Introduction
Most of the cancer deaths result from metastasis. In
many cases, survival rate of cancer patients with metastatic tumors
is much lower than those of patients with localized tumors.
Metastasis of breast cancer, the most common form of cancer in
woman, is responsible for nearly 90% of deaths from breast cancer.
Although breast cancer comprises about 25% of all cancers, the
survival rates of patients with breast cancer are relatively high
as long as cancer is detected and treated before it metastasizes.
Therefore, better understanding of the mechanism that promotes
metastasis of breast cancer would be helpful to design effective
therapies.
The phosphoinositol-3-kinase (PI3K)/AKT pathway is
involved in multiple cellular processes including cell
differentiation, proliferation, survival, angiogenesis, invasion
and metastasis (1–8). PI3K phosphorylates
phosphatidylinositol 4,5-bisphosphate (PIP2), yielding
phosphatidylinositol 3,4,5-triphosphate (PIP3), which
recruits and activates phosphatidylinositol-dependent kinase 1
(PDK1). Activated PDK1 phosphorylates serine/threonine-specific
protein kinase (AKT), also known as protein kinase B (PKB). AKT
activation can be induced by phosphatase and tensin homolog (PTEN)
abnormality (9–11). PTEN tumor suppressor is a negative
regulator of the PI3K/AKT pathway, in which PTEN dephosphorylates
PIP3 to PIP2 (12). Mutation of deletion of PTEN is
frequently found to be mutated or deleted in a broad range of
cancers, resulting in the hyper-activation of the PI3K/AKT
signaling pathway. In addition, the HER2/neu proto-oncogene (also
known as ErbB-2, CD340 or p185) is the most oncogenic signaling
activator of PI3K/AKT, in which HER2/neu can phosphorylate
PIP2 and in turn induce AKT activation (13,14).
HER2/neu plays important roles in the development and progression
of certain aggressive types of breast cancer and is found at a
relatively high level in most aggressive tumors. Activated AKT acts
as a key regulator for many events related with tumor malignancies
including cell survival, proliferation, growth, angiogenesis and
metastasis (7,12,14–18).
The critical steps to initiate the metastasis are the
phosphorylation of AKT on serine 473 residue and regulation of its
downstream target proteins. GSK3β is one of important downstream
target proteins involved in metastasis. GSK3β inhibition promotes
metastasis by affecting β-catenin, a dual function protein that
regulates the coordination of cell-cell adhesion and gene
transcription (19–22). Deregulated β-catenin expression is
associated with many cancers including malignant breast tumors
(21,22). GSK3β can be inhibited in at least
two ways in PI3K/AKT signaling pathway: activation of AKT to
directly phosphorylate GSK3β on serine 9 residue (19) and directly/indirectly by
integrin-liked kinase (ILK) (23).
ILK is a multifunctional serine/threonine protein kinase that
mediates a variety of cellular responses to integrin stimulation by
extracellular matrix proteins. The mechanism of ILK activation is
not fully understood but it is reported that p21 activated kinase
(PAK) can phosphorylate ILK on threonine 173 for its activation
(24). It was demonstrated that
purified recombinant ILK phosphorylates recombinant GSK3β in
vitro (25). It means that ILK
can directly phosphorylate GSK3β. Furthermore, inhibition of ILK
suppresses tumorigenesis and tumor growth (24,26).
Exposure to ILK inhibitor suppresses the snail and β-catenin
protein stability and transcriptional activity as well as GSK3β
phosphorylation indicating that ILK activity affects the
epithelial-mesenchymal transition (EMT) process. Despite these
facts, its effect on cancer metastasis still remains unclear and
controversial. It has been believed that ILK is a component of the
PI3K-AKT pathway by phosphorylating AKT. However, it has been
reported that ILK acts as an inhibitor of AKT phosphorylation even
though cell proliferation was induced by stably overexpression of
ILK (26,27).
With the goal of elucidating the roles of PI3K/AKT
signaling pathway in cancer metastasis, we have previously reported
the participation of the p34SEI-1 oncoprotein in this
pathway. p34SEI-1 enhances cancer cell survival and
promotes tumorigenesis by inducing NEDD4-1-mediated PTEN
degradation (28). NEDD4-1
negatively regulates PTEN as a proto-oncogenic E3 ubiquitin ligase
for PTEN and in turn activates the PI3K/AKT signaling pathway
(29,30). Considering the vital roles of
PI3K/AKT signaling pathway in metastasis and the indirect effect of
p34SEI-1 on this pathway, it was suspected that
p34SEI-1 may play an important role in the development
of cancer metastasis. p34SEI-1 has multiple biological
functions including transcription regulation, cell cycle
regulation, inhibition of apoptosis and tumorigenesis (31–33).
Furthermore, p34SEI-1 increases chromosomal instability,
which is closely related to cancer invasiveness (34,35).
We previously showed that the expression level of
p34SEI-1 is significantly increased in cancer tissue
compared to normal tissues, in which p34SEI-1 stabilizes
X-linked inhibitor of apoptosis protein (XIAP) leading to an
anti-apoptotic effect (33). XIAP
also promotes progression of metastasis by activating the oncogenic
NF-κB transcriptional factor, fibronectin-related gene expression
and cell motility kinase such as focal adhesion kinase (FAK) or Src
(36,37).
All these data implicate that p34SEI-1
may be involved in the progression of metastatic cancers. We
therefore investigated whether or not p34SEI-1 has
metastatic potential and the nature of the mechanism.
Materials and methods
Cell lines, cell culture and
materials
Five cancer cell lines were used in this study.
MCF7, T47D, HEK293T and MDA-MB-231 cancer cells were cultured in
DMEM medium and SKBR3 cells were grown in RPMI medium (Welgene
Inc., Daegu, Korea). All media were supplemented with 10% FBS
(Gibco-BRL, Carlsbad, CA, USA) and 1% antibiotic-anti-mycotic
(Gibco-BRL). All cultures were grown at 37°C in a humidified
atmosphere composed of 95% air and 5% CO2. LY294004 (cat
no. 440202) and Cpd22 (cat no. 407331) were purchased from
Calbiochem (La Jolla, CA, USA).
Reverse transcription (RT)-PCR
Total RNA was extracted from MDA-MB-231 cells with
the RNeasy mini kit (cat no. 74106; Qiagen, Hilden, Germany)
following the manufacturer’s instructions. For reverse
transcription, 1 μg RNA of each sample was subjected to cDNA
synthesis using an oligo (dT) primer and the ImProm-II™ Reverse
Transcription System (A3800; Promega, Madison, WI, USA). PCR
amplification was performed using 10 ng cDNA, different sets of
primers, and AccuPower PCR PreMix system (Bioneer, Daejeon, Korea).
The amplification reaction was carried out using a PCR Thermal
Cycler Dice (Applied Biosystems, Foster City, CA, USA). Each gene
product was amplified using corresponding pairs of primers, in
which β-actin gene product was used as an internal control. The
oligonucleotide sequences for RT-PCR analysis were:
pRT-p34SEI-1-RT forward, 5′-AGGACCTCAGCCACAT TGAG-3′ and
reverse, 5′-GGTGCCCAAAGTTCATTGTC-3′; pRT-HER2/neu-RT forward,
5′-CTGAACTGGTGTATGC AGAT-3′ and reverse, 5′-CCACACAGTCACACCATAA-3′;
pRT-NEDD4-1 forward, 5′-TGGGACATCACTTTGT GATC-3′ and reverse,
5′-TGAGGCTTTTACTGGGGTC-3′; pRT-β-catenin forward,
5′-CATTTCCAATCTACTAATGC-3′ and 5′-CTGCATTCTGACTTTCAGTA-3′;
pRT-c-MYC forward, 5′-ACCAGCAGCGACTCTGAGGA-3′ and reverse,
5′-TGACCCTCTTGGCAGCAGGATAGTCC-3′; pRT-ACTB forward,
5′-AGGTCGGAGTCAACGGATTTG-3′ and reverse,
5′-GTGATGGCATGGACTGTGGT-3′.
Western blot analysis
Cells were recovered from culture by centrifugation
at 3,000 rpm for 1 min and washed twice in an ice-cold
phosphate-buffered saline (PBS) buffer. The cells were then lysed
in RIPA lysis buffer and the protein was quantified using a protein
assay kit (Bio-Rad, Hercules, CA, USA). Approximately 25 μg
of total protein per sample was subjected to 12% SDS-PAGE and the
resolved proteins were transferred to Immobilon transfer membranes
(cat no. IPVH00010, Millipore, Billerica, MA, USA). The filter was
blocked in 5% non-fat dry milk/0.1% Tween-20/Tris-buffered saline
(TBS) followed by incubation with each corresponding antibody and
immune-detection was accomplished using the Power Opti-ECL Western
blotting detection reagent (Bionote, Hwaseong, Korea). Antibodies
used in this study were purchased as follows: p34SEI-1
(ALX-804-645; Enzo Life Sciences, Farmingdale, NY, USA), NEDD4-1
(sc-25508) and PTEN (sc-7974; from Santa Cruz Biotechnology, Santa
Cruz, CA, USA), E-cadherin (cat no. 610181) and N-cadherin (cat no.
610921; from BD Biosciences, Franklin Lakes, NJ, USA), vimentin
(sc-7557), fibronectin (sc-8422), HER2/neu (sc-33684; from Santa
Cruz Biotechnology), pAKTser473 (cat no. 9271; Cell
Signaling, Danvers, MA, USA), pILKthr178 (sc-130196;
Santa Cruz Biotechnology), pGSK3βser9 (cat no. 2435-1;
Epitomics, Burlingame, CA, USA) and γ-tubulin (sc-7396; Santa Cruz
Biotechnology).
Overexpression or suppression of
p34SEI-1 or HER2/neu gene
For overexpression of p34SEI-1, cells
were plated at 1×106 cells in a 60-mm-diameter culture
dish and transfected with 4 μg of either the control empty
vector (pEF-BOS-EX) or the C-terminally Flag-tagged human
p34SEI-1 protein (pEF-p34SEI-1-Flag) for 6 h
in serum free medium using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA). For knockdown of p34SEI-1
expression, MDA-MB231 cells were plated as above and transfected
with 4 μg of either empty control vector (pLKO.1) or
p34SEI-1 directed shRNA expression vector
[pLKO.1/p34SEI-1-short hairpin (sh) RNA] for 6 h in
serum free medium using Lipofectamine 2000. After replacing the
DNA-Lipofectamine complex-containing medium with complete,
antibiotic-free growth medium, transfected cells were incubated for
48 h. For knockdown of endogenous HER2/neu, SKBR3 cells were plated
4×105 in a 60-mm-diameter culture dish and transiently
transfected with 200 pmol of either scramble control RNA (scRNA) or
a HER2/neu silencing siRNA (siHER2/neu) for 2 h in serum free
medium using Lipofectamine 2000. After replacing the
RNA-Lipofectamine complex-containing medium with complete growth
medium, transfected cells were incubated for 48 h. pEF-BOS-EX and
pEF-p34SEI-1-Flag plasmids were kindly provided by Dr
Rikiro Fukunaga (Osaka University, Osaka, Japan) and
pLKO.1/p34SEI-1-shRNA plasmid was obtained from OriGene
(http://www.origene.com). HER2/neu
oligonucleotides were chemically synthesized by ST Pharm Co. Ltd
(Seoul, Korea).
Wound healing migration assay
Cell migration was estimated by a wound-healing
migration assay and monitored by microscopy. MCF7 and MDA-MB231
cells were transfected with p34SEI-1 overexpressing
pEF-p34SEI-1-Flag or p34SEI-1 suppressing
pLKO.1/p34SEI-1-shRNA vector with each corresponding
control vector, respectively. Each cell line was fully cultured in
a 60-mm-diameter culture dish and a scratch was made on the
monolayer using a sterile white tip. The distance of migration by
cancer cells was measured after 48 h.
Matrigel invasion assay
In vitro invasion assay was performed using a
Transwell membrane apparatus (Corning Life Sciences, Tewksbury, MA,
USA) and Matrigel (BD Biosciences, Seoul, Korea). SKBR3, MCF7 and
MDA-MB-231 breast cancer cells (2×105 cells) were placed
in the Matrigel-coated upper chamber of the apparatus. Medium to
each cell line was placed in the lower chamber as a source of
chemo-attractants. Incubation was carried out for 24 h at 37°C. The
capacity of these cells to invade through the semi-solid Matrigel
was estimated by fluorescence.
Immunohistochemistry
IHC data using human tissue samples were kindly
provided by Dr Chang-Jin Kim at Soonchunhyang University Hospital
(Chonan, Korea) and it was obtained as previously described
(28).
Results
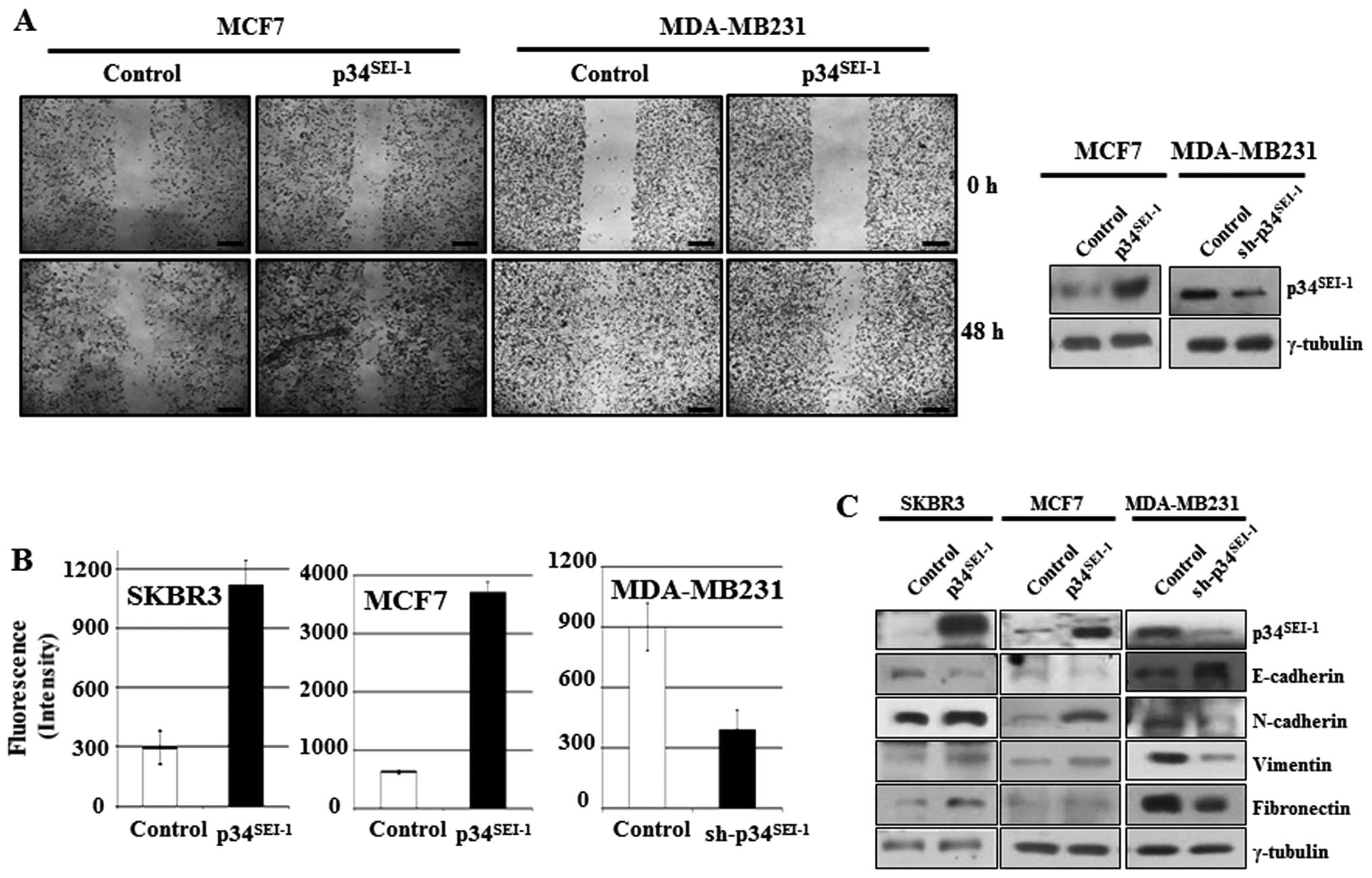
Enhanced cancer cell migration and
invasion by p34SEI-1 overexpression
To investigate whether p34SEI-1
oncoprotein is involved in the development of metastasis, the
effect of p34SEI-1 on migration and invasion was tested
by wound healing migration and Matrigel invasion assays. In the
wound healing assay, cell mobility was enhanced in MCF7 cells
transfected with p34SEI-1 overexpressing
pEF-p34SEI-1-Flag vector, while it was reduced in
MDA-MB-231 cells transfected with p34SEI-1 suppressing
pLKO.1/p34SEI-1-shRNA vector compared to control cells
(Fig. 1A). The Matrigel invasion
assay showed that overexpression of p34SEI-1 in SKBR3
and MCF7 cells increased the invasiveness compared to control
cells, while knockdown of p34SEI-1 in MDA-MB 231 cells
decreased invasiveness (Fig. 1B).
These data strongly suggest that p34SEI-1 exerts a
positive effect on the cell migration and invasion in vitro
implying the involvement of p34SEI-1 in metastasis.
During the EMT process, epithelial markers like E-cadherin are
diminished, while expression of mesenchymal markers including
N-cadherin, vimentin and fibronectin increase due to activation of
matrix metalloproteinases (MMPs) inducing metastasis (38). Accordingly, the expression levels
of EMT-related proteins were checked using western blot analysis.
The representative epithelial marker E-cadherin was decreased but
its antagonist N-cadherin was increased in SKBR3 and MCF7 cells
after transfection with pEF-p34SEI-1-Flag vector
(Fig. 1C). Furthermore, vimentin
another mesenchymal marker, was also increased in SKBR3 and MCF7
cells. The opposite result was obtained in MDA-MB-231 cells
transfected with pLKO.1/p34SEI-1-shRNA vector compared
to control (Fig. 1C). Taken
together, the findings indicate that p34SEI-1 promotes
metastasis by enhancing migration and invasion of cancer cells.
Change of p34SEI-1 expression
level in tissue samples with different degrees of tumor
invasiveness
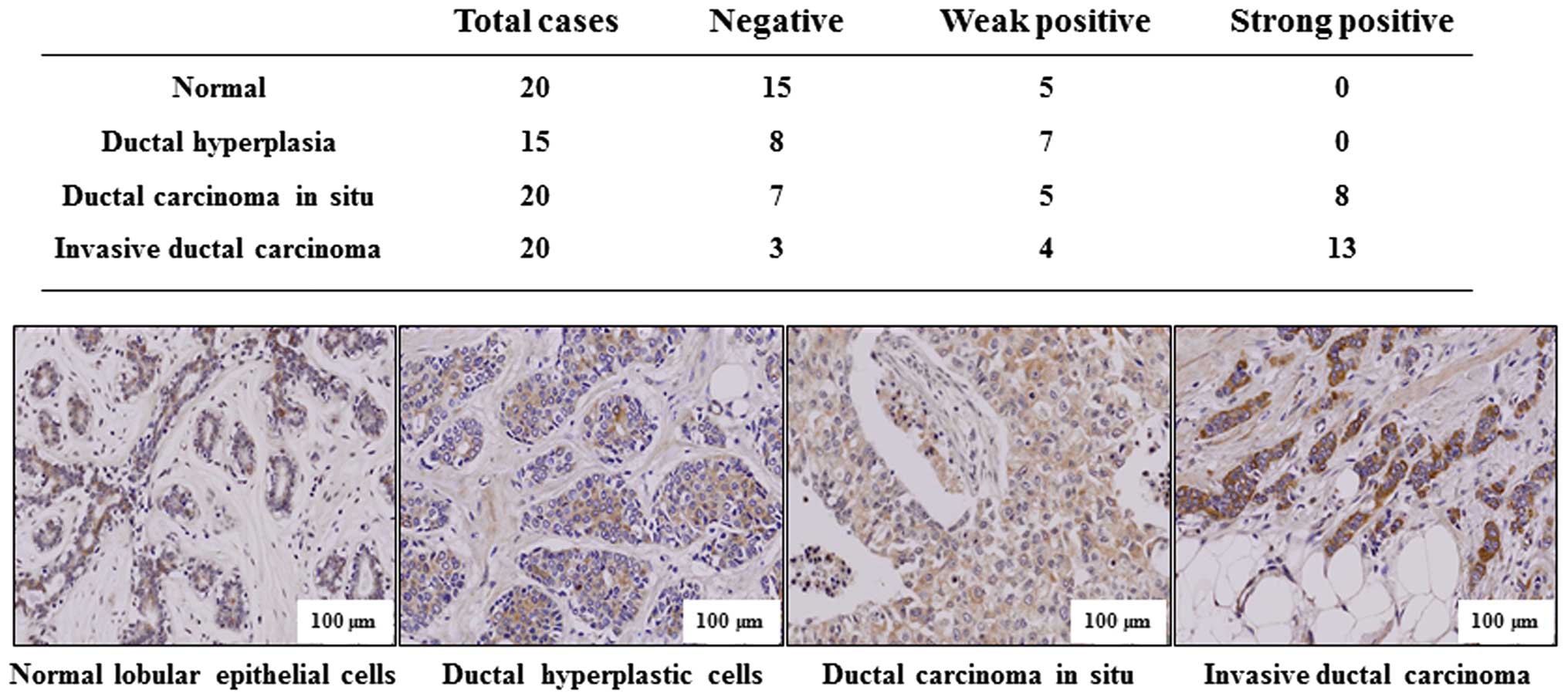
To support the conclusion that p34SEI-1
has metastatic potential, immunohistochemistry was performed to
test whether p34SEI-1 expression is clinically related
with the metastasis of breast cancer. Indicated tissue samples were
stained with p34SEI-1 antibody and the degree of
positive signals was estimated. Normal lobular and ductal
hyperplastic cells showed no strong positive signal (0 of 20
samples) while 8 of 20 (40%) ductal carcinoma in situ and 13
of 20 (70%) invasive ductal carcinoma samples displayed strong
positive signals (Fig. 2). The
invasive carcinoma showed more strong expression than non-invasive
ductal carcinoma in situ. The representative
immunohistochemistry data are shown in Fig. 2, in which p34SEI-1 was
not or very weakly expressed in normal lobular epithelial or ductal
hyperplasia breast cancer tissues, but was more strongly expressed
in invasive ductal carcinoma tissue samples than in ductal
carcinoma in situ. The data indicate that
p34SEI-1 expression increases as the tumor invasiveness
progresses in human breast tissues, strongly relating
p34SEI-1 with breast cancer metastasis.
Positive effect of p34SEI-1 on
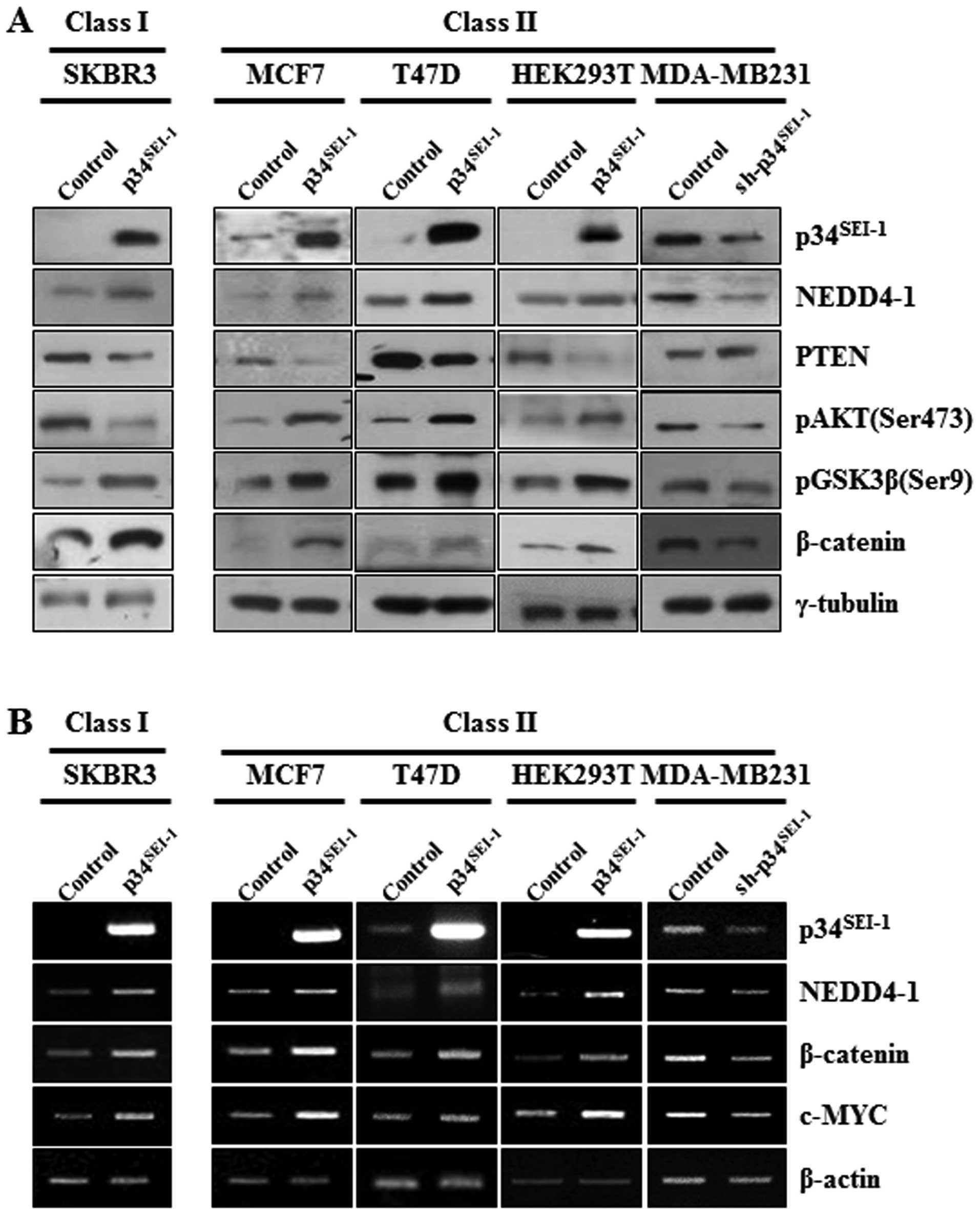
metastasis via regulation of PI3K/AKT signaling pathway
To elucidate the mechanism of how
p34SEI-1 promotes metastasis during cancer cell
tumorigenesis, western blot analysis was employed to check the
expression levels of the main components of the PI3K-AKT pathway,
NEDD4-1, PTEN, phosphorylation of AKT on serine 473 residue
(pAKTser473) and phosphorylation of GSK3β on serine 9
residue (pGSK3βser9) after overexpression or suppression
of p34SEI-1. In MCF7, T47D and HEK293T cells transfected
with pEF-p34SEI-1-Flag, p34SEI-1 induced
increased pAKTser473 and pGSK3βser9 protein
levels at least partly by inducing NEDD4-1-mediated PTEN
degradation as we previously reported (20). In MDA-MB231 cells transfected with
pLKO.1/p34SEI-1-shRNA vector, suppression of
p34SEI-1 resulted in a decrease of NEDD4-1 and an
increase of PTEN compared to the control. Consequently, the protein
levels of pAKTser473 and pGSK3βser9 decreased
(Fig. 3A). However, SKBR3 cells
transfected with pEF-p34SEI-1-Flag revealed a different
expression pattern, in which pAKTser473 was unexpectedly
diminished. More interestingly, expression levels of GSK3β
phosphorylation and β-catenin, downstream target of GSK3β, were not
affected by the AKT inactivation (Fig.
3A). This fact implies the presence of another upstream kinase
regulating GSK3β phosphorylation regardless of AKT inactivation.
This speculation was supported by RT-PCR results showing that both
classes had very similar expression pattern to the NEDD4-1,
β-catenin, and its downstream target, c-MYC, at the transcriptional
level. Overexpression of p34SEI-1 produced an increase
of NEDD4-1, β-catenin and c-MYC in SKBR3, MCF7 and HEK293T cells,
whereas, in MDA-MB231 cells p34SEI-1 was suppressive
(Fig. 3B). The data implicated an
unknown kinase in this pathway downstream of NEDD4-1 and upstream
of GSK3β. Collectively, the data indicate that p34SEI-1
may promote cancer metastasis using distinct signaling pathways in
two different types of cancer cell lines with different genetic
background.
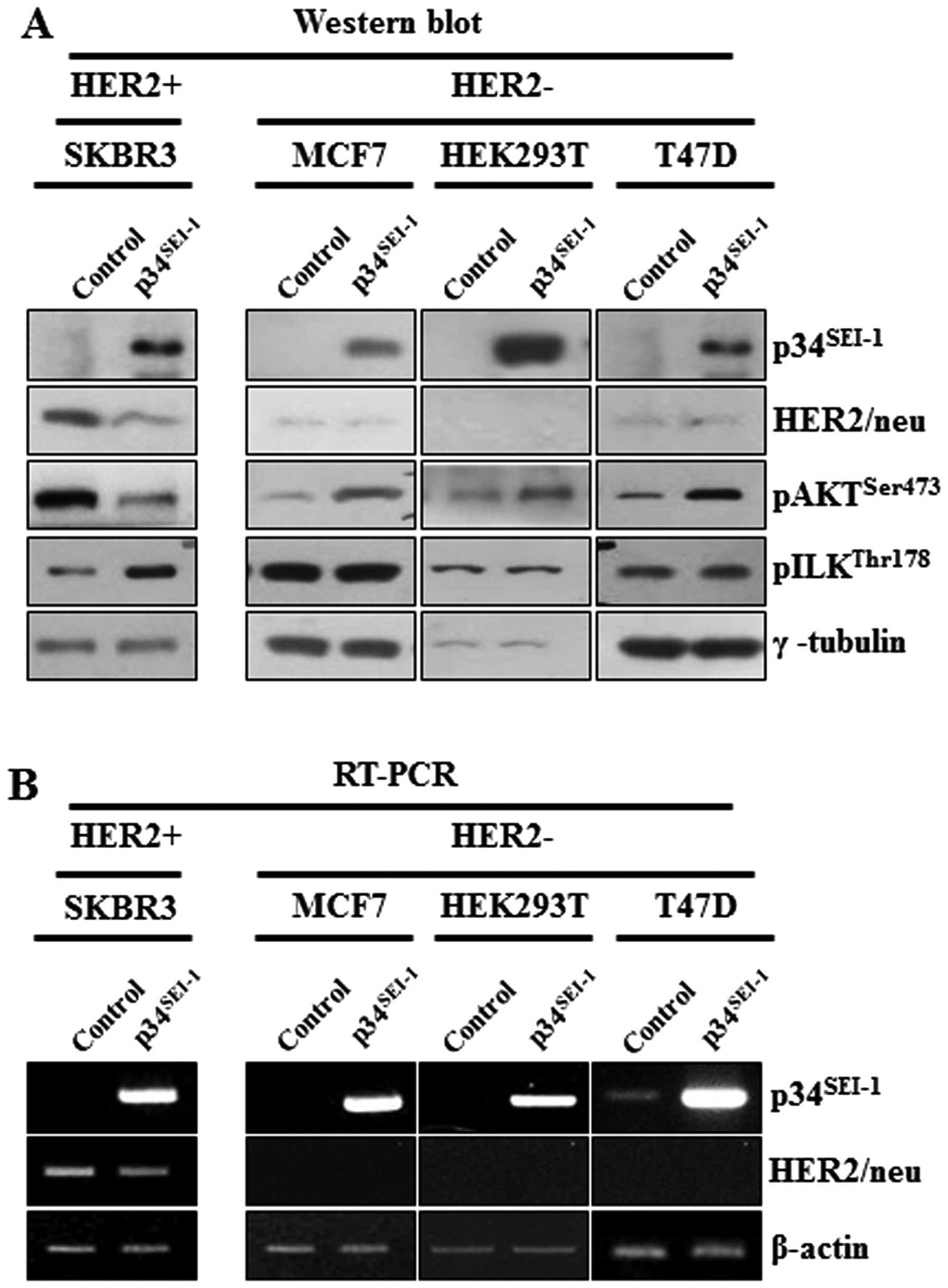
Effect of p34SEI-1
overexpression on the activation of AKT and ILK in
HER2/neu-positive and -negative cancer cell lines
p34SEI-1 seems to promote metastasis by
activating the PI3K/AKT signaling pathway. During this process,
overexpression of p34SEI-1 decreased the phosphorylation
of AKT on 473 serine residue in SKBR3 cells but increased
phosphorylation in MCF7, T47D and HEK293T cells. Considering that
SKBR3 is HER2/neu-positive cell line, but MCF7, T47D and HEK293T
are HER2/neu-negative cell lines, it was assumed that HER2/neu
might be responsible for the decrease of pAKTser473
protein level in p34SEI-1 overexpressing SKBR3 cells,
since HER2/neu is a positive regulator of the PI3K/AKT signaling
pathway and its expression was significantly decreased by
p34SEI-1 overexpression (Fig. 4A). This result was consistent with
the view that p34SEI-1 suppresses HER2/neu expression
and downregulated HER2/neu inhibits the phosphorylation of AKT at
the 473 serine residue. Unexpectedly, the phosphorylation level of
GSK3β on 9 serine residue was increased despite AKT inactivation
(Fig. 3A). This finding indicated
that GSK3β might be phosphorylated by another factor rather than
AKT in SKBR3 cells. To elucidate the mechanism, ILK was at first
suspected to be responsible for GSK3β phosphorylation because ILK
is known to affect PI3K/AKT signaling pathway by directly
phosphorylating the 9 serine residue of GSK3β (23). The phosphorylation levels of ILK on
the 178 threonine residue were checked in both HER2/neu-positive
and -negative cell lines. pILKthr178 protein level was
significantly increased in HER2/neu-positive SKBR3 cells while no
change was found in HER2/neu-negative MCF7, HEK293 and T47D cells
(Fig. 4A). The protein level of
pILKthr178 was significantly induced when
phosphorylation of pAKTser473 was inhibited by decreased
HER2/neu. However, it was not changed in HER2/neu-negative cells,
in which AKT phosphorylation was induced (Fig. 4A). This observation indicates that
p34SEI-1 overexpression promotes cancer metastasis by
inducing ILK instead of AKT when HER2/neu is diminished or
depleted. Further RT-PCR analysis showed that the negative effect
of p34SEI-1 on HER2/neu expression occurred both at the
transcriptional and the translational levels (Fig. 4B).
Taken together, the data demonstrates that
p34SEI-1 activates the PI3K/AKT signaling pathway using
at least two different types of signaling pathways depending on
HER2/neu expression status.
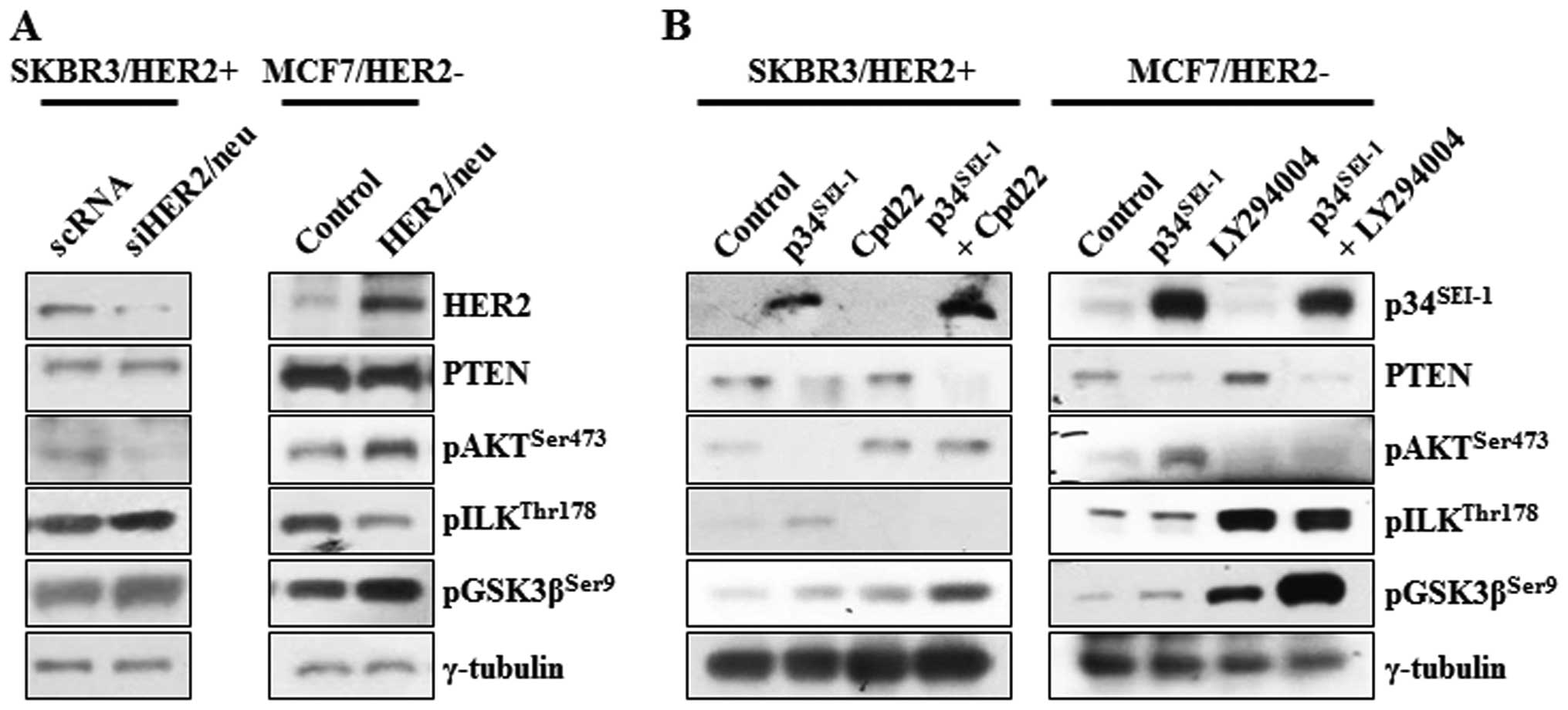
HER2/neu-dependent switching relationship
between AKT and ILK signaling pathways
Decreased HER2/neu activity by p34SEI-1
overexpression might be responsible for the activation of ILK
signaling in SKBR3 cells. This idea prompted the assessment of the
dependence of the activation of ILK on HER2/neu. The
phosphorylation of AKT, ILK and GSK3β was checked at the protein
level after SKBR3 and MCF7 cells were transfected with HER2/neu
specific siRNA or overexpressing pHER2/neu vector. In HER2/neu
silenced SKBR3 cells, pAKTser473 protein level was
decreased, probably due to HER2/neu mediated inhibition of PI3K, a
direct downstream target of HER2/neu. Surprisingly, both
pILKthr178 and pGSK3βser9 protein levels were
increased after treatment of HER2/neu silencing siRNA, in which
GSK3β was thought to be phosphorylated by the ILK rather than AKT
(Fig. 5A). On the other hand, the
expression levels of same proteins were also checked after MCF7 was
transfected with HER2/neu overexpressing vector. Overexpression of
HER2/neu highly promoted the phosphorylation of AKT on 473 serine
residue but reduced that of ILK on the 178 threonine residue. PTEN
was not affected by neither HER2/neu inhibition or overexpression
(Fig. 5A). In an extended
experiment, the switching relationship between AKT and ILK was
investigated using Cpd22 of ILK inhibitor and LY29002 of PI3K/AKT
inhibitor. In both groups, PTEN was decreased whenever
p34SEI-1 was overexpressed probably due to
p34SEI-1 mediated NEDD4-1 activation as we showed before
(28). Very importantly, both
groups showed an inverse relationship between pAKTser473
and pILKthr178 expression levels. When HER2/neu-positive
SKBR3 cells were treated with Cpd22, pAKTser473 protein
level increased even under p34SEI-1 overexpression
(Fig. 5B). When HER2/neu-negative
MCF7 cells were treated with LY29002, the ratio of ILK
phosphorylation was significantly elevated (Fig. 5B). LY29002 treatment after
p34SEI-1 overexpression produced an even higher level of
pILKthr178 expression (Fig.
5B). It may be explained by the strong switching relationship
between AKT and ILK. In both cases, p34SEI-1
overexpression and treatment of Cpd22 and LY29002 tremendously
increased the phosphorylation of GSK3β on the 9 serine residue in
both HER2/neu-positive and -negative cell lines. This may be the
basis for progression of metastasis because either AKT or ILK
phosphorylation could promote the metastasis under the circumstance
that one of them is inhibited (Fig.
5B).
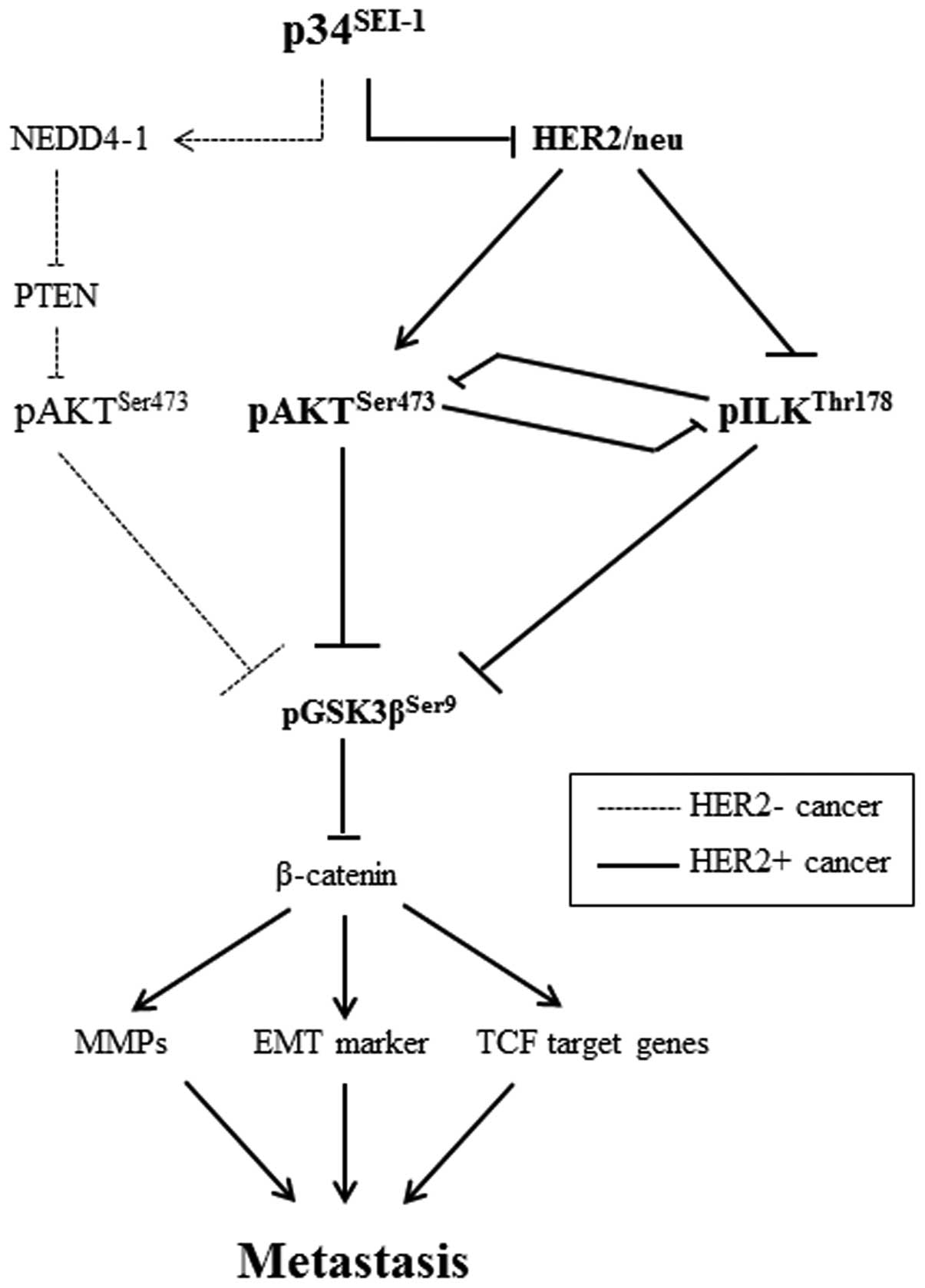
Taken together, the data demonstrate that
p34SEI-1 induces the activation of either AKT or ILK
signaling in a HER2/neu-dependent manner (Fig. 6).
Discussion
The present study shows that p34SEI-1
exerts a positive effect on cancer metastasis by inducing migration
and invasion of cancer cells. p34SEI-1 appears to
promote metastasis by activating the PI3K/AKT signaling pathway, in
which two different serine/threonine kinases, AKT and ILK, are
alternatively activated depending on HER2/neu expression. In
HER2/neu suppressed cells, p34SEI-1 overexpression
increased pAKTser473 protein level and in turn activated
PI3K/AKT signaling pathway at least partly via NEDD4-1 mediated
PTEN ubiquitination. However, pAKTser473 protein was
unexpectedly diminished despite exuberant p34SEI-1
overexpression in HER2/neu expressing SKBR3 cancer cells. In
HER2/neu strongly positive SKBR3 cells, p34SEI-1 reduced
HER2/neu, leading to the inhibition of AKT phosphorylation.
Instead, p34SEI-1 activated another multifunctional
serine/threonine protein kinase ILK to promote metastasis. This
result suggests that p34SEI-1 affects cancer metastasis
in a HER2/neu-dependent manner. Interestingly, our data also showed
the inverse relationship in the expression levels of AKT and ILK
proteins, implying that AKT and ILK have a switching relationship
with each other in HER2/neu-dependent manner. Considering all these
results, we suggest that drug resistance or recurrence of
metastatic cancer may be caused by the switching relationship
between AKT and ILK after the treatment of HER2/neu overexpressing
breast cancers with a monoclonal antibody targeting HER2/neu
oncogene (39). However, the exact
mechanism of HER2/neu inhibition by p34SEI-1
overexpression is still not clear and needs further study.
We showed that p34SEI-1 can downregulate
HER2/neu at the transcriptional and protein levels. We also found
that p21 enhancer activator 3 (PEA3) is at least partly responsible
for the p34SEI-1 mediated HER2/neu downregulation at the
transcription level (data not shown). Interestingly,
p34SEI-1 overexpression increased the expression levels
of PEA3 gene at the transcription level (data not shown). PEA3 can
bind to the promoter region of HER2/neu directly (40). PEA3 facilitates cancer invasion via
regulation of PI3K/AKT-related proteins and MMP13. Inhibition of
PEA3 diminishes the non-adherent tumor growth, migration and
invasion via downregulation of EMT markers in a variety of tumors
including breast cancer (41).
Therefore, we assumed that p34SEI-1 might negatively
affect HER2/neu expression by regulating PEA3 gene expression or by
interacting with it. However, more precise experiments such as ChIP
assay need to be performed to make the mechanism clear.
In summary, our data demonstrate that
p34SEI-1 activates the PI3K/AKT signaling pathway by
positively regulating at least two different types of AKT or
ILK-mediated signaling pathways depending on HER2/neu expression
status. Taken together, p34SEI-1 would be considered as
blocking metastatic breast cancer, and it might be used for the
prevention and treatment of metastatic breast cancer.
Abbreviations:
|
p34SEI-1
|
34-kD protein encoding SEI-1
(selected with Ink4a-1 as bait) gene;
|
|
HER2/neu
|
human epidermal growth factor receptor
2;
|
|
ILK
|
integrin-linked kinase;
|
|
PI3K
|
phosphoinositide-3 kinase;
|
|
AKT
|
serine/threonine-specific protein
kinase
|
Acknowledgements
This study was supported by Sookmyung
Women’s University (2011).
References
|
1.
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
progress, pitfalls, and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar
|
|
3.
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
DeFeo-Jones D, Barnett SF, Fu S, et al:
Tumor cell sensitization to apoptotic stimuli by selective
inhibition of specific Akt/PKB family members. Mol Cancer Ther.
4:271–279. 2005.PubMed/NCBI
|
|
6.
|
Qiao M, Sheng S and Pardee AB: Metastasis
and AKT activation. Cell Cycle. 7:2991–2996. 2008. View Article : Google Scholar
|
|
7.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Brader S and Eccles SA: Phosphoinositide
3-kinase signalling pathways in tumor progression, invasion and
angiogenesis. Tumori. 90:2–8. 2004.
|
|
9.
|
Stambolic V, Suzuki A, de la Pompa JL, et
al: Negative regulation of PKB/Akt-dependent cell survival by the
tumor suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Akca H, Demiray A, Tokgun O and Yokota J:
Invasiveness and anchorage independent growth ability augmented by
PTEN inactivation through the PI3K/AKT/NFκB pathway in lung cancer
cells. Lung Cancer. 73:302–309. 2011.PubMed/NCBI
|
|
11.
|
Carver BS, Chapinski C, Wongvipat J, et
al: Reciprocal feedback regulation of PI3K and androgen receptor
signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011. View Article : Google Scholar
|
|
12.
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: the PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Moasser MM: The oncogene HER2: its
signaling and transforming functions and its role in human cancer
pathogenesis. Oncogene. 26:6469–6487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Baselga J and Swain SM: Novel anticancer
targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer.
9:463–475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Grille SJ, Bellacosa A, Upson J, et al:
The protein kinase Akt induces epithelial mesenchymal transition
and promotes enhanced motility and invasiveness of squamous cell
carcinoma lines. Cancer Res. 63:2172–2178. 2003.PubMed/NCBI
|
|
16.
|
Toker A and Yoeli-Lerner M: Akt signaling
and cancer: surviving but not moving on. Cancer Res. 66:3963–3966.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yoeli-Lerner M and Toker A: Akt/PKB
signaling in cancer: a function in cell motility and invasion. Cell
Cycle. 5:603–605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Gagnon V, St-Germain ME, Parent S and
Asselin E: Akt activity in endometrial cancer cells: Regulation of
cell survival through cIAP-1. Int J Oncol. 23:803–810.
2003.PubMed/NCBI
|
|
19.
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Luo J: Glycogen synthase kinase 3beta
(GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett.
273:194–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Morin PJ: beta-catenin signaling and
cancer. Bioessays. 21:1021–1030. 1999. View Article : Google Scholar
|
|
22.
|
Guturi KK, Mandal T, Chatterjee A, et al:
Mechanism of beta-catenin-mediated transcriptional regulation of
epidermal growth factor receptor expression in glycogen synthase
kinase 3 beta-inactivated prostate cancer cells. J Biol Chem.
287:18287–18296. 2012. View Article : Google Scholar
|
|
23.
|
Hannigan GE, McDonald PC, Walsh MP and
Dedhar S: Integrin-linked kinase: not so ‘pseudo’ after all.
Oncogene. 30:4375–4385. 2011.
|
|
24.
|
Acconcia F, Barnes CJ, Singh RR, Talukder
AH and Kumar R: Phosphorylation-dependent regulation of nuclear
localization and functions of integrin-linked kinase. Proc Natl
Acad Sci USA. 104:6782–6787. 2007. View Article : Google Scholar
|
|
25.
|
Wani AA, Jafarnejad SM, Zhou J and Li G:
Integrin-linked kinase regulates melanoma angiogenesis by
activating NF-kappaB/interleukin-6 signaling pathway. Oncogene.
30:2778–2788. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Tan C, Cruet-Hennequart S, Troussard A, et
al: Regulation of tumor angiogenesis by integrin-linked kinase
(ILK). Cancer Cell. 5:79–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Taylor CJ, Qiao J, Colon NC, Schlegel C,
Josifi E and Chung DH: Integrin-linked kinase regulates phosphatase
and tensin homologue activity to promote tumorigenesis in
neuroblastoma cells. Surgery. 150:162–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Jung S, Li C, Jeong D, et al: Oncogenic
function of p34SEI-1 via NEDD41 mediated PTEN
ubiquitination/degradation and activation of the PI3K/AKT pathway.
Int J Oncol. 43:1587–1595. 2013.
|
|
29.
|
Wang X, Trotman LC, Koppie T, et al:
NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell.
128:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Amodio N, Scrima M, Palaia L, et al:
Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative
regulator, in non-small-cell lung carcinomas. Am J Pathol.
177:2622–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Hayashi R, Goto Y, Ikeda R, Yokoyama KK
and Yoshida K: CDCA4 is an E2F transcription factor family-induced
nuclear factor that regulates E2F-dependent transcriptional
activation and cell proliferation. J Biol Chem. 281:35633–35648.
2006. View Article : Google Scholar
|
|
32.
|
Hsu SI, Yang CM, Sim KG, Hentschel DM,
O’Leary E and Bonventre JV: TRIP-Br: a novel family of PHD zinc
finger- and bromodomain-interacting proteins that regulate the
transcriptional activity of E2F-1/DP-1. EMBO J. 20:2273–2285. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hong SW, Kim CJ, Park WS, et al:
p34SEI-1 inhibits apoptosis through the stabilization of
the X-linked inhibitor of apoptosis protein: p34SEI-1 as
a novel target for anti-breast cancer strategies. Cancer Res.
69:741–746. 2009.
|
|
34.
|
Li Y, Nie CJ, Hu L, et al:
Characterization of a novel mechanism of genomic instability
involving the SEI1/SET/NM23H1 pathway in esophageal cancers. Cancer
Res. 70:5695–5705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Tang DJ, Hu L, Xie D, et al: Oncogenic
transformation by SEI-1 is associated with chromosomal instability.
Cancer Res. 65:6504–6508. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Van Themsche C, Leblanc V, Parent S and
Asselin E: X-linked inhibitor of apoptosis protein (XIAP) regulates
PTEN ubiquitination, content, and compartmentalization. J Biol
Chem. 284:20462–20466. 2009.PubMed/NCBI
|
|
37.
|
Mehrotra S, Languino LR, Raskett CM,
Mercurio AM, Dohi T and Altieri DC: IAP regulation of metastasis.
Cancer Cell. 17:53–64. 2010. View Article : Google Scholar
|
|
38.
|
Arias AM: Epithelial mesenchymal
interactions in cancer and development. Cell. 105:425–431. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Knuefermann C, Lu Y, Liu B, et al:
HER2/PI-3K/Akt activation leads to a multidrug resistance in human
breast adenocarcinoma cells. Oncogene. 22:3205–3212. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Xing X, Wang SC, Xia W, et al: The ets
protein PEA3 suppresses HER-2/neu overexpression and inhibits
tumorigenesis. Nat Med. 6:189–195. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Yuen HF, McCrudden CM, Chan KK, et al: The
role of Pea3 group transcription factors in esophageal squamous
cell carcinoma. Am J Pathol. 179:992–1003. 2011. View Article : Google Scholar : PubMed/NCBI
|