Introduction
Ovarian cancer is the second most common gynecologic
cancer in women and the leading cause of cancer death of
gynecologic malignancy in the world (1). More than 90% ovarian cancers are
believed to arise from the surface epithelium of the ovary and
frequently absent of early signs and symptoms; thus most ovarian
cancer patients are diagnosed at advanced stage of disease, which
makes curable surgery infrequent and has a relatively poor
prognosis. For past decades, chemotherapy has been a general
standard of care for ovarian cancer with highly variable protocols
and used alone or after surgery to treat any residual disease
(2). Cisplatin-based chemotherapy
is used as a primary treatment of ovarian cancer and its
combination with other agents has become standard chemotherapy for
treatment of advanced ovarian cancer, but prolonged drug treatment
results in development of acquired drug resistance impeding
successful treatment (3). The
antineoplastic effect of cisplatin is mediated by formation of DNA
adducts and inter- and intra-strand crosslinks (4). These adducts distort the DNA template
with deceleration of cells in S phase followed with G2 phase arrest
(5) and also result in diverse
effects, including DNA synthesis inhibition, RNA transcription
suppression, cell cycle arrest and apoptosis. Cisplatin resistance
is multifactorial and rather complicated, including reduced
platinum accumulation and enhanced platinum detoxification and
metabolism in cells, altered DNA damage repair, activation of
phospholipid kinase and phosphatidyl inositol 3-kinase and other
signaling pathways ultimately causing dysregulation of apoptotic
pathway (6). Thus, this
disappointing outcome strongly suggests that a better understanding
of the mechanisms of chemoresistance could lead to novel
therapeutic strategies for effective control of ovarian cancer.
Our study is focusing on the Notch signaling. A
previous study demonstrated that activation of the Notch signaling
was linked to chemoresistance of pancreatic cancer to cisplatin
(7) and inhibition of Notch
signaling with γ-secretase inhibitors could sensitize colon cancer
cells to chemotherapy and was synergistic with some antineoplastic
agents (8). Indeed, the Notch
signaling pathway plays a key role in the proliferation and
differentiation of many tissues. This evolutionarily conserved
pathway can regulate critical cell fate decision (9). In mammals, the Notch family consists
four receptors (i.e., Notch1 to Notch4) and five ligands (Jagged-1,
Jagged-2, Delta-like-1, Delta-like-3 and Delta-like-4) (10). Notch ligands and receptors are type
I membrane proteins that regulate cell fate during cell-cell
contact (10–12). Receptor-ligand interaction between
two neighboring cells leads to γ-secretase-mediated proteolytic
release of the Notch intracellular domain (NICD) (13). NICD then translocates into the
nucleus, and in turn interacts with the transcriptional cofactor
CBF1 and transactivates target genes, such as Hes and Hey families
to affect numerous pathways involving cell-fate determination
(14,15). Abnormal Notch signaling has been
documented in many cancers, including ovarian cancer (16,17).
Overexpression of Notch proteins was associated with poor prognosis
of different cancer patients (18)
and with tumor de-differentiation in ovarian cancer (19). Molecularly, activation of Notch
proteins are triggered by γ-secretase, which cleaves the Notch
receptor to activate the pathway (13). Previous studies showed that
γ-secretase inhibitors were able to inhibit tumor cell viability
and induced apoptosis in different cancer cell lines (20,21).
We found that γ-secretase inhibitor (DAPT)-blocked Notch signaling
reduced viability of ovarian cancer A2780 cells but induced them to
undergo apoptosis (22). In this
study, we hypothesized that inhibition of Notch signaling by DAPT
could sensitize drug-resistant ovarian cancer cells to cisplatin
chemotherapy. We assessed the effects of DAPT on sensitizing
cisplatin-resistant ovarian cancer A2780/CP70 and OV2008/C13 cells
to cisplatin-induced cell death and the underling molecular
events.
Materials and methods
Cell culture and reagents
Two pairs of cisplatin-sensitive and
cisplatin-resistant human ovarian cancer cell lines A2780,
A2780/CP70 and OV2008, OV2008/C13 were kindly provided by Dr Jun Hu
(The Third Military Medical University, Chongqing, China) and
maintained in RPMI-1640 medium containing 10% heat-inactivated
fetal bovine serum (FBS; all from Xin Xing Tang Biotechnology
Company, Beijing, China) at 37°C in a humidified 5% CO2
atmosphere. For cisplatin treatment, cells were maintained in
medium with the desired doses of cisplatin for 1 h and then washed
with PBS and followed by incubation in fresh drug-free medium for
varying times post-treatment.
Cell viability MTT assay
Cells (5×103) were seeded in 96-well cell
culture plates, treated with different concentrations of
γ-secretase inhibitor
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl
ester (DAPT), cisplatin (all from Sigma-Aldrich, St. Louis, MO,
USA), or combination for 72 h. For combination experiments, ovarian
cancer A2780/CP70 and OV2008/C13 cells were treated for 72 h with
DAPT (30 μmol/l) either 24 h before or after cisplatin (3 or
6 μmol/l) treatment and then subjected to the MTT assay.
Specifically, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MMT) was added to the cultures and the cells were
incubated for an additional 4 h. The resulting formazan crystals
were solubilized by addition of 150 μl dimethyl sulfoxide
(DMSO) to each well. The optical density was measured at 570 nm
using a microplate reader and cell viability was determined by the
formula: cell viability (%) = (absorbance of the treated wells -
absorbance of the blank control wells) / (absorbance of the
negative control wells - absorbance of the blank control wells) ×
100%. All experiments were performed in triplicate and repeated at
least three times. Drug interactions and isobologram were analyzed
using CalcuSyn software (Biosoft, Beijing, China).
Colony formation assay
Ovarian cancer A2780/CP70 and OV2008/C13 cells
(5×104/ml) were seeded into 6-well plates according to
the manufacturer’s instructions. Cells were treated with DAPT (30
μmol/l) either 24 h before or after cisplatin (3 or 6
μmol/l) treatment. After 14 days of incubation at 37°C in a
humidified atmosphere containing 5% CO2 in air, colonies
were counted using an inverted microscope (Leica, Heidelberg,
Germany).
Flow cytometric cell cycle assay
The cell cycle was analyzed using flow cytometry.
Briefly, cells (1×106) were collected and washed in
phosphate-buffered saline (PBS), then fixed in 75% ice-cold alcohol
for 30 min at 4°C. After washing with ice-cold PBS three times,
cells were resuspended in 1 ml of PBS containing 40 μg of
propidium iodide (Sigma-Aldrich) and 100 μg of RNase A
(Sigma-Aldrich) and incubated for 30 min at 37°C. Samples were then
analyzed by FACS (BD Immunocytometry Systems, San Jose, CA, USA).
Each experiment was repeated for at least three times.
ELISA apoptosis assay
A Cell Death Detection ELISA kit (Roche, Shanghai,
China) was used to detect apoptosis in treated cells according to
the protocol provided by the manufacturer. Briefly, cell culture
supernatants were washed away to remove fragmented DNA from
necrotic cells, and then cells were lysed and loaded into
microtiter plate modules coated with an anti-histone antibody for
incubation for 45 min at room temperature. Next, samples were
incubated with the anti-DNA peroxidase followed by color
development with ABTS substrate. After that, the absorbance rates
of these samples were measured using a microplate reader (SLT,
Spectra LabInstruments Deutschland GmbH) at 405 and 490 nm
(reference wavelength).
RNA isolation and quantitative
RT-PCR
Total RNA was isolated from ovarian cancer cells
using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according
to the manufacturer’s instructions and then reverse transcribed
into cDNA using an PrimeScript™ RT reagent kit (Takara, Dalian,
China). These cDNA samples were then amplified in the ABI 7500
system (Applied Biosystems) using SYBR Premix Ex Taq™ II (Takara).
Primers used for Hes1 were 5′-TGGAAAT GACAGTGAAGCACCTC-3′ and
5′-TCGTTCATGCACTC GCTGAAG-3′. The internal control β-actin primers
were 5′-TGGCACCCAGCACAATGAA-3′ and 5′-CTAAGTCATAG
TCCGCCTAGAAGCA-3′. Thermocycling was set as follows: 94°C for 5
min, 40 cycles of 94°C for 30 min, 55°C for 30 min and 72°C for 60
min, and a final extension at 72°C for 10 min and then permanently
stored at 4°C. Relative quantitation of mRNA expression levels was
determined using the relative standard curve method according to
the manufacturer’s instructions (Applied Biosystems).
Protein extraction and western blot
analysis
Total proteins from ovarian cancer A2780/CP70 and
OV2008/C13 cells were lysed in a lysis buffer containing NaCl,
sodium desoxycholate, sodium dodecyl sulfate and Tris and incubated
at 4°C for 15 min. The protein concentrations were determined using
the Bio-Rad assay system (Bio-Rad, Hercules, CA, USA). These
protein samples were then fractionated using sodium dodecyl sulfate
polyacrylamide (10%) gels for electrophoresis (SDS-PAGE) and then
transferred onto a nitrocellulose membrane (Kenker, USA). For
western blotting, the membranes were blocked in 5% non-fat milk in
Tris-buffered saline containing 0.1% Tween-20 (TBS-T) and then
incubated with appropriate primary antibodies overnight at 4°C.
Horseradish peroxidaseconjugated anti-goat IgG was used as the
secondary antibody, and the protein bands were visualized using the
enhanced chemiluminescence (ECL) method (GE Healthcare, USA) and
quantified by using laser densitometry. The data were summarized as
the mean of 3 independent experiments with the standard deviation.
The membranes were then stripped by incubated for 30 min at 50°C in
a buffer that contained 2% SDS, 62.5 mmol/l Tris (pH 6.7), and 100
mmol/l 2-mercaptoethanol and further washed and incubated with the
desired primary antibody. Antibodies against Hes1, cyclin B1,
caspase-3, Bcl-2, and β-actin were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA).
Statistical analysis
The data were expressed as means ± standard error.
The Student’s t-test was performed to analyze the data between
groups. A P-value <0.05 was considered as statistically
significant.
Results
DAPT potentiates cisplatin-reduced
viability of ovarian cancer cells in a drug sequence-dependent
manner
In this study, we first determined whether
γ-secretase inhibitor DAPT was able to sensitize ovarian cancer
cells to low-dose cisplatin-reduced cell viability. We treated
A2780/CP70 and OV2008/C13 cells with various concentrations of DAPT
up to 90 μmol/l and cisplatin up to 9 μmol/l under
two different drug administration scenarios, i.e., an initial 24-h
DAPT exposure followed by 72-h cisplatin treatment or vice versa.
Cell viability was assessed and the values of inhibiting
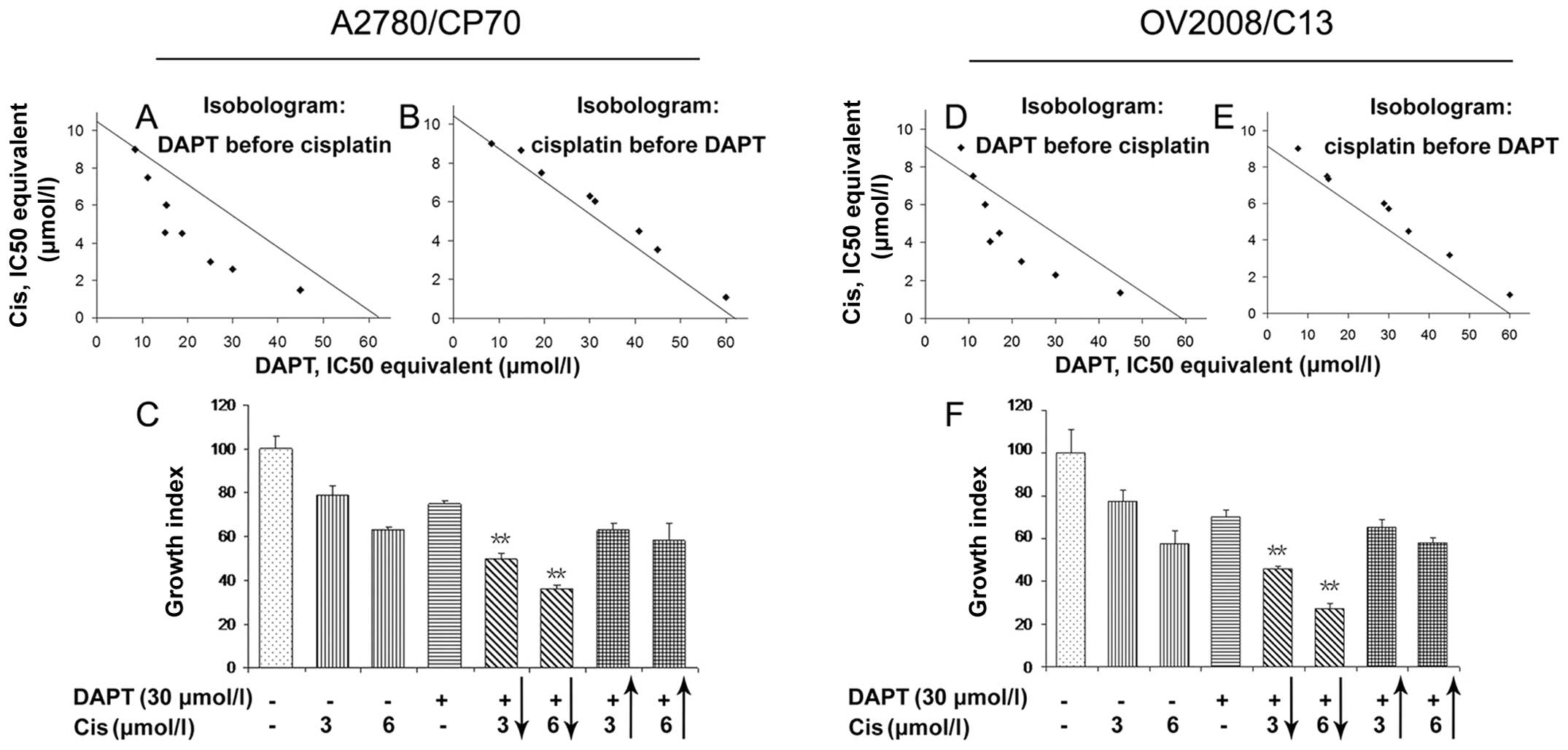
concentration (IC), were calculated. As shown in Fig. 1A and D, if DAPT was administered
before cisplatin, DAPT can synergistically sensitize cisplatin
anti-tumor activity in cisplatin-resistant ovarian cancer cell
lines, i.e., most of IC50 were below the
combination-isobol line. On the other hand, if DAPT was
administered after cisplatin, DAPT was additive or antagonistic
rather than synergistic effects with cisplatin, i.e., most of
IC50 were above the combination-isobol line (Fig. 1B and E). After that, we selected
two cisplatin doses (3 and 6 μmol/l) that kill ∼20–40% of
ovarian cancer cells and combined them with 30 μmol/l DAPT.
Data showed that pretreatment of tumor cells with DAPT increased
the potency of cisplatin-reduced cell viability, e.g., treatment of
tumor cells with 3 and 6 μmol/l cisplatin for 72 h caused
∼21 and 38% reduction of viability of A2780/CP70 cells,
respectively. In contrast, tumor cell viability was reduced to 51
and 64% when pretreated with DAPT (Fig. 1C and F and Table I), whereas tumor cell viability was
only reduced to 37 and 42% for 3 and 6 μmol/l cisplatin,
respectively, if cisplatin was administrated before DAPT (Table II).
 | Table I.Reduced cell viability (%) by DAPT
pretreatment and then cisplatin addition. |
Table I.
Reduced cell viability (%) by DAPT
pretreatment and then cisplatin addition.
| DAPT
(μmol/l)
|
|---|
| Cis
(μmol/l) | 0 | 15 | 30 | 45 | 60 | 75 | 90 |
IC50 |
|---|
| 0 | 0 | 13 | 25 | 35 | 50 | 57 | 65 | 61.88 |
| 1.5 | 15 | 31 | 42 | 55 | 62 | 67 | 70 | 36.93 |
| 3 | 21 | 38 | 51 | 67 | 73 | 77 | 81 | 24.97 |
| 4.5 | 27 | 46 | 58 | 75 | 78 | 83 | 86 | 18.79 |
| 6 | 38 | 52 | 64 | 82 | 86 | 89 | 90 | 15.28 |
| 7.5 | 43 | 61 | 73 | 88 | 89 | 92 | 93 | 11.13 |
| 9 | 49 | 70 | 81 | 91 | 92 | 94 | 96 | 8.33 |
|
IC50 | 10.39 | 4.56 | 2.58 | 1.47 | 1.09 | 0.93 | 0.85 | |
| Table II.Reduced cell viability (%) by
cisplatin pretreatment and then DAPT addition. |
Table II.
Reduced cell viability (%) by
cisplatin pretreatment and then DAPT addition.
| DAPT
(μmol/l)
|
|---|
| Cis
(μmol/l) | 0 | 15 | 30 | 45 | 60 | 75 | 90 |
IC50 |
|---|
| 0 | 0 | 13 | 25 | 35 | 50 | 57 | 65 | 61.88 |
| 1.5 | 15 | 22 | 30 | 38 | 54 | 59 | 66 | 55.61 |
| 3 | 21 | 28 | 37 | 44 | 55 | 60 | 67 | 48.33 |
| 4.5 | 27 | 32 | 41 | 56 | 61 | 63 | 69 | 40.83 |
| 6 | 38 | 39 | 42 | 59 | 63 | 67 | 72 | 31.25 |
| 7.5 | 43 | 48 | 54 | 62 | 65 | 68 | 73 | 19.41 |
| 9 | 49 | 57 | 63 | 64 | 66 | 70 | 75 | 8.32 |
|
IC50 | 10.39 | 8.65 | 6.27 | 3.51 | 1.09 | 0.53 | 0.13 | |
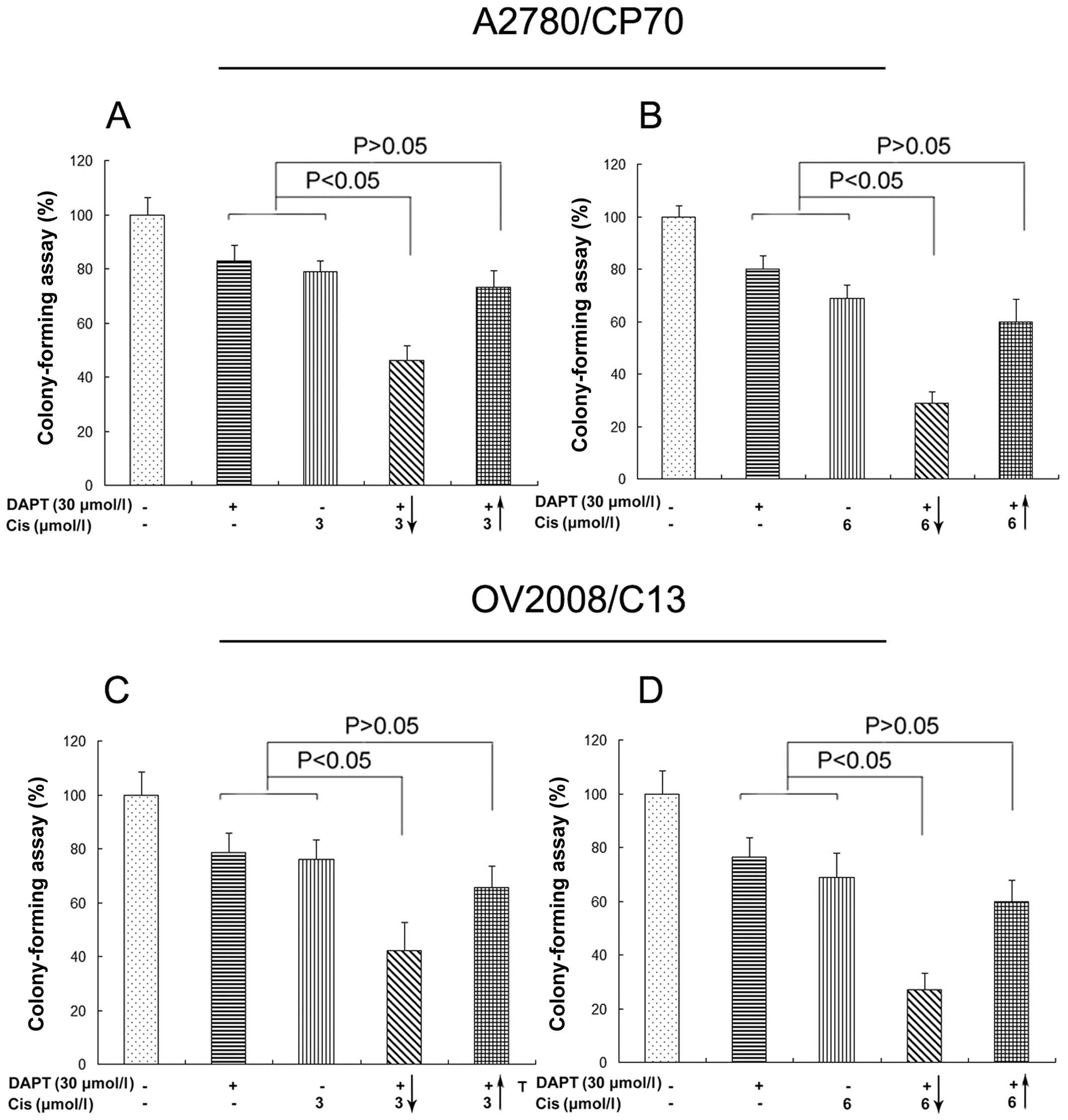
Furthermore, we also determined the effects of their
combination in regulation of tumor cell colony formation capacity.
Our data showed that pretreatment of tumor cells with DAPT
increased the potency of cisplatin-reduced colony formation in
vitro (Fig. 2).
Combination of DAPT with cisplatin
arrests cisplatin-resistant ovarian cancer cells in G2 phase of
cell cycle
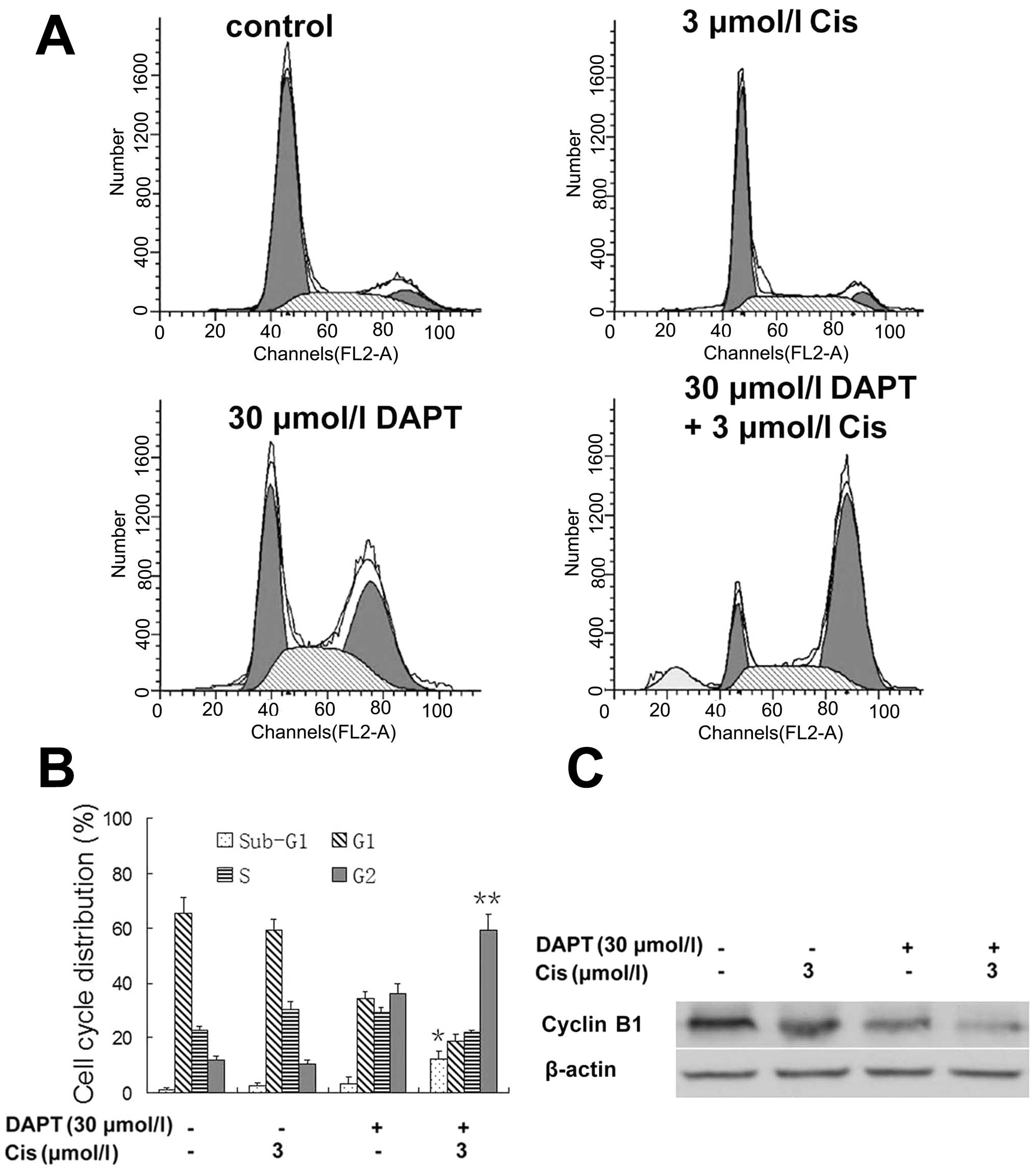
We next assessed the changed cell cycle after their
treatment. In untreated control cells, the percentage of cells in
G1, S, and G2 phases were 65.42, 22.73 and 11.85%, respectively,
while 3 μmol/l cisplatin treatment had no significant effect
on changes in the cell cycle distributions, whereas 30
μmol/l DAPT alone caused cell cycle redistribution to G2
phase. In contrast, DAPT-potentiated cisplatin treatment at the
above named dose resulted in a pronounced G2 arrest (% of G1, S,
and G2 phase cells was 18.90, 21.85 and 59.25%, respectively). We
also found a high proportion of sub-G1 phase population (apoptotic
cells) in the DAPT-cisplatin treated tumor cells (Fig. 3A and B). Molecularly,
DAPT-cisplatin treatment reduced the levels of cyclin B1 protein
(Fig. 3C).
DAPT enhances cisplatin-induced apoptosis
in ovarian cancer cells
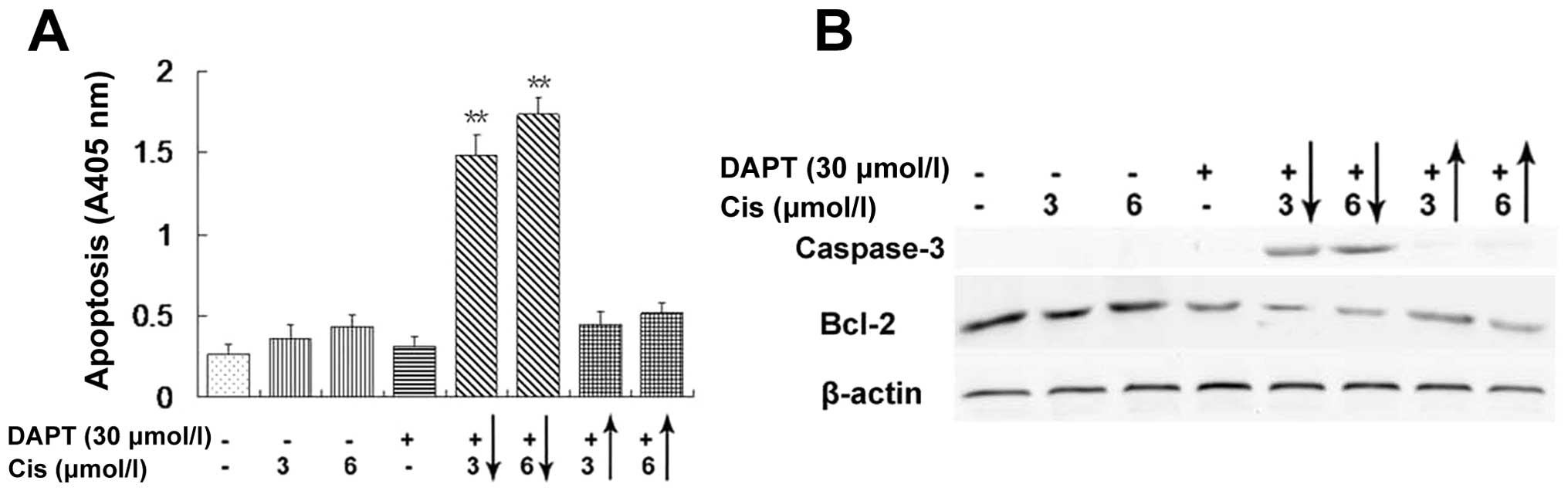
Since DAPT combination with low dose of cisplatin
had a high proportion of sub-G1 phase population, we determined
apoptosis levels in A2780/CP70 cells. As shown in Fig. 4A, treatment of A2780/CP70 cells
with 30 μmol/l DAPT or 3 or 6 μmol/l cisplatin caused
negligible increase in tumor cell apoptosis over the background.
However, pretreatment of tumor cells with DAPT for 24 h and then
with cisplatin for 72 h caused a significant increase in apoptosis
(Fig. 4A, columns 5 and 6),
whereas cisplatin treatment before DAPT addition at the same dose
did not show a significant increase in cell death (Fig. 4A, columns 7 and 8). These findings
further confirmed that the combination of DAPT with low dose of
cisplatin resulted in induction of apoptosis in A2780/CP70 cells in
a drug sequence-dependent manner. At the molecular level,
expression of apoptosis-related genes, such as caspase-3 and Bcl-2
proteins was also altered, i.e., caspase-3 was significantly higher
and Bcl-2 was lower in DAPT before cisplatin treatment (Fig. 4B, lanes 5 and 6), which were in
accordance with the cell death data in Fig. 4A.
DAPT treatment downregulated Notch
signaling and its target gene Hes1 expression in ovarian cancer
cells
We investigated potential targets of DAPT in ovarian
cancer cells. qRT-PCR data revealed that different concentrations
of DAPT (30 or 45 μmol/l) resulted in significant
downregulation of Notch1 mRNA (Fig. 5A
and B) and western blot analysis showed that levels of Notch1
protein were also downregulated by DAPT treatment in A2780/CP70 and
OV2008/C13 cells (Fig. 5A and B).
These findings indicated that the Notch1 signaling pathway was
efficiently suppressed by DAPT treatment in A2780/CP70 and
OV2008/C13 cells in a dose-dependent manner. The concentration of
30 μM was therefore used subsequently to effectively inhibit
the Notch1 pathway.
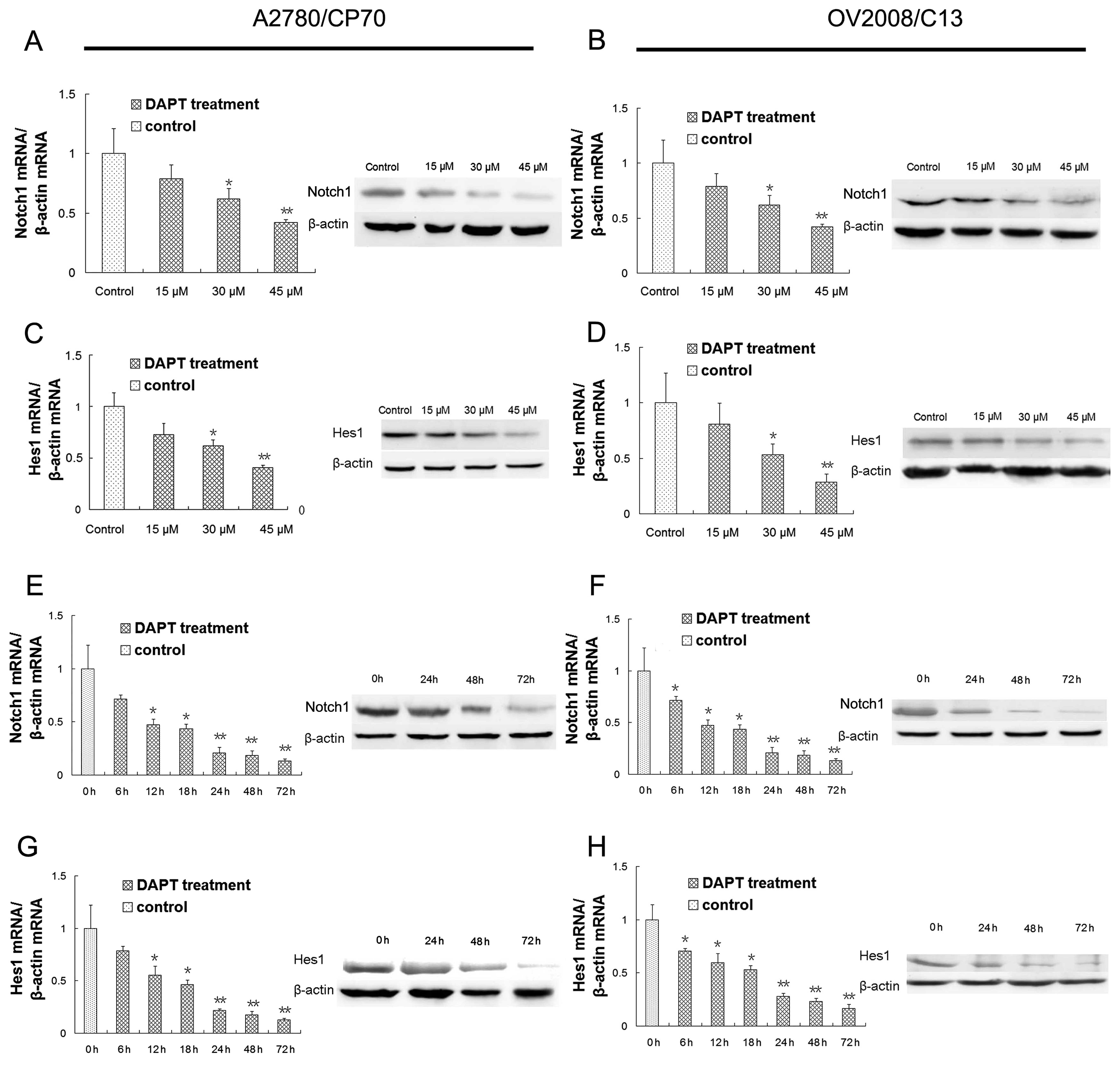 | Figure 5.Effects of DAPT and cisplatin on
regulation of gene expression. (A) Inhibition of Notch1 mRNA and
protein expression after 72 h DAPT treatment in ovarian cancer
cisplatin-resistant A2780/CP70 cells. (B) Inhibition of Notch1 mRNA
and protein expression after 72 h DAPT treatment in ovarian cancer
cisplatin-resistant OV2008/C13 cells. (C) Inhibition of Hes1 mRNA
and protein expression after 72 h DAPT treatment in A2780/CP70
cells. (D) Inhibition of Hes1 mRNA and protein expression after 72
h DAPT treatment in OV2008/C13 cells. (E) Inhibition of Notch1 mRNA
after 6, 12, 18, 24, 48 and 72 h and protein expression after 24,
48 and 72 h with 30 μM/l DAPT treatment in A2780/CP70 cells.
(F) Inhibition of Notch1 mRNA after 6, 12, 18, 24, 48 and 72 h and
protein expression after 24, 48 and 72 h with 30 μM/l DAPT
treatment in OV2008/C13 cells. (G) Inhibition of Hes1 mRNA after 6,
12, 18, 24, 48 and 72 h and protein expression after 24, 48 and 72
h with 30 μM/l DAPT treatment in A2780/CP70 cells. (H)
Inhibition of Hes1 mRNA after 6, 12, 18, 24, 48 and 72 h and
protein expression after 24, 48 and 72 h with 30 μM/l DAPT
treatment in OV2008/C13 cells. Control cells were treated with
dimethyl sulfoxide (DMSO). *P<0.05;
**P<0.01 compared to the controls. |
To further determine if DAPT could downregulate
expression of Notch1 downstream gene Hes1, we performed qRT-PCR and
western blot analyses and found that different concentrations of
DAPT (30 and 45 μmol/l) significantly inhibited levels of
Hes1 mRNA and protein (Fig. 5C and
D). These findings indicated that DAPT treatment downregulated
expression of Hes1 dose-dependently.
Moreover, we also performed time course treatments
for changed expression of these genes. Our data showed that the
altered expression of Notch1 was observed as early as 6 h after
DAPT (30 μmol/l) treatment and was more pronounced with a
longer period of treatment in A2780/CP70 and OV2008/C13 cells
(Fig. 5E and F). These data
suggested that the Notch1 signaling pathway was efficiently blocked
by DAPT treatment in A2780/CP70 and OV2008/C13 cells in a
time-dependent manner. Similarly, the altered expression of Hes1
gene was observed as early as 6 h after DAPT (30 μmol/l)
treatment and was more pronounced with a longer period of treatment
in A2780/CP70 and OV2008/C13 cells (Fig. 5G and H).
Discussion
Notch signaling is implicated in ovarian cancer
tumorigenesis (19,23). Our previous studies showed that
inhibition of Notch signaling with γ-secretase inhibitor DAPT
resulted in reduced tumor cell viability and induction of apoptosis
in ovarian cancer cells (22). In
this study, we assessed whether DAPT has the same effect on ovarian
cancer cells that are resistant to cisplatin and the underlying
molecular events. We found that pretreatment of ovarian cancer cell
lines with DAPT and then with cisplatin can synergistically
sensitize cisplatin antitumor activity in cisplatin-resistant
ovarian cancer cell lines, while cisplatin treatment with a delayed
DAPT treatment had an additive or antagonistic effects on these
cisplatin-resistant ovarian cancer cells. Similarly, these
treatments also inhibited tumor cell colony formation capacity,
arrested tumor cells at G2 phase of the cell cycle, but induced
apoptosis. Expression of the cell cycle-related gene cyclin B1 and
apoptosis-related gene Bcl-2 was suppressed but apoptosis-related
gene caspase-3 was activated by these treatments. Molecularly,
pretreatment of ovarian cancer cell lines with DAPT and then with
cisplatin downregulated Notch1 and Hes1 expression dose- and
time-dependently. This study demonstrated that DAPT pretreatment
could sensitize cisplatin-resistant human ovarian cancer cells to
cisplatin by downregulation of Notch signaling. Future in
vivo study need to confirm our current data before translating
into clinical trials.
To date, cisplatin is still widely used in the
treatment of various human cancers, including testicular, ovarian,
cervical, bladder, head and neck, esophageal and lung cancers
(24). In spite of the efficacy of
cisplatin-based treatment regimens, long-term cure is difficult to
obtain due to drug resistance (3),
although great efforts have been made to develop combining
chemotherapeutic agents to potentiate the effectiveness of current
cytostatic drugs and to overcome chemotherapy resistance (8). Thus, our present study could provide
a novel strategy by using combined agents to treat ovarian cancer.
Our data showed pretreatment of ovarian cancer cell lines with DAPT
and then with cisplatin synergistically sensitized cisplatin
antitumor activity in cisplatin-resistant ovarian cancer cell
lines. Our treatment regime could reduce the toxic dose of
cisplatin but achieved synergistic effects on ovarian cancer
cells.
Indeed, γ-secretase is a critical proteinase for
Notch protein activation via nicastrin ectodomain binding to the
N-terminus of Notch protein and cleavage of NICD (25). Thus, γ-secretase inhibitors can
prevent generation of the intracellular domain of Notch protein and
suppress the Notch activity (26).
Notch signaling is implicated in ovarian cancer tumori-genesis
(19,23). Recently, there has been increased
enthusiasm in targeting this pathway using γ-secretase inhibitors
for novel and effective cancer therapy strategy (27). For example, treatment of leukemia
using this strategy revealed a better efficacy but less side
effects of γ-secretase inhibitors (28). A γ-secretase inhibitor GSI could be
helpful in treating human T-cell acute lymphoblastic leukemia
(T-ALL) by inhibiting the Notch signaling (29) and combination therapy of
γ-secretase inhibitors with glucocorticoids improved the
anti-leukemic effects of γ-secretase inhibitors and reduced their
gut toxicity in vivo (30).
It has also been reported that combination of γ-secretase
inhibitors with chemotherapy might represent a novel approach for
treating metastatic colon cancers (8). In the present study, we investigated
the potential use of DAPT to sensitize the cisplatin-resistant
ovarian cancer A2780/CP70 and OV2008/C13 cells to cisplatin. We
found that DAPT pretreatment reduced cisplatin-resistant ovarian
cancer cells to low-dose cisplatin-induced cell death by
downregulation of the Notch signaling.
Previous studies reported that DAPT had
anti-proliferative activity against several human cancer cell lines
(22,31). This anti-proliferative effect was
due to induction of apoptosis and cell cycle arrest. Molecularly,
Bcl-2 and caspase-3 play an importance role in regulation of
apoptosis (32,33) and cyclin B1 is one of the key
molecules in the cell cycle modulation (34). Thus, our present study revealed an
accumulation of G2 cell cycle arrest after DAPT treatment and
DAPT-cisplatin treatment in cisplatin-resistant human ovarian
cancer A2780/CP70 cells. As G2 arrest is typically linked to DNA
damage response, we postulate that DAPT pretreatment-sensitized
A2780/CP70 cells to respond to DNA damage. Moreover, overexpression
of caspase-3 could sensitize breast cancer cells to drug-induced
apoptosis and enhance chemosensitivity (35). In contrast, Bc1-2 expression can
confer cisplatin-induced apoptosis in ovarian cancer cells
(36). Our current data showed
that inhibition of Notch signaling by DAPT sensitized
cisplatin-resistant human ovarian cancer cells and enhances
chemosensitivity associated with modulation of apoptosis-related
gene expression.
Nevertheless, γ-secretase inhibitors can cleave a
number of proteins, such as ErbB-4, CD44, E-cadherin and Notch
family proteins (37). In the
present study, we observed that DAPT resulted in downregulation of
Hes1 in ovarian cancer cells in a dose-dependent manner by
confirmed DAPT-inhibited Notch expression. However, it is unknown
whether there are other signaling pathways or other important
events in this process and further study is needed. Apoptosis
induced by cisplatin is another feature of cellular response to
combined DAPT and cisplatin treatment, which manifests as the
synergetic inhibitory effect on cisplatin-resistant ovarian cancer
cells, although the underlying molecular mechanisms mediating this
synergistic antitumor effect are not completely understood. Further
studies are required to better understand how the combination of
γ-secretase inhibitors with cisplatin treatment can sensitize
cisplatin-resistant ovarian cancer cells, especially the sequential
use of DAPT and cisplatin in combination for control of ovarian
cancer.
Acknowledgements
We thank Medjaden Bioscience Limited,
Hong Kong, China, for assistance in preparation of this
manuscript.
References
|
1.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2.
|
McGuire WP III and Markman M: Primary
ovarian cancer chemotherapy: current standards of care. Br J
Cancer. 89(Suppl 3): S3–S8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Stewart JJ, White JT, Yan X, et al:
Proteins associated with Cisplatin resistance in ovarian cancer
cells identified by quantitative proteomic technology and
integrated with mRNA expression levels. Mol Cell Proteomics.
5:433–443. 2006. View Article : Google Scholar
|
|
4.
|
Mellish KJ, Barnard CF, Murrer BA and
Kelland LR: DNA-binding properties of novel cis- and trans
platinum-based anticancer agents in 2 human ovarian carcinoma cell
lines. Int J Cancer. 62:717–723. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
O’Neill CF, Koberle B, Masters JR and
Kelland LR: Gene-specific repair of Pt/DNA lesions and induction of
apoptosis by the oral platinum drug JM216 in three human ovarian
carcinoma cell lines sensitive and resistant to cisplatin. Br J
Cancer. 81:1294–1303. 1999.
|
|
6.
|
Ohmichi M, Hayakawa J, Tasaka K, Kurachi H
and Murata Y: Mechanisms of platinum drug resistance. Trends
Pharmacol Sci. 26:113–116. 2005. View Article : Google Scholar
|
|
7.
|
Wang Z, Li Y, Kong D, et al: Acquisition
of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar
|
|
8.
|
Meng RD, Shelton CC, Li YM, et al:
gamma-Secretase inhibitors abrogate oxaliplatin-induced activation
of the Notch-1 signaling pathway in colon cancer cells resulting in
enhanced chemosensitivity. Cancer Res. 69:573–582. 2009. View Article : Google Scholar
|
|
9.
|
Kuroda Y, Murakami N, Morota M, et al:
Impact of concurrent chemotherapy on definitive radiotherapy for
women with FIGO IIIb cervical cancer. J Radiat Res. 53:588–593.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Lai EC: Notch signaling: control of cell
communication and cell fate. Development. 131:965–973. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Nickoloff BJ, Osborne BA and Miele L:
Notch signaling as a therapeutic target in cancer: a new approach
to the development of cell fate modifying agents. Oncogene.
22:6598–6608. 2003. View Article : Google Scholar
|
|
13.
|
Suwannarurk K, Bhamarapravatana K,
Thaweekul Y, Mairaing K, Poomtavorn Y and Pattaraarchachai J: A
1-year experience with liquid-based and conventional papanicolaou
smear in Thammasat University Hospital. J Med Assoc Thai. 94(Suppl
7): S47–S51. 2011.PubMed/NCBI
|
|
14.
|
Wagner U, Marth C, Largillier R, et al:
Final overall survival results of phase III GCIG CALYPSO trial of
pegylated liposomal doxorubicin and carboplatin vs paclitaxel and
carboplatin in platinum-sensitive ovarian cancer patients. Br J
Cancer. 107:588–591. 2012. View Article : Google Scholar
|
|
15.
|
Doll CM, Aquino-Parsons C, Pintilie M, et
al: The significance of tumoral ERCC1 status in patients with
locally advanced cervical cancer treated with chemoradiation
therapy: a multicenter clinicopathologic analysis. Int J Radiat
Oncol Biol Phys. 85:721–727. 2013. View Article : Google Scholar
|
|
16.
|
Miyamura T, Masuzaki H and Ishimaru T:
Conservative treatment of a cervical pregnancy with local
methotrexate injection. Int J Gynaecol Obstet. 45:62–63. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Rose SL, Kunnimalaiyaan M, Drenzek J and
Seiler N: Notch 1 signaling is active in ovarian cancer. Gynecol
Oncol. 117:130–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Chu D, Wang W, Xie H, et al: Notch1
expression in colorectal carcinoma determines tumor differentiation
status. J Gastrointest Surg. 13:253–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wang M, Wang J, Wang L, Wu L and Xin X:
Notch1 expression correlates with tumor differentiation status in
ovarian carcinoma. Med Oncol. 27:1329–1335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yoo J, Choi JY, Moon SH, et al: Prognostic
significance of volume-based metabolic parameters in uterine
cervical cancer determined using 18F-fluorodeoxyglucose positron
emission tomography. Int J Gynecol Cancer. 22:1226–1233. 2012.
|
|
21.
|
Sorosky JI: Endometrial cancer. Obstet
Gynecol. 120:383–397. 2012. View Article : Google Scholar
|
|
22.
|
Wang M, Wu L, Wang L and Xin X:
Down-regulation of Notch1 by gamma-secretase inhibition contributes
to cell growth inhibition and apoptosis in ovarian cancer cells
A2780. Biochem Biophys Res Commun. 393:144–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Hopfer O, Zwahlen D, Fey MF and Aebi S:
The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer.
93:709–718. 2005. View Article : Google Scholar
|
|
24.
|
Tsang RY, Al-Fayea T and Au HJ: Cisplatin
overdose: toxicities and management. Drug Saf. 32:1109–1122. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Jones ME, van Leeuwen FE, Hoogendoorn WE,
et al: Endometrial cancer survival after breast cancer in relation
to tamoxifen treatment: pooled results from three countries. Breast
Cancer Res. 14:R912012. View
Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ma K, Wen HW and Liao QP: Expression of
Notch3 and Notch intracellular domain in ovarian carcinoma and
effect of N-[N-(3,5-difluorophenyl) acetyl-L-alanyl]-S-phenyl
glycine t-butyl ester on ovarian carcinoma cell. Zhonghua Fu Chan
Ke Za Zhi. 45:921–926. 2010.In Chinese.
|
|
27.
|
Ma XG, Wang YM, Xue FX, et al:
Clinicalpathological characteristics of Lynch syndrome related
epithelial ovarian cancer. Zhonghua Fu Chan Ke Za Zhi. 47:201–204.
2012.In Chinese.
|
|
28.
|
van den Akker T, Beltman J, Leyten J, et
al: The WHO maternal near miss approach: consequences at Malawian
District level. PLoS One. 8:e548052013.PubMed/NCBI
|
|
29.
|
Masuda S, Kumano K, Suzuki T, et al: Dual
antitumor mechanisms of Notch signaling inhibitor in a T-cell acute
lymphoblastic leukemia xenograft model. Cancer Sci. 100:2444–2450.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Kohorn EI: Imaging practices in the
diagnosis and management of gestational trophoblastic disease: an
assessment. J Reprod Med. 57:207–210. 2012.PubMed/NCBI
|
|
31.
|
Sun XM, Wen HW, Chen CL and Liao QP:
Expression of Notch intracellular domain in cervical cancer and
effect of DAPT on cervical cancer cell. Zhonghua Fu Chan Ke Za Zhi.
44:369–373. 2009.In Chinese.
|
|
32.
|
Korsmeyer SJ: Regulators of cell death.
Trends Genet. 11:101–105. 1995. View Article : Google Scholar
|
|
33.
|
Drexler HC: Programmed cell death and the
proteasome. Apoptosis. 3:1–7. 1998. View Article : Google Scholar
|
|
34.
|
Ito M: Factors controlling cyclin B
expression. Plant Mol Biol. 43:677–690. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Friedrich K, Wieder T, Von Haefen C, et
al: Overexpression of caspase-3 restores sensitivity for
drug-induced apoptosis in breast cancer cell lines with acquired
drug resistance. Oncogene. 20:2749–2760. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Williams J, Lucas PC, Griffith KA, et al:
Expression of Bcl-xL in ovarian carcinoma is associated with
chemoresistance and recurrent disease. Gynecol Oncol. 96:287–295.
2005. View Article : Google Scholar
|
|
37.
|
Beel AJ and Sanders CR: Substrate
specificity of gamma-secretase and other intramembrane proteases.
Cell Mol Life Sci. 65:1311–1334. 2008. View Article : Google Scholar : PubMed/NCBI
|