Introduction
Rhabdomyosarcoma (RMS) is the most common
soft-tissue sarcoma of infancy with an incidence of approximately
4–7 cases per million children (1). As RMS can arise from any striated
muscle in the body, the localization is very diverse. The most
common site of origin is the head and neck region (35%), but also
the genitourinary tract (24%) and the extremities (20%) are
favoured afflicted spots (2).
Histologically, RMS can be divided into two major subtypes.
Embryonal RMS is the most common variant and is characterized by an
early onset in children <10 years of age (3). Because of a fairly low tendency of
metastasis, its prognosis, especially of the botryoid variant, is
quite good (4). On the other hand,
there is the alveolar RMS known as the second most common RMS
subtype (3). Typical features of
this subtype are a later onset, mainly in adolescence, the
localization in extremities, and a worse prognosis, because of
rapid metastasis and poor response to treatment (5). The RMS subtypes are also
distinguishable on the molecular level. Embryonal RMS are commonly
characterized by loss of heterozygosity of the chromosomal region
11p15, which affects the locus of the insulin-like growth factor
II (IGF2) gene and leads to its overexpression (6). Alveolar RMS can bear two typical
reciprocal translocations, one between chromosome 1 and 13
(t(1;13)(p36;q14)), the other between chromosome 2 and 13
(t(2;13)(p35;q14)). This can fuse the PAX3 or the
PAX7 gene with the FKHR gene, which subsequently
results in abnormal activation of target genes and oncogenic
deregulation (7,8).
Currently, epigenetic alterations are discussed as
an important element in the regulation of various factors
implicated in tumor development. One of the most common molecular
features of cancer cells is aberrant DNA methylation of usually
unmethylated CpG islands located in the promoter regions of tumor
suppressor genes, which could lead to their transcriptional
suppression (9). Although a whole
plethora of epigenetically silenced genes has been described for
most cancers, the methylation status of RMS is still fairly
unknown. One of the few genes investigated in this tumor is Ras
association (RalGDS/AF-6) domain family member 1 (RASSF1A)
(10). The epigenetic
downregulation of RASSF1A is a common phenomenon in a
variety of childhood cancers and associated with poorer prognosis
in some of them (11,12). Nevertheless, because of the
important link between promoter methylation and the development and
progression of cancer the identification of epigenetically
regulated genes in RMS is of utmost importance.
A candidate gene for epigenetic regulation encodes
the bone morphogenetic protein 2 (BMP2), which belongs to the
transforming growth factor β superfamily. Although BMP2 was first
described as an initiator of enchondral bone formation (13), its function is way more
comprehensive playing important roles in embryonic development
during the formation and differentiation of various organs
(14–16). BMPs act via activation of
transmembrane serine/threonine-kinase-receptors, which are composed
of type I and type II subunits, namely BMPR1A, BMPR1B and BMPR2
(17). This heterodimerization
leads to phosphorylation and grouping of SMAD proteins and their
translocation to the nucleus. After binding to specific
DNA-sequences, several target genes, such as ID1-3
(inhibitor of DNA binding protein 1-3) are either going to
be activated or repressed (18).
Besides these physiological mechanisms of action, BMP2 is
also involved in tumorigenesis. There is growing evidence that
suggests BMP2 as an important tumor suppressor gene. It was
shown that different tumors often feature a genetic loss of
BMP2 expression (19).
Reconstitution of BMP2 function through recombinant BMP2 treatment
resulted in a reduced growth and even partially increased apoptosis
rate of tumor cells, as described for prostate carcinoma,
medulloblastoma, and myeloma (20–22).
Here, we aimed at identifying epigenetically
silenced genes in RMS in order to further the understanding of
tumor development and progression. Our finding that BMP2 is
heavily methylated and the gene thus transcriptionally inactive in
RMS cell lines along with the anti-proliferative effect mediated by
BMP2 reconstitution in vitro suggests a strong tumor
suppressive role of this protein, which might be of use to develop
future treatment options.
Materials and methods
Tumor cell lines
We used the three different RMS cell lines RD
(embryonal RMS, ATCC, Manassas, VA, USA), RH30 (alveolar RMS, DSMZ,
Braunschweig, Germany), and RMS13 (alveolar RMS, ATCC). The
embryonal RUCH2 as well as the alveolar RH4 and RH18 cell lines
were generously provided by Dr Beat Schäfer. All cell lines were
maintained as the suppliers recommended.
Real-time reverse transcription
polymerase chain reaction (RT-PCR)
Total RNA was extracted from fresh-frozen healthy
muscle tissue and RMS cell lines in TRI Reagent® RNA
isolation reagent (Molecular Research Center, Cincinnati, OH, USA).
Total RNA was depleted from DNA and subsequently purified using
RNeasy Mini Kit in combination with DNase set (Qiagen, Hilden,
Germany). Reverse transcription of total RNA was performed using
random hexamers (Roche Diagnostics, Penzberg, Germany) and
SuperScript® II reverse transcriptase (Invitrogen,
Karlsruhe, Germany). RT-PCR amplifications of the selected genes
were carried out with 40 ng complementary DNA, 10 μM forward
and reverse primer, and 2X iTaq SYBR-Green Supermix with ROX
(Bio-Rad Laboratories GmbH, München, Germany) in a final volume of
20 μl. RT-PCR reactions started with a primary denaturation
for 2 min at 95°C and thereupon were run for 40 cycles consisting
of 15-sec denaturation at 95°C, primer annealing for 15 sec at
55°C, and extension for 20 sec at 68°C. We used the following
primer pairs (5′→3′ orientation): BMP2, CAGCCAG CCGAGCCAA,
AATCTCCGGGTTGTTTTCCC; BMPR1A, CTACCAAACTGTGCTAATGCGC,
CAAATAGAGCTGAG TCCAGGAACC; BMPR1B, CCATTGTCCAGAAGACTC
AGTCAA, CACAGGCAACCCAGAGTCATC; BMPR2, TGGACGCATGGAATATTTGC,
GCTTACCCAGTCACTT GTGTGG; CDKN1A, AGGCTGAAGGGTCCCCAG, CGGC
GTTTGGAGTGGTAGAA; CDKN2A, AGGCAGTAACCA TGCCCG,
TTCCCGAGGTTTCTCAGAGC; CDKN2B, AGCTGAGCCCAGGTCTCCTAG,
CACCGTTGGCCGTAA ACTTAAC; ID1, AACGGCTGTTACTCACGCCT, CGATGA
CGTGCTGGAGAATCT; ID3, CCTGGACCCCCTGATGG,
GGCAAAAGCTCCTTTTGTCGT; RASSF1A, GGCGTCG TGCGCAAA,
GATCTTCTGCTCAATCTCAGCTTG; TBP, GCCCGAAACGCCGAATAT,
CCGTGGTTCGTGGCTC TCT. Primer sequences of the 92-gene screening
approach are available upon request. RT-PCR amplifications were
performed on a Mastercycler ep realplex2 S
(Eppendorf, Hamburg, Germany) and all experiments were performed in
doublets. Amplification of the housekeeping gene
TATA-box-binding-protein (TBP) was performed to
standardize the amount of sample RNA. Relative quantification of
gene expression was performed using the ΔΔct method as described
earlier (23).
Methylation analyses
Genomic DNA of RMS cell lines was extracted with
phenol and chloroform, ethanol precipitated and dissolved in TE
buffer following standard procedures. In order to get fully
methylated DNA as a positive control for methylation analyses, 10
μg genomic DNA extracted from blood of a healthy individual
was incubated with CpG methyltransferase M. SssI (4 U/μl,
New England Biolabs, Frankfurt, Germany) in 10X NE buffer 2 and SAM
(1:20) for 4 h at 37°C. After enzyme inactivation at 65°C, DNA was
precipitated with 0.3 μM sodium acetate and 100% ethanol.
Genomic DNA was bisulfite-treated using EpiTect®
Bisulfite Kit (Qiagen GmbH).
For bisulfite sequencing, we amplified
bisulfite-modified genomic DNA by using primers not specific for
methylation status (BMP2-BS-fw, CAAAGGGCACTTGGCCCCAGGG;
BMP2-BS-rev, CAAGTTATTCTCCCTGCAAGTTCA). We cloned the PCR products
into the pCR2.1 vector and transformed them into TOP10F’ (both from
Invitrogen). After the preparation of plasmid DNA by the use of
Qiagen plasmid mini kit (Qiagen), the clones were sent for
sequencing (MWG Biotech, Ebersberg, Germany).
For methylation-specific-PCR (MSP), we used
primer-pairs specific for either methylated or unmethylated CpGs
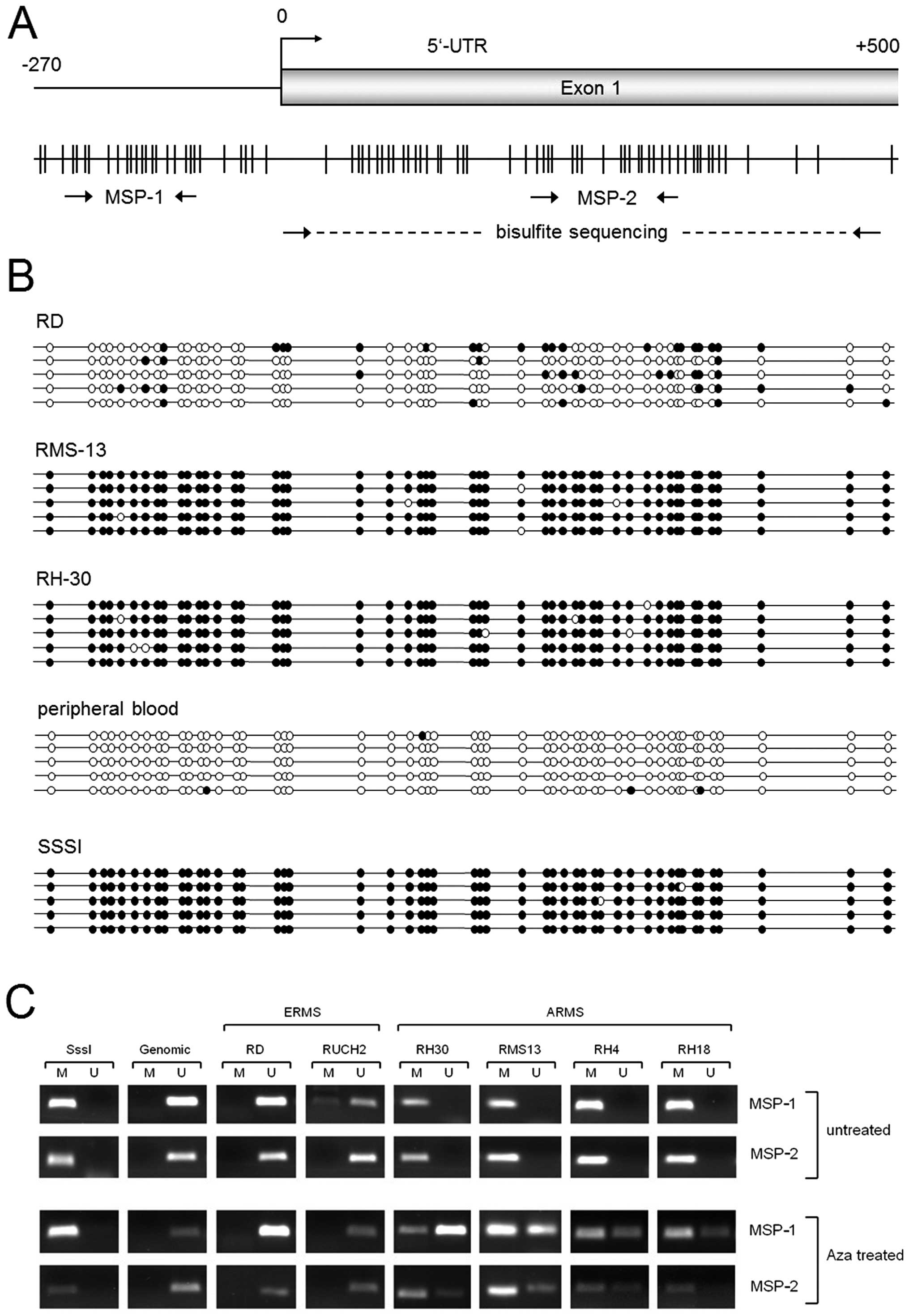
that bind to two different regions of the BMP2 promoter (Fig. 2A). The following primer sets were
used (5′→3′-orientation): BMP2 region 1 methylated,
GTAGAGCGCGTTATAGCGTC, CATC GCGACGCTAAAAAT; BMP2 region 1
unmethylated, GGT AGAGTGTGTTATAGTGTTGTGG, ACATCACAACACTA
AAAATCAACTC; BMP2 region 2 methylated, TGGTATCG AGATCGTCGTC,
TCCGAAACTCGAAAATCG; BMP2 region 2 unmethylated,
GTTTGGTATTGAGATTGTTGT TGT, AACTCCAAAACTCAAAAATCA. Primer design for
MSP was accomplished using Methyl Primer Express (Applied
Biosystems, Foster City, CA, USA) considering the following
criteria: primers cover at least three CpG dinucleotides in its
sequence, CpG percentage >55%, observed/expected CpG >65%,
and CpG island length between 300–2,000 bp. The MSP reaction
contained 225 ng DNA, 10 μM forward and reverse primer, 10
mM deoxyribonucleotide triphosphates, 25 mM MgCl2, 1 U
Hot Start Taq DNA polymerase, and 10X Hot Start PCR buffer
(Fermentas, St. Leon-Rot, Germany). Conditions for MSP were 95°C
for 4 min, followed by 38 cycles of 94°C for 30 sec, 60°C (BMP2
region 1) or 59°C (BMP2 region 2) for 30 sec and 72°C for 45 sec,
with a final extension cycle of 72°C for 10 min. The PCR products
were resolved by electrophoresis in a 2% agarose gel.
For DNA demethylation experiments, cells were seeded
in 5 or 25 cm2 cell flasks and incubated with 1
μM 5-aza-2′-deoxycytidine (5-Aza-dC; Sigma-Aldrich,
Steinheim, Germany) for 5 days. Demethylating agent-containing
media were changed every day.
Cell viability assay
For proliferation assays 5×103
cells/96-well plates were treated with recombinant human BMP2
(rhBMP2, R&D Systems, Minneapolis, MN, USA) in concentrations
of 50, 75, 100, and 200 ng/ml. The viability was assessed at 0, 24,
48 and 72 h using the Cell Proliferation Kit I (Roche Diagnostics)
according to the manufacturer’s protocol. Optical density was
measured after the addition of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
labeling reagent on the GENios microplate reader (Tecan Deutschland
GmbH, Crailsheim, Germany) at a wavelength of 595 nm.
Apoptosis analyses
For Annexin V-based apoptosis analysis, cells were
seeded in 6-well plates (5×105 cells/well) and treated
with 75 ng/ml rhBMP2 for 24, 48 and 72 h. Thereafter, the cells
were trypsinized, washed with PBS, and suspended in 500 μl
of calcium-containing binding buffer. Cy5-conjugated Annexin V
(1:100; BioVision, Mountain View, CA, USA) and 1 mM calcein AM
(Invitrogen) were added to the cell suspension. Early apoptotic
cells (Annexin V and calcein-positive) were detected using cell
fluorescence assays on a 2100 Bioanalyzer (Agilent Technologies,
Santa Clara, CA, USA).
Statistical analysis
Data were expressed as means plus standard
deviations and statistically subjected to Student’s unpaired
t-test. A level of p<0.05 was considered significant. BMP2
expression values and clinical data of the Williamson and
colleagues study (24) were
retrieved from the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/experiments/ETABM-1202/)
and analyzed with Mann-Whitney test and Spearman correlation using
GraphPad Prism 5.0.
Results
Identification of BMP2 as an
epigenetically silenced gene in RMS cell lines
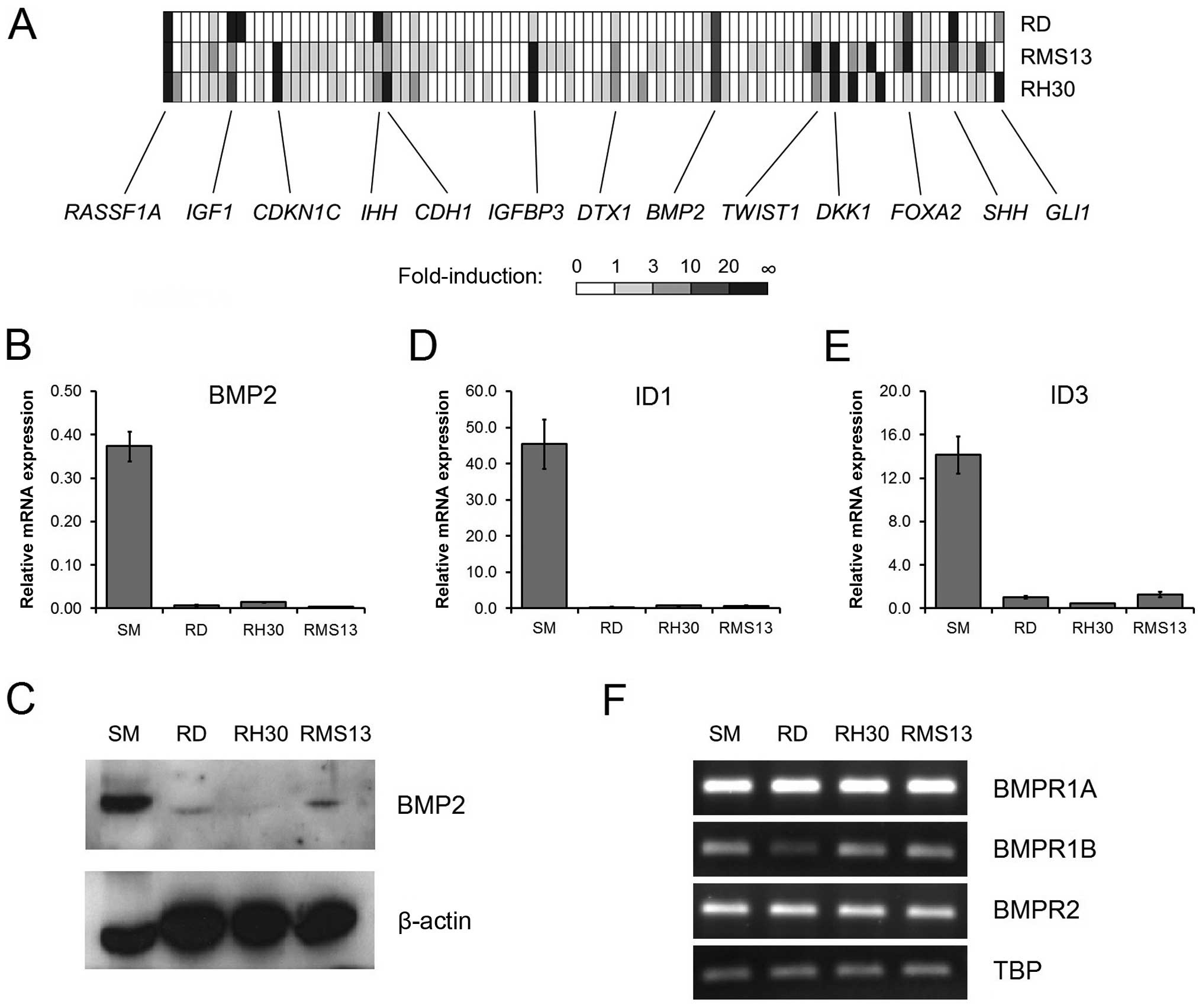
In a first step, we performed an expression
profiling analysis of three RMS tumor cell lines that were treated
with the demethylating agent 5-Aza-dC, which should result in the
re-expression of epigenetically silenced genes. By using an
in-house developed RT-PCR-based assay that simultaneously measures
92 genes implicated in different embryonal signaling pathways, we
found 11 genes to be upregulated ≥3-fold in two 5-Aza-dC treated
RMS cell lines (GLI1, SHH, FOXA2, DKK1, TWIST1, DTX1, IGFBP3,
CDH1, IHH, CDKN1C and IGF1). Importantly, strong
increased mRNA expression levels of BMP2 (up to 20-fold) and
RASSF1A (up to 100-fold) were detected in all treated RMS
cell lines (Fig. 1A), the latter
confirming earlier results (10).
This suggests that BMP2 is epigenetically silenced in
RMS.
BMP2 signaling is inactive in RMS cell
lines
BMP2 is known to activate a signalling cascade via
binding to BMP receptor complexes, which leads to the transcription
of the downstream target genes ID1 and ID3 (17). As expected from the demethylation
experiments, all RMS cell lines expressed very low levels of BMP2
mRNA (Fig. 1B) and protein
(Fig. 1C), as compared to normal
skeletal muscle. In line with the absence of the BMP2 ligand,
transcriptional activity of ID1 and ID3 was very low
in the RMS cell lines (Fig. 1D and
E). By measuring the expression of all three BMP receptor
subtypes we could rule out that a lack of receptor expression might
be the cause for pathway inactivity (Fig. 1F).
CpG island methylation is present in RMS
tumor cell lines
To investigate whether the suppression of the
BMP2 gene is caused by DNA hypermethylation, we first
screened the BMP2 locus for the occurrence of CpG-islands.
We found a CpG-rich region of approximately 500 bp in the
5′-untranslated region of the BMP2 gene, spanning 50
CpG-dinucleotides (Fig. 2A).
Bisulfite sequencing of this region revealed extremely high
methylation rates for the two alveolar RMS cell lines RH30 (97.2%)
and RMS13 (98.0%) cells, whereas the embryonal RMS cell line RD was
less methylated and featured a methylation rate of only 18.4%
(Fig. 2B). In contrast, DNA
isolated from peripheral blood displayed no methylation of the
whole region under investigation (1.6%). In vitro methylated
DNA (SssI-treated) served as an internal control for detecting full
methylation (99.2%).
In a next step we analyzed if the increase in
BMP2 expression upon 5-Aza-dC treatment is associated with
DNA demethylation. We therefore performed methylation specific PCR
(MSP) of two regions (Fig. 2A),
one covering the central part of the CpG islands screened before by
bisulfite sequencing (MSP-2), the other a CpG-rich region in the
putative BMP2 promoter region (MSP-1). In untreated cells,
MSP assays showed strong methylation of both regions in RH30 and
RMS13 cells, whereas RD cells had a band only for the unmethylated
state (Fig. 2C). Upon 5-Aza-dC
treatment, RH30 and RMS13 cells exhibited bands for both the
methylated and the unmethylated state, thereby suggesting
demethylation of large proportions of the BMP2 locus. In
contrast, the methylation status of 5-Aza-dC treated RD cells
remained unchanged (Fig. 2C). As
BMP2 promoter methylation was more pronounced in the two
alveolar RMS cell lines we performed further MSP analysis before
and after 5-Aza-dC treatment using additional RMS cell lines
(embryonal, RUCH2; alveolar, RH4 and RH18) and detected a similar
pattern (Fig. 2C).
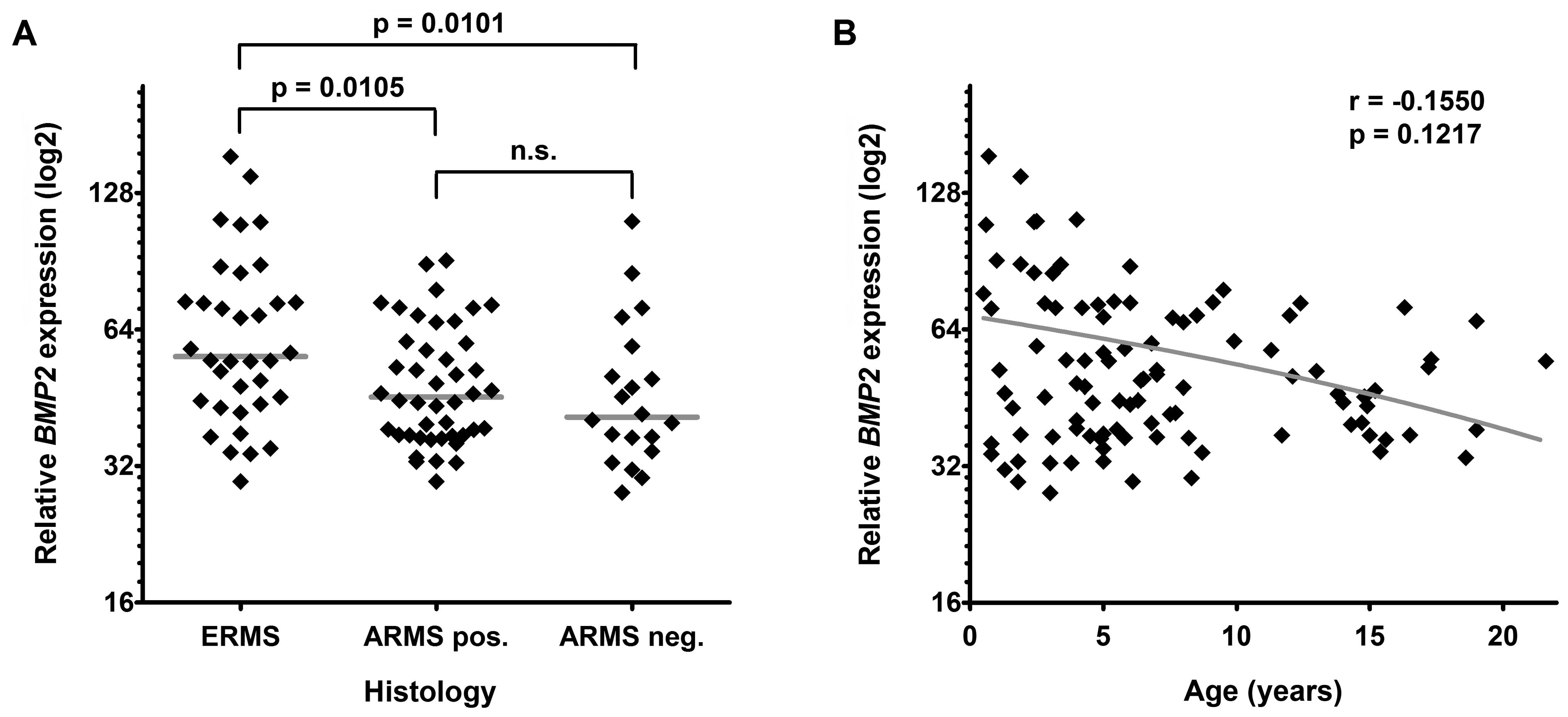
BMP2 expression is preferentially
downregulated in alveolar RMS
In order to see whether BMP2 expression
differs between the RMS subtypes not only in tumor cell lines but
also in primary tissue, we studied the BMP2 mRNA levels in a
large cohort of human RMS samples that have previously been
analyzed in a microarray-based expression profiling study (25). As expected, we found significantly
decreased expression levels of BMP2 in alveolar RMS compared
to embryonal RMS (Fig. 3A).
However, there was no gross difference between
translocation-positive and -negative alveolar RMS. In line with the
increased age of RMS patients with the alveolar subtype we found a
trend towards lower BMP2 expression levels in older patients
(Fig. 3B). These data suggest that
methylation-dependent BMP2 suppression predominantly occurs
in alveolar RMS.
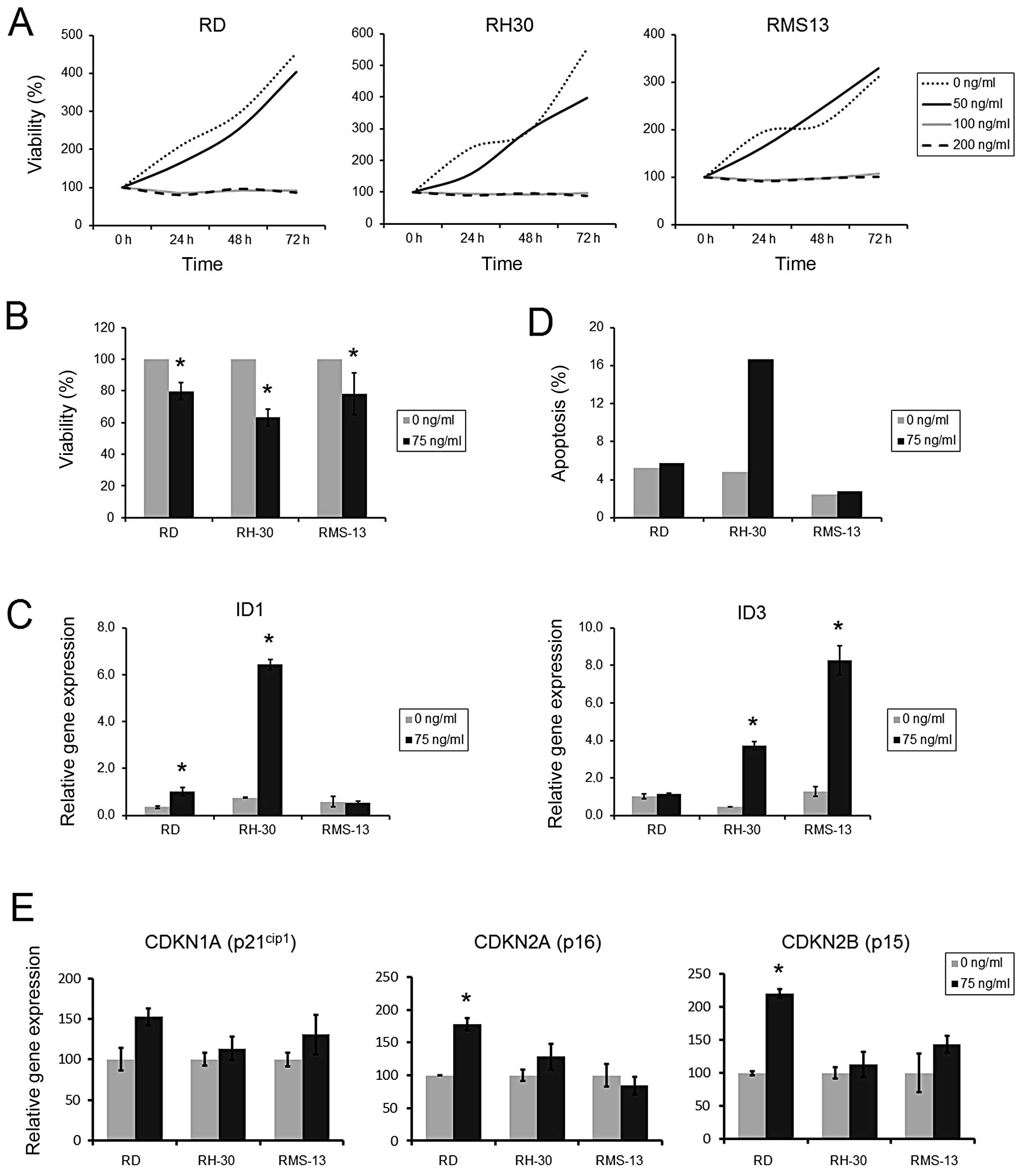
BMP2 reconstitution leads to reduced RMS
cell viability
Having shown that BMP2 expression can be
reactivated by demethylation, we determined the effects of BMP2
reconstitution in RMS cells by supplementation of recombinant human
BMP2 (rhBMP2). Using cell viability as readout, we detected a
complete block of proliferation in all three RMS cell lines at
doses of 100 and 200 ng/ml rhBMP2, an effect that was already
amenable after 24 h (Fig. 4A). In
contrast, doses of ≤50 ng/ml rhBMP2 had no effect on RMS cell
growth, even after 72 h of incubation. By choosing an intermediate
concentration of 75 ng/ml rhBMP2 we found a significantly decreased
cell viability of RD, RH30 and RMS13 cells after 72 h, with RH30
being the most sensitive cell line (Fig. 4B). To ensure BMP2 pathway induction
by this rhBMP2 concentration, we checked transcriptional activation
of the target genes ID1 and ID3 upon treatment.
Indeed, significantly induced mRNA expression of at least one of
the target genes was found in each cell line, which was most
prominent in RH30 cells (Fig.
4C).
Next, we analyzed if reduced cell growth is
associated with an enhanced apoptosis. Using 75 ng/ml rhBMP2 and
Annexin V staining as readout for early apoptosis, we found no
significant influence of rhBMP2 treatment on the apoptosis rate at
24 and 48 h in the three RMS cell lines (data not shown). After 72
h of treatment, only RH30 cells showed an increase in apoptosis,
whereas RD and RMS13 showed no response (Fig. 4D).
To obtain insight into the mechanism of cell
proliferation inhibition, we furthermore examined mRNA levels of
the growth inhibitor genes CDKN1A, CDKN2A and CDKN2B.
Treatment of RMS tumor cell lines with 75 ng/ml rhBMP2 led to
significant upregulation of CDKN2A and CDKN2B mRNA
levels in RD, but not in RH30 and RMS13 cells (Fig. 4E).
Taken together, these results suggest that
reconstitution of BMP2 in RMS cells leads to the inhibition of cell
proliferation, presumably caused by either the induction of
apoptosis (RH30) or cell cycle arrest (RD).
Discussion
Epigenetic silencing of tumor suppressor genes via
CpG hypermethylation is a well-known phenomenon fostering tumor
growth and progression in a variety of cancers (9,26).
In RMS, however, very little is known about the epigenetic basis of
tumor development. In order to detect epigenetically silenced genes
in RMS, we performed an expression profiling analysis of three RMS
cell lines treated with the demethylating agent 5-Aza-dC. Besides
others, our approach identified two candidate genes, RASSF1A
and BMP2, which displayed the most significant
transcriptional upregulation in all three cell lines.
RASSF1A has already been shown to be
epigenetically silenced in RMS (10). As RASSF1A is thought to act
as a tumor suppressor in other tumors including lung, breast,
kidney, bladder and ovarian cancer (27–31),
it has been suggested that loss of RASSF1A through
epigenetic silencing is also critical in RMS development (10). Although this has not yet been
proven experimentally, the identification of RASSF1A in our
approach at least underscores the reliability of our experimental
setup to identify epigenetically silenced genes.
BMP2 has been suggested to be a negative regulator
in the growth of skeletal muscle, which can be deduced from the
finding that BMP2 overexpression inhibits injury-induced
intimal hyperplasia (32).
Furthermore, overexpression of the positive regulator of myogenesis
follistatin leads to an exorbitant increase of skeletal muscle in
mice by neutralizing the inhibitory action of activin, myostatin
and BMP2 (33,34). In line with this, BMP2 expression
was very high in fully differentiated normal muscle tissue, both on
the mRNA and protein level, whereas the levels in all RMS cell
lines were significantly low. Besides the impaired BMP2
expression it has been reported that the BMP2/SMAD pathway itself
could be defective and contributes to tumor growth, as described
for colon cancer (35,36). By detecting all three BMP-receptor
components (BMPR1A, BMPR1B and BMPR2) in RMS and in normal muscle
tissue as well as by revealing the induction of the target genes
ID1 and ID3 after rhBMP2 treatment we could avert the
suspicion that an impaired BMP2/SMAD pathway might be the reason
for non-activated BMP2 signaling.
Interestingly, BMP2 expression in RMS was
increased after 5-Aza-dC treatment to levels found in healthy
muscle tissue. These results suggested that DNA methylation might
be the major cause of BMP2 suppression in RMS cells, a
scenario that has earlier been described in gastric cancer
(37). Indeed, using bisulfite
sequencing of a CpG-island located in the promoter region of
BMP2, we found an almost complete methylation of all
CpG-dinucleotides in the two investigated alveolar RMS cell lines,
whereas the embryonal RMS cell line RD showed a distinct
methylation of ∼20%. In clear contrast is the methylation pattern
of DNA isolated from whole blood, which was completely
unmethylated. These findings were verified by methylation specific
PCR, also in additional RMS cell lines. Furthermore, significantly
lower BMP2 expression levels in primary ARMS compared to ERMS
together with the differential methylation patterns in alveolar
compared to embryonal RMS cell lines strengthen previous findings
of diverse methylation patterns in RMS subtypes (38). Therefore, the mechanism of gene
silencing via promoter hypermethylation, which is well known from a
variety of different cancers (9,26,39,40),
can also be anticipated of being important in RMS. In view of only
moderate methylation patterns in the embryonal RMS cell line, there
may also be mechanisms other than promoter hypermethylation causing
BMP2 silencing. Accordingly, it has been shown that the
transcription of BMP2 in osteoblasts is regulated via
enhancers, regulatory DNA elements located far upstream of the
promoter (41,42). Thus, methylation of these elements
or other unknown regions at the BMP2 locus not covered by
our assays may be responsible for BMP2 silencing.
In order to illuminate whether demethylation is the
cause of BMP2 reexpression after 5-Aza-dC treatment, we determined
the methylation pattern in treated RMS cell lines. However, the
assumed demethylating effect of 5-Aza-dC causing the re-expression
was only obvious in the two RMS cell lines showing strong CpG
methylation, although all three cell lines showed a significant
reactivation of BMP2 gene expression after treatment. This
discrepancy could be explained by the finding that 5-Aza-dC is
known of being a relatively unspecific agent, which can also change
histone modifications which might be important in the
transcriptional regulation of BMP2 in RD cells. Furthermore,
5-Aza-dC leads to global demethylation and thereby transcriptional
regulation of many different genes (43,44).
In colon cancer it has been shown that 5-Aza-dC rather led to
global demethylation than specific demethylation in CpG-rich
promoter regions, which results in a strong gene re-expression
(45). Similar results have been
described for the CDH1 gene in malignant liver tumors
(46). Thus, it can be suggested
that not only strong demethylation in promoter regions can cause
re-expression of genes, but also demethylation on distant parts of
the gene. In addition, histone modifications or methylation of
other parts of the BMP2 promoter, which were not included in
our analysis, could be responsible for changes in BMP2
expression.
In a variety of tumors with loss of BMP2
expression, e.g., medulloblastoma (22), gastric, breast, colon and prostatic
cancer (19,36,37,47–49),
the reconstitution of BMP2 resulted in reduced tumor proliferation.
In addition, research on smooth muscle cells, whose growth were
restricted by rhBMP2 and BMP2 gene transfer (32,50),
gave reason to expect a similar function in RMS. After having
proved the induction of the BMP2 pathway in RMS cells upon rhBMP2
treatment by the transcriptional activation of the well-known BMP2
target genes ID1 and ID3 (18,51,52),
we could show that rhBMP2 treatment significantly reduced RMS cell
proliferation in all cell lines in a dose-dependent manner, thereby
suggesting that BMP2 acts as a tumor suppressor gene by the
inhibition of RMS tumor growth.
The ability of rhBMP2 to reduce proliferation has
been already ascribed to a raised G1-arrest with consecutive
apoptosis, as described for myeloma cells (21). Moreover, smooth muscle cells have
been reported to react on BMP2 by reduced proliferation and
increased apoptosis (53).
Nevertheless, there are also reports of cell protective effects,
such as a reduced apoptosis rate in neuroectodermal cells (54). Accordingly, the BMP2 function on
apoptosis seems to be tissue- or at least cell type-dependent. Our
apoptosis analysis on BMP2-treated RMS cells only detected an
enhanced apoptosis rate in one of three cell lines (RH30), despite
reduced proliferation in all RMS cell lines. In order to illuminate
this divergence between proliferation and apoptosis, we examined
mRNA levels of CDKN1A (p21), CDKN2A (p16) and CDKN2B
(p15), important genes for the control of the cell cycle and
the G1-phase (55). CDKN1A
has been described earlier to be activated via rhBMP2 treatment in
gastric cancer and smooth muscle leading to reduced cell
proliferation (49,50). However, a significant increase of
these genes after rhBMP2 treatment was only detected in one cell
line (RD). Thus, we assume that the inhibition of proliferation
through BMP2 is not due to a single mechanism, but rather that cell
cycle-arrest and in addition the induction of apoptosis are
relevant, which has to be addressed in future experiments.
Collectively, our study shows that BMP2 is
silenced in RMS via promoter hypermethylation and that the
reconstitution of BMP2 significantly decreases tumor cell
proliferation. However, the underlying molecular mechanisms for the
growth suppressive effect of BMP2 are not entirely clear, as the
induction of apoptosis or cell cycle-inhibitory genes is not
evident in all RMS cell lines. Nevertheless, our data warrant
further investigations on the use of BMP2 reconstitution for
therapeutic purposes in RMS.
Acknowledgements
We are grateful to Fatemeh Promoli,
Anett Domokos, and Nicole Stadler for technical assistance. Dr Beat
Schäfer (University of Zurich, Switzerland) provided the RMS cell
lines. This study was supported by the FöFoLe program ‘Molecular
Medicine’ of the Medical Faculty of the Ludwig-Maximilians
University of Munich.
References
|
1.
|
Paulino AC and Okcu MF: Rhabdomyosarcoma.
Curr Probl Cancer. 32:7–34. 2008. View Article : Google Scholar
|
|
2.
|
Crist W, Gehan EA, Ragab AH, Dickman PS,
Donaldson SS, Fryer C, Hammond D, Hays DM, Herrmann J, Heyn R, et
al: The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol.
13:610–630. 1995.PubMed/NCBI
|
|
3.
|
Meyer WH and Spunt SL: Soft tissue
sarcomas of childhood. Cancer Treat Rev. 30:269–280. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Newton WA Jr, Soule EH, Hamoudi AB, Reiman
HM, Shimada H, Beltangady M and Maurer H: Histopathology of
childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II:
clinicopathologic correlation. J Clin Oncol. 6:67–75.
1988.PubMed/NCBI
|
|
5.
|
Sorensen PH, Lynch JC, Qualman SJ,
Tirabosco R, Lim JF, Maurer HM, Bridge JA, Crist WM, Triche TJ and
Barr FG: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic
indicators in alveolar rhabdomyosarcoma: a report from the
children’s oncology group. J Clin Oncol. 20:2672–2679.
2002.PubMed/NCBI
|
|
6.
|
Loh WE Jr, Scrable HJ, Livanos E, Arboleda
MJ, Cavenee WK, Oshimura M and Weissman BE: Human chromosome 11
contains two different growth suppressor genes for embryonal
rhabdomyosarcoma. Proc Natl Acad Sci USA. 89:1755–1759. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Barr FG: Gene fusions involving PAX and
FOX family members in alveolar rhabdomyosarcoma. Oncogene.
20:5736–5746. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Fredericks WJ, Galili N, Mukhopadhyay S,
Rovera G, Bennicelli J, Barr FG and Rauscher FJ III: The PAX3-FKHR
fusion protein created by the t(2;13) translocation in alveolar
rhabdomyosarcomas is a more potent transcriptional activator than
PAX3. Mol Cell Biol. 15:1522–1535. 1995.PubMed/NCBI
|
|
9.
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Harada K, Toyooka S, Maitra A, Maruyama R,
Toyooka KO, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Minna
JD and Gazdar AF: Aberrant promoter methylation and silencing of
the RASSF1A gene in pediatric tumors and cell lines. Oncogene.
21:4345–4349. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Wong IH, Chan J, Wong J and Tam PK:
Ubiquitous aberrant RASSF1A promoter methylation in childhood
neoplasia. Clin Cancer Res. 10:994–1002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Sugawara W, Haruta M, Sasaki F, Watanabe
N, Tsunematsu Y, Kikuta A and Kaneko Y: Promoter hypermethylation
of the RASSF1A gene predicts the poor outcome of patients with
hepatoblastoma. Pediatr Blood Cancer. 49:240–249. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wozney JM, Rosen V, Celeste AJ, Mitsock
LM, Whitters MJ, Kriz RW, Hewick RM and Wang EA: Novel regulators
of bone formation: molecular clones and activities. Science.
242:1528–1534. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lyons KM, Hogan BL and Robertson EJ:
Colocalization of BMP 7 and BMP 2 RNAs suggests that these factors
cooperatively mediate tissue interactions during murine
development. Mech Dev. 50:71–83. 1995. View Article : Google Scholar
|
|
15.
|
Duprez DM, Coltey M, Amthor H, Brickell PM
and Tickle C: Bone morphogenetic protein-2 (BMP-2) inhibits muscle
development and promotes cartilage formation in chick limb bud
cultures. Dev Biol. 174:448–452. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Narita T, Saitoh K, Kameda T, Kuroiwa A,
Mizutani M, Koike C, Iba H and Yasugi S: BMPs are necessary for
stomach gland formation in the chicken embryo: a study using
virally induced BMP-2 and Noggin expression. Development.
127:981–988. 2000.PubMed/NCBI
|
|
17.
|
Chen D, Zhao M and Mundy GR: Bone
morphogenetic proteins. Growth Factors. 22:233–241. 2004.
View Article : Google Scholar
|
|
18.
|
Hollnagel A, Oehlmann V, Heymer J, Ruther
U and Nordheim A: Id genes are direct targets of bone morphogenetic
protein induction in embryonic stem cells. J Biol Chem.
274:19838–19845. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Horvath LG, Henshall SM, Kench JG, Turner
JJ, Golovsky D, Brenner PC, O’Neill GF, Kooner R, Stricker PD,
Grygiel JJ and Sutherland RL: Loss of BMP2, Smad8, and Smad4
expression in prostate cancer progression. Prostate. 59:234–242.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Soda H, Raymond E, Sharma S, Lawrence R,
Cerna C, Gomez L, Timony GA, Von Hoff DD and Izbicka E:
Antiproliferative effects of recombinant human bone morphogenetic
protein-2 on human tumor colony-forming units. Anticancer Drugs.
9:327–331. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kawamura C, Kizaki M and Ikeda Y: Bone
morphogenetic protein (BMP)-2 induces apoptosis in human myeloma
cells. Leuk Lymphoma. 43:635–639. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Zhao H, Ayrault O, Zindy F, Kim JH and
Roussel MF: Post-transcriptional down-regulation of Atoh1/Math1 by
bone morphogenic proteins suppresses medulloblastoma development.
Genes Dev. 22:722–727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Williamson D, Missiaglia E, de Reynies A,
Pierron G, Thuille B, Palenzuela G, Thway K, Orbach D, Lae M,
Freneaux P, Pritchard-Jones K, Oberlin O, Shipley J and Delattre O:
Fusion gene-negative alveolar rhabdomyosarcoma is clinically and
molecularly indistinguishable from embryonal rhabdomyosarcoma. J
Clin Oncol. 28:2151–2158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Missiaglia E, Dalai I, Barbi S, Beghelli
S, Falconi M, della Peruta M, Piemonti L, Capurso G, Di Florio A,
delle Fave G, Pederzoli P, Croce CM and Scarpa A: Pancreatic
endocrine tumors: expression profiling evidences a role for
AKT-mTOR pathway. J Clin Oncol. 28:245–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Esteller M: Epigenetic gene silencing in
cancer: the DNA hypermethylome. Hum Mol Genet. 16(Spec No 1):
R50–R59. 2007. View Article : Google Scholar
|
|
27.
|
Burbee DG, Forgacs E, Zochbauer-Muller S,
Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S,
Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI,
Zabarovsky E, White M and Minna JD: Epigenetic inactivation of
RASSF1A in lung and breast cancers and malignant phenotype
suppression. J Natl Cancer Inst. 93:691–699. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Dammann R, Li C, Yoon JH, Chin PL, Bates S
and Pfeifer GP: Epigenetic inactivation of a RAS association domain
family protein from the lung tumour suppressor locus 3p21.3. Nat
Genet. 25:315–319. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Morrissey C, Martinez A, Zatyka M,
Agathanggelou A, Honorio S, Astuti D, Morgan NV, Moch H, Richards
FM, Kishida T, Yao M, Schraml P, Latif F and Maher ER: Epigenetic
inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both
clear cell and papillary renal cell carcinoma. Cancer Res.
61:7277–7281. 2001.PubMed/NCBI
|
|
30.
|
Yoon JH, Dammann R and Pfeifer GP:
Hypermethylation of the CpG island of the RASSF1A gene in ovarian
and renal cell carcinomas. Int J Cancer. 94:212–217. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dammann R, Yang G and Pfeifer GP:
Hypermethylation of the cpG island of Ras association domain family
1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3
locus, occurs in a large percentage of human breast cancers. Cancer
Res. 61:3105–3109. 2001.PubMed/NCBI
|
|
32.
|
Nakaoka T, Gonda K, Ogita T,
Otawara-Hamamoto Y, Okabe F, Kira Y, Harii K, Miyazono K, Takuwa Y
and Fujita T: Inhibition of rat vascular smooth muscle
proliferation in vitro and in vivo by bone morphogenetic protein-2.
J Clin Invest. 100:2824–2432. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lee SJ and McPherron AC: Regulation of
myostatin activity and muscle growth. Proc Natl Acad Sci USA.
98:9306–9311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Suryawan A, Frank JW, Nguyen HV and Davis
TA: Expression of the TGF-beta family of ligands is developmentally
regulated in skeletal muscle of neonatal rats. Pediatr Res.
59:175–179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Howe JR, Bair JL, Sayed MG, Anderson ME,
Mitros FA, Petersen GM, Velculescu VE, Traverso G and Vogelstein B:
Germline mutations of the gene encoding bone morphogenetic protein
receptor 1A in juvenile polyposis. Nat Genet. 28:184–187. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Kodach LL, Wiercinska E, de Miranda NF,
Bleuming SA, Musler AR, Peppelenbosch MP, Dekker E, van den Brink
GR, van Noesel CJ, Morreau H, Hommes DW, Ten Dijke P, Offerhaus GJ
and Hardwick JC: The bone morphogenetic protein pathway is
inactivated in the majority of sporadic colorectal cancers.
Gastroenterology. 134:1332–1341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Wen XZ, Akiyama Y, Baylin SB and Yuasa Y:
Frequent epigenetic silencing of the bone morphogenetic protein 2
gene through methylation in gastric carcinomas. Oncogene.
25:2666–2673. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Mahoney SE, Yao Z, Keyes CC, Tapscott SJ
and Diede SJ: Genome-wide DNA methylation studies suggest distinct
DNA methylation patterns in pediatric embryonal and alveolar
rhabdomyosarcomas. Epigenetics. 7:400–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar
|
|
40.
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Pregizer S and Mortlock DP: Control of BMP
gene expression by long-range regulatory elements. Cytokine Growth
Factor Rev. 20:509–515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Chandler RL, Chandler KJ, McFarland KA and
Mortlock DP: Bmp2 transcription in osteoblast progenitors is
regulated by a distant 3′ enhancer located 156.3 kilobases from the
promoter. Mol Cell Biol. 27:2934–2951. 2007.PubMed/NCBI
|
|
43.
|
Jabbour E, Issa JP, Garcia-Manero G and
Kantarjian H: Evolution of decitabine development: accomplishments,
ongoing investigations, and future strategies. Cancer.
112:2341–2351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Cameron EE, Bachman KE, Myohanen S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Mossman D, Kim KT and Scott RJ:
Demethylation by 5-aza-2′-deoxycytidine in colorectal cancer cells
targets genomic DNA whilst promoter CpG island methylation
persists. BMC Cancer. 10:3662010.
|
|
46.
|
Tachibana K, Takeda K and Shiraishi M:
5-Aza-2′-deoxycytidine reactivates the CDH1 gene without
influencing the methylation of the entire CpG island or histone
modification in a human cancer cell line. Proc Japan Acad.
80:342–348. 2004.
|
|
47.
|
Ghosh-Choudhury N, Woodruff K, Qi W,
Celeste A, Abboud SL and Ghosh Choudhury G: Bone morphogenetic
protein-2 blocks MDA MB 231 human breast cancer cell proliferation
by inhibiting cyclin-dependent kinase-mediated retinoblastoma
protein phosphorylation. Biochem Biophys Res Commun. 272:705–711.
2000. View Article : Google Scholar
|
|
48.
|
Beck SE, Jung BH, Fiorino A, Gomez J,
Rosario ED, Cabrera BL, Huang SC, Chow JY and Carethers JM: Bone
morphogenetic protein signaling and growth suppression in colon
cancer. Am J Physiol Gastrointest Liver Physiol. 291:G135–G145.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Wen XZ, Miyake S, Akiyama Y and Yuasa Y:
BMP-2 modulates the proliferation and differentiation of normal and
cancerous gastric cells. Biochem Biophys Res Commun. 316:100–106.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Wong GA, Tang V, El-Sabeawy F and Weiss
RH: BMP-2 inhibits proliferation of human aortic smooth muscle
cells via p21Cip1/Waf1. Am J Physiol Endocrinol Metab.
284:E972–E979. 2003.PubMed/NCBI
|
|
51.
|
Du Y and Yip H: Effects of bone
morphogenetic protein 2 on Id expression and neuroblastoma cell
differentiation. Differentiation. 79:84–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Ogata T, Wozney JM, Benezra R and Noda M:
Bone morphogenetic protein 2 transiently enhances expression of a
gene, Id (inhibitor of differentiation), encoding a
helix-loop-helix molecule in osteoblast-like cells. Proc Natl Acad
Sci USA. 90:9219–9222. 1993. View Article : Google Scholar
|
|
53.
|
Zhang S, Fantozzi I, Tigno DD, Yi ES,
Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ and
Yuan JX: Bone morphogenetic proteins induce apoptosis in human
pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol
Physiol. 285:L740–L754. 2003.PubMed/NCBI
|
|
54.
|
Iantosca MR, McPherson CE, Ho SY and
Maxwell GD: Bone morphogenetic proteins-2 and -4 attenuate
apoptosis in a cerebellar primitive neuroectodermal tumor cell
line. J Neurosci Res. 56:248–258. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|