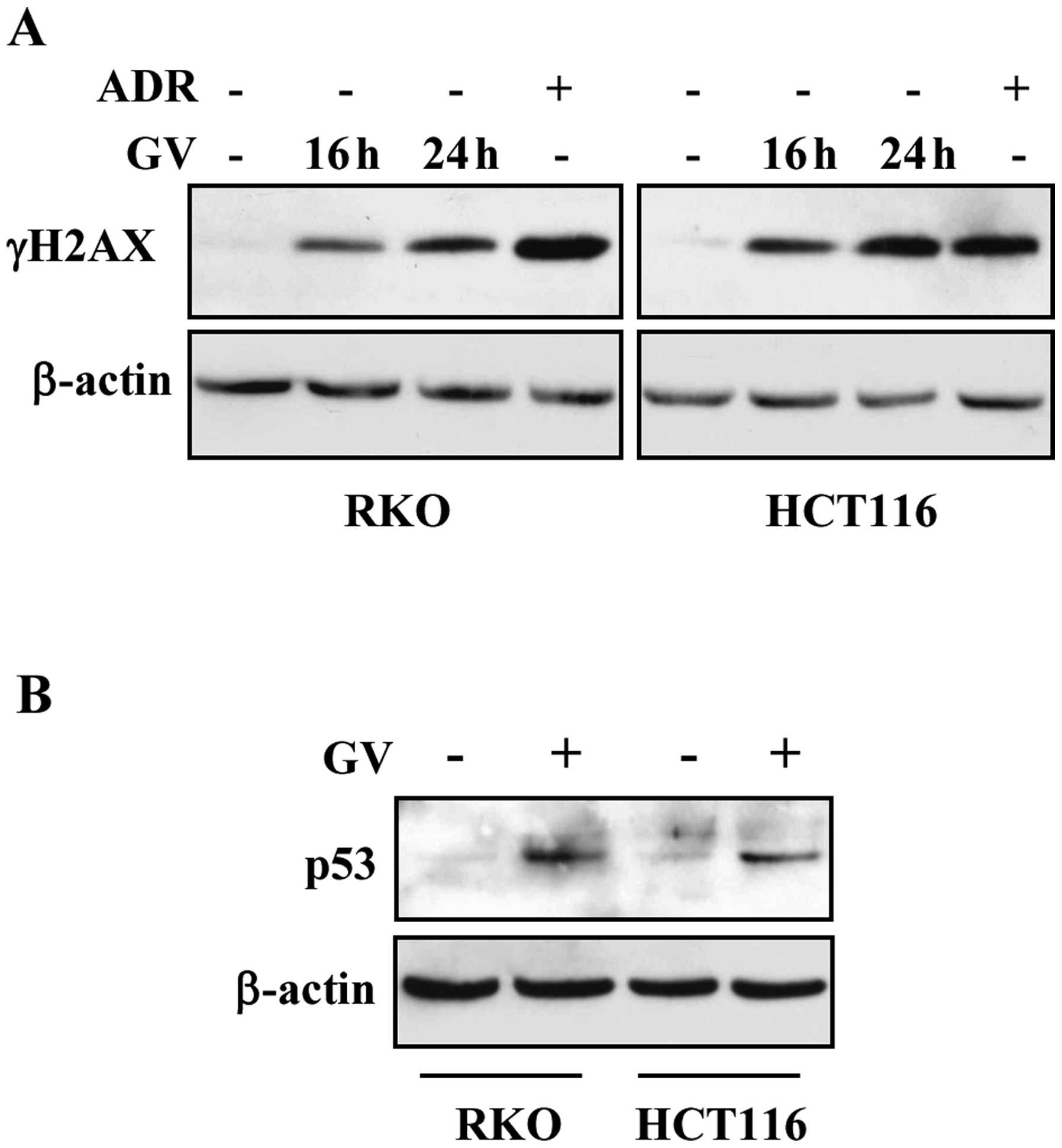
produced γH2AX expression in both RKO and HCT116 cells (Fig. 2A). As a positive control we treated
cells with the chemotherapeutic adriamycin (ADR) that indeed
efficiently phosphorylated histone H2AX (Fig. 2A). Next, western immunoblotting
showed that GV treatment induced p53 protein stabilization
(Fig. 2B), suggesting that the
effect induced on DNA by GV may be responsible for p53
activation.
GV induces p53/DNA binding and
transcriptional activity
We then explored whether GV could directly induce
p53 transcriptional activity. To this aim, in vivo p53-DNA
binding activity was analysed by chromatin immunoprecipitation
(ChIP) technique. Cells untreated or treated with GV were
cross-linked with formaldehyde, p53 was immunoprecipitated and
co-precipitated p53-bound elements were analysed by PCR. The
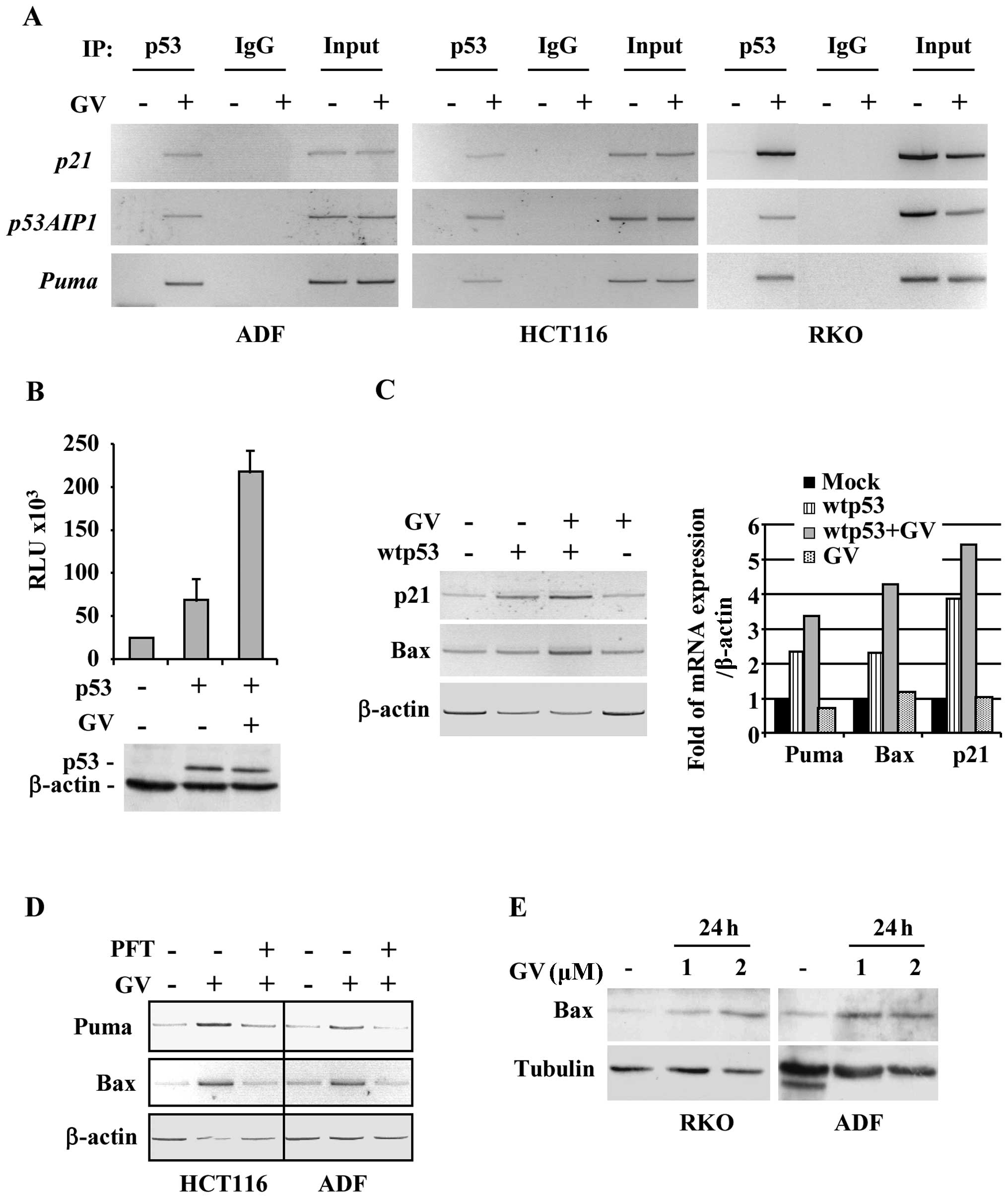
results show that p53 was efficiently recruited onto canonical
target promoters, such as p21, p53AIP1 and Puma, after GV treatment
in all cancer cell lines analysed (Fig. 3A). Then, H1299 cells were
co-transfected with Noxa-luc reporter plasmid and wtp53 expression
vector and treated with GV. As shown in Fig. 3B, GV remarkably increased wtp53
transcriptional activity. In agreement with the luciferase results,
the in vivo wtp53-induced p21 and Bax gene transcription was
further increased after GV treatment, as also indicated by
densitometric analyses (Fig. 3C).
The role of p53 in transcriptional activation of target genes was
confirmed by the use of pifithryn-α (PFT-α), an inhibitor of p53
transactivation function (21). As
shown in Fig. 3D, the GV-induced
p53 target gene transcription was efficiently impaired by PFT-α
co-treatment. Then, the effect of GV on Bax protein expression was
evaluated by western immunoblotting. As shown in Fig. 3E, GV caused an increase of Bax
protein levels in RKO and ADF cells. These data demonstrate that GV
is able to directly induce p53/DNA binding and transactivation
activities.
p53 is involved in GV-induced cancer cell
death
Finally, the GV effect on wtp53-carrying cancer cell
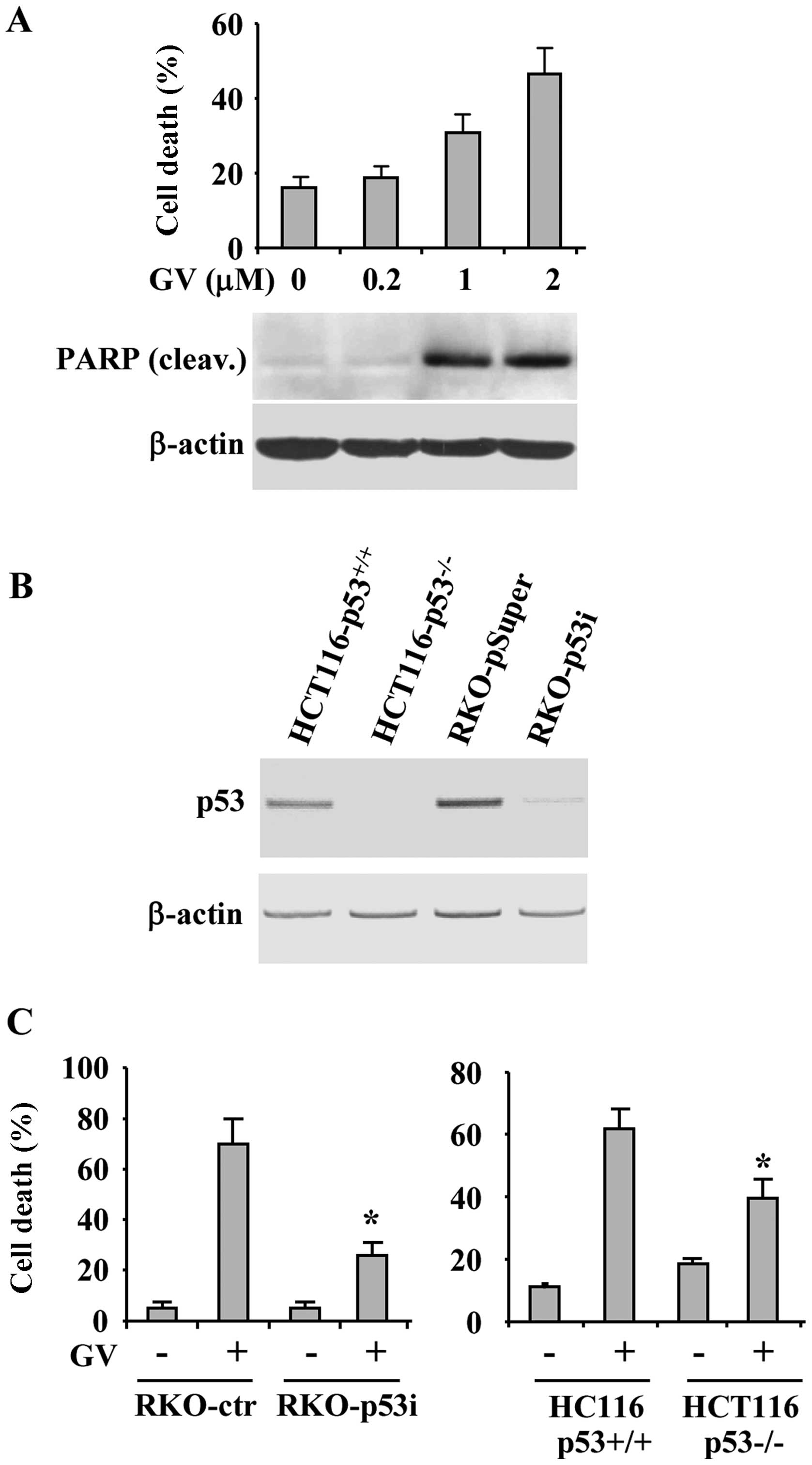
death was tested by viability assay. Increasing concentration of GV
progressively augmented cancer cell death with concomitant PARP
cleavage (Fig. 4A), a marker of
apoptotic cell death. To evaluate the role played by p53 in
GV-induced cancer cell death we took advantage of a cell line
stably interfered for p53 function (RKO-p53i) (18) and of HCT116-p53−/− cells
(Fig. 4B). RKO and HCT116 control
and p53 negative cells were treated with GV and 24 h later cell
viability was measured by trypan blue exclusion. As reported in
Fig. 4C, cells deprived of p53
function showed reduced sensitivity to GV, compared to
wtp53-carrying cells, with significant reduction of cell death,
suggesting that the GV-induced cell death depends in part by p53
activation.
Discussion
In this study we aimed at evaluating whether
GV-induced cancer cell death could depend on p53 activation. GV was
shown to induce the DNA binding and transactivation activities of
p53 in the human colon cancer cells RKO and HCT116 and in
glioblastoma ADF cells. The GV-induced p53 activation correlated
with cancer cell death. Thus, this biological outcome was reduced
in wtp53-negative cells as compared to wtp53-carrying cells. This
implies that the GV anticancer activity was in part dependent on
wtp53 activation.
GV has a long history of human use as
anti-bacterial, antimycotic and antiparasitic agent. More recently,
GV has been used also in different research fields (10) and has been shown to have strong
anticancer activities in mice and human without having mutagen
effect in humans (11–13). Thus, GV has been shown to be safe
for human treatment (25). GV may
have strong inhibitory activity against NADPH oxidases (Nox genes)
(15). Among Nox genes, Nox1 has
been reported to generate reactive oxygen (ROS) which in turn
triggers the angiogenic switch (7), a hallmark of rapidly growing solid
tumors (26). Moreover, we
previously showed that Nox1 inhibits p53 transcriptional activity
and oncosuppressor function by impairing p53 acetylation (6). Herein we found that GV was able to
overcome the Nox1-induced p53 inhibition and restore p53
transcriptional activity. Nox1 is often overexpressed in tumors
(27,28) and induces genome instability
(29) which may account for p53
inhibition even in the absence of p53 mutation (30). Inactivation of p53 function by gene
mutation or deregulation of wild-type p53 protein are common in
human cancers and are indeed associated with increased cancer
resistance to chemo- and radiotherapy (30). That is why significant efforts
towards reactivation of defective p53 are underway, because
functional p53 is considered a key factor for efficient antitumor
drug response and apoptotic clearance of cancer cells (31). In this respect, a molecule such as
GV that inhibits Nox1 activity may be an interesting agent to
restore p53 function for anticancer activity.
More interestingly, though, we found that GV could
directly induce p53/DNA binding and transactivation activities.
Although GV stimulated the transactivation function of p53, the
precise mechanism responsible for this activation needs to be
determined. Established pathways of p53 activation that involve
increased p53 stability or enhanced site-specific DNA binding
(32–34) appeared to play a role in the p53
activation response to GV-treatment. The stabilization of p53 could
depend on GV-induced DNA damage, as evidenced by H2AX
phosphorylation in the position of Ser139 producing γH2AX that in
general occurs in response to formation of double strand brakes
(DSB) and is an early sign of replication stalling (24). Thus, the oncosuppressor p53 is
activated in response to a variety of cellular stress signals,
including DNA damage (1). However,
different mechanisms cannot be excluded and need further
evaluation.
In previous studies we reported that ADF
glioblastoma cells carried an endogenous wild-type p53 whose
transcriptional activity could not be activated by chemotherapeutic
agent such as ADR, although the molecular mechanisms of such
inhibition are still elusive (19,20).
In the present study, GV was able to activate endogenous p53 in ADF
cells, suggesting that different mechanisms other than DNA damage
might be responsible of p53 activation in this cell line. In
summary, our results indicate that while p53 induction by GV
appears to be mediated to a large extent by increased p53
stability, it is unclear whether GV can have additional direct or
indirect effects that induce p53 transactivation activity, such as
protein phosphorylation and acetylation that may modulate the
binding of p53 binding to DNA (35). Future experiments will be required
to clarify this issue.
The role of p53 in GV-induced antitumor activity was
evaluated by using cells with lack of (HCT116-p53−/−) or
reduced p53 activity (RKO-sip53). GV was able to efficiently induce
apoptotic cell death in wtp53-carrying cells and such biological
outcome was reduced in cells deprived of p53 function.
In conclusion, the present study, shows for the
first time the effect of GV on p53 activation, suggesting that the
GV-mediated anticancer effect may in part depend on induction of
the DNA binding and transactivation functions of p53. Future in
vitro and in vivo studies will help to give more insight
into unveiling the molecular mechanisms of GV activity and the
potential role of GV alone or in combination with chemotherapeutic
agents in cancer therapy.
Acknowledgements
This study was supported by grants
from the Italian Association for Cancer Research (AIRC, to G.D.O.)
and NIHAR47901, The Rabinowitch-David Foundation, Margolis
Foundation and Minsk Foundation (to J.L.A.). We thank G. Piaggio
for sharing reagents and M.P. Gentileschi for technical
assistance.
References
|
1.
|
Vousden KH and Lane DP: P53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar
|
|
2.
|
Schuler M and Green DR: Transcription,
apoptosis and p53: catch-22. Trends Genet. 2:182–187. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Vazquez A, Bond EE, Levine AJ and Bond LG:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Disc. 7:979–984. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Muller PAJ and Vousden KH: P53 mutations
in cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar
|
|
5.
|
Puca R, Nardinocchi L, Sacchi A, Rechavi
G, Givol D and D’Orazi G: HIPK2 modulates p53 activity towards
pro-apoptotic transcription. Mol Cancer. 8:1–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Puca R, Nardinocchi L, Starace G, Rechavi
G, Sacchi A, Givol D and D’Orazi G: Nox1 is involved in p53
deacetylation and suppression of its transcriptional activity and
apoptosis. Free Rad Biol Med. 48:1338–1346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Arbiser JL, Petros J, Kiafter R,
Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy
S, Arnold RS and Lambeth JD: Reactive oxygen generated by Nox1
triggers the angiogenic switch. Proc Natl Acad Sci USA. 99:715–720.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Block K and Gorin Y: Aiding and abetting
roles of NOX oxidases in cellular transformation. Nat Rev Cancer.
12:627–637. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Docampo R and Moreno SN: The metabolism
and mode of action of gentian violet. Drug Metab Rev. 22:161–178.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Maley AM and Arbiser JL: Gentian Violet: A
19th century drug re-emerges in the 21st century. Exp Dermatol. Oct
7–2013. View Article : Google Scholar
|
|
11.
|
Perry BN, Govindarajan B, Bhandarkar SS,
Knaus UG, Valo M, Sturk C, Carrillo CO, Sohn A, Cerimele F, Dumont
D, Losken A, Williams J, Brown LF, Tan X, Yancopoulos GD and
Arbiser JL: Pharmacologic blockade of angiopoietin-2 is efficacious
against model hemangiomas in mice. J Invest Dermatol.
126:2316–2322. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lapidoth M, Ben-Amitai D, Bhandarakar S,
Fried J and Arbiser JL: Efficacy of topical application of eosin
for ulcerated hemangiomas. J Am Acad Dermatol. 60:350–351. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Arbiser JL, Bips M, Seidler A, Bonner MY
and Kovach C: Combination therapy of imiquimod and gentian violet
for cutaneous melanoma metastases. J Am Acad Dermatol. 67:e81–e33.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ingraham MA: The bacteriostatic action of
Gentian Violet and dependence on the oxidation-reduction potential.
J Bacteriol. 26:573–598. 1933.PubMed/NCBI
|
|
15.
|
Bhandarkar SS, Jaconi M, Fried LE, Bonner
MY, Lefkove B, Govindarajan B, Perry BN, Parhar R, Mackelfresh J,
Sohn A, Stouffs M, Knaus U, Yancopoulos G, Reiss Y, Benest AV,
Augustin HG and Arbiser JL: Fulvene-5 potently inhibits NAPDH
oxidase 4 and blocks the growth of endothelial tumors in mice. J
Clin Invest. 119:2359–2365. 2009.PubMed/NCBI
|
|
16.
|
Zhang X, Zheng Y, Fried LE, Du Y, Montano
SJ, Sohn A, Lefkove B, Holmgren L, Arbiser JL, Holmgren A and Lu J:
Disruption of the mitochondrial thioredoxin system as a cell death
mechanism of cationic triphenylmethanes. Free Rad Biol Med.
50:811–820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Newick K, Cunniff B, Preston K, Held P,
Arbiser JL, Pass H, Mossmann B, Shukla A and Heintz N:
Peroxiredoxin 3 is a redox-dependent target of thiostrepton in
malignant mesothelioma cells. PloS One. 7:e394042012.PubMed/NCBI
|
|
18.
|
Puca R, Nardinocchi L, Gal H, Rechavi G,
Amariglio N, Domany E, Notterman DA, Scarsella M, Leonetti C,
Sacchi A, Blandino G, Givol D and D’Orazi G: Reversible dysfunction
of wild-type p53 following homeodomain-interacting protein kinase-2
knockdown. Cancer Res. 68:3707–3714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Biroccio A, Del Bufalo D, Ricca A,
D’Angelo C, D’Orazi G, Sacchi A, Soddu S and Zupi G: Increase of
BCNU sensitivity by wt-p53 gene therapy in glioblastoma lines
depends on the administration schedule. Gene Ther. 6:1064–1072.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
D’Avenia P, Porrello A, Berardo M,
D’Angelo M, Soddu S, Arcangeli G, Sacchi A and D’Orazi G: TP53-gene
transfer induces hupersensitivity to low-dose of X-rays in
glioblastoma cells: a strategy to convert a radio-resistant
phenotype into a radiosensitive one. Cancer Lett. 231:102–112.
2006.PubMed/NCBI
|
|
21.
|
Komarov PG, Komarova EA, Kondratov RV,
Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV: A chemical
inhibitor of p53 that protects mice from the side effects of cancer
therapy. Science. 285:1733–1737. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chen C and Hokayama H: High-efficiency
transformation of mammalian cells by plasmid DNA. Mol Cell Biol.
7:2745–2752. 1987.PubMed/NCBI
|
|
23.
|
Di Stefano V, Soddu S, Sacchi A and
D’Orazi G: HIPK2 contributes to PCAF-mediated p53 acetylation and
selective transactivation of p21Waf1 after non-apoptotic DNA
damage. Oncogene. 24:5431–5442. 2005.PubMed/NCBI
|
|
24.
|
Podhorecka M, Skladanowski A and Bozko P:
H2AX phosphorylation: its role in DNA damage response and cancer
therapy. J Nucl Acids ID. 9201612010.PubMed/NCBI
|
|
25.
|
Arbiser JL: Gentian violet is safe. J Am
Acad Derm. 61:3592009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lim SD, Sun C, Lambeth JD, Marshall F,
Amin M, Chung L, Petros JA and Arnold RS: Increased Nox1 and
hydrogen peroxide in prostate cancer. Prostate. 62:200–207. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Laurent E, McCoy JW, Macinz RA, Liu W,
Cheng G, Robine S, Papkoff J and Lambeth JD: Nox1 is over-expressed
in human colon cancers and correlates with activating mutations in
K-Ras. Int J Cancer. 123:100–107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Chiera F, Meccia E, Degan P, Aquilina G,
Pietraforte D, Minetti M, Lambeth JD and Bignami M: Overexpression
of human NOX1 complex induces genome instability in mammalian
cells. Free Rad Biol Med. 44:332–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Brown CJ, Lain S, Verma CS, Fersht AR and
Lane DP: Awakening guardian angels: drugging the p53 pathway. Nat
Rev Cancer. 9:862–873. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Selivanova G and Wiman KG: Reactivation of
mutant p53: molecular mechanisms and therapeutical potential.
Oncogene. 26:2243–2254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Oren M: Regulation of the p53 tumor
suppressor protein. J Biol Chem. 274:36031–36034. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Meek DW: Mechanisms of switching on p53: a
role for covalent modification? Oncogene. 18:7666–7675. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Hirao A, Kong YY, Matsuoka S, Wakeham A,
Ruland J, Yoshida H, Liu D, Elledge SJ and Mak TW: DNA
damage-induced activation of p53 by the checkpoint kinase Chk2.
Science. 287:1824–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hecker D, Page G, Lohrum M, Weiland S and
Scheidtmann KH: Complex regulation of the DNA-binding activity of
p53 by phosphorylation: differential effects of individual
phosphorylation sites on the interaction with different binding
motifs. Oncogene. 12:953–961. 1996.
|