Introduction
The peroxiredoxins (Prdxs) are a ubiquitous family
of evolutionarily conserved antioxidant proteins that reduce
aqueous and lipid peroxides associated with normal metabolism
(1–3). There are six members of the mammalian
peroxiredoxin family, which can be subdivided into three classes:
the 2-Cys Prdxs (Prdx1–4), the atypical 2-Cys Prdx (Prdx5) and the
1-Cys Prdx (Prdx6). These proteins reduce cellular substrates by
converting their active-site cysteines to sulfenic acid, which can
be re-reduced by thiols such as ascorbic acid or glutathione
through different mechanisms for different classes. These proteins
are highly abundant in mammalian cells, suggesting an important
role in anti-oxidant defense. In addition, peroxiredoxins
participate in redox-sensitive signal transduction pathways, and
are know to have effects on cell growth, proliferation,
differentiation and apoptosis (4,5).
It has long been recognized that cancer cells harbor
elevated levels of highly reactive oxygen species (ROS) such as
hydrogen peroxide and superoxide (6,7),
exhibiting many signs of an oxidatively stressed environment. There
is evidence that this oxidative stress may be a factor in both
cancer initiation and/or cancer progression, depending on the tumor
(8). Unlike normal cells, which
are susceptible to cytotoxicity from such high levels of ROS,
cancer cells are relatively resistant and can evade cell death, yet
the precise mechanisms are not clear. Over the past several years,
studies have reported overexpression of Prdxs in several types of
cancer (9–14), and there is mounting evidence that
Prdxs play a role in carcinogenesis (9,15–19).
These data suggest that cancer cell resistance to ROS may be
provided, at least in part, through peroxiredoxin overexpression,
leading to increased antioxidant activity and/or alteration in key
redox-regulated growth and death pathways. However, there is very
little understanding of the precise role peroxiredoxins play in
this protection or the mechanism that governs increased
peroxiredoxin expression in cancer cells.
Many studies have reported that breast tumors
exhibit elevated levels of Prdxs compared to normal breast
epithelial tissue (10,12,16,20).
Furthermore, based on evidence that an adaptive oxidative stress
response is critical to chemoresistance, it was recently suggested
that peroxiredoxins are likely to be key players in chemoresistance
in breast cancer, and may be potential targets for intervention
(21). While clinical studies are
important, a comparison of relevant cell lines can be a valuable
tool in understanding the role of Prdxs in breast cancer biology
and drug resistance. We and others have shown significant
overexpression of Prdxs in the MCF-7 adenocarcinoma cell line, as
compared to the non-cancerous MCF-10A breast epithelial line
(22–24). We previously demonstrated that
MCF-7 cells are much more resistant to
H2O2-induced apoptosis than the non-malignant
MCF-10A breast cells (22). Bae
et al also reported elevated Prdx levels in MCF-7 cells, and
went on to show that overexpression of either Prdx1 or Prdx2 in
MCF-10A cells conferred resistance to
H2O2-induced apoptosis (22). Furthermore, radiation-resistant
lines derived from the MCF-7 cells have elevated Prdx2 levels, and
Prdx2 suppression in these cells partially reversed the radiation
resistance (25). Taken together,
these studies suggest a protective role for Prdxs in breast cancer
and suggest that use of these cell lines may be an important tool
in understanding the function of Prdxs in breast cancer.
Based on these data and implications, we sought to
examine the role of Prdxs in MCF-7 cell survival and
doxorubicin-resistance using siRNA-mediated protein suppression.
Doxorubicin is an anthracycline antibiotic that has been used as an
effective chemotherapy agent in the treatment of breast cancer in
patients, although many patients develop resistance to the drug
leading to aggressive relapse (26). Doxorubicin induces oxidative stress
and apoptosis in MCF-7 cells (27), suggesting that antioxidants may be
an important line of defense in this and other breast cancer cells.
We hypothesized that Prdx suppression in MCF-7 cells would decrease
the viability of these cells and increase their susceptibility to
doxorubicin-induced toxicity. Likewise, we hypothesized that
prolonged exposure of MCF-7 cells to doxorubicin would lead to
induction of Prdx expression.
Materials and methods
Cell culture
MCF-7 cells were cultured in ATCC-formulated Eagle’s
minimum essential medium containing bovine insulin (0.01 mg/ml) and
10% fetal bovine serum. MCF-10A cells were cultured in MEBM medium,
supplemented with BPE (13 mg/ml), hydrocortisone (0.5 mg/ml), hEGF
(10 μg/ml), insulin (5 mg/ml), and cholera toxin (100
ng/ml). Both cell lines were cultured at 37°C in a humidified 5%
CO2 atmosphere.
Doxorubicin treatments
To determine resistance to doxorubicin, MCF-7 and
MCF10-A cells were cultured in 48-well plates and treated the
following day with 0.1 or 0.5 μM doxorubicin for 48 h. To
determine the effect of Prdx suppression on doxorubicin-resistance,
MCF-7 cells were allowed to grow for 48 h after siRNA transfection
and then were treated with 0.5 μM doxorubicin for an
additional 24 h. For generation of a doxorubicin-resistant culture,
MCF-7 cells were subcultured for 14 days in T-75 flasks in the
presence of 0.1 μM doxorubicin, with media and treatment
replacement every 3–4 days.
Measurement of viable cell density
Cell viability was determined using an MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
MCF-7 and MCF-10A cells were seeded into separate 48-well plates in
replicate groups of four and transfection/treatment experiments
conducted. After treatment incubation, cells were assayed for MTT
at a confluency less than 80%. Cells were rinsed with phenol
red-free medium and incubated with 0.5 mg/ml MTT (diluted in phenol
red-free medium) for 1.5 h at 37°C. This medium was then removed
and replaced with 200 μl of acidic isopropanol and the plate
was rocked for 5 min at room temperature. The absorbance of the
solubilized product was read at 570 nm using the corresponding
absorbance of cell-free wells or the absorbance at 650 for
background subtraction.
Suppression of Prdx1–6 by siRNA
MCF-7 cells were seeded into 24-well plates at
50,000 cells per well. Twenty-four hours after seeding, cells were
transfected using Silencer Select siRNA (Ambion, Austin, TX, USA)
for Prdx1-6 (or a negative control siRNA) at a final concentration
of 33 nM using the Lipofectamine 2000 reagent (Invitrogen,
Carlsbad, CA, USA). The siRNA ID#s are as follows: Prdx1 (s10007),
Prdx2 (s13959), Prdx3 (s21507), Prdx4 (s20686), Prdx5 (s24559), and
Prdx6 (s18429). Briefly, for each well 20 pmoles of siRNA was mixed
with 1 μl Lipofectamine 2000 reagent and 98 μl
Opti-MEM I serum-free media (Invitrogen), allowed to precipitate
for 20 min, and subsequently added to wells. Transfected cells were
cultured for 48 h followed by protein extraction and western blot
analysis to determine levels of Prdx suppression. Transfected cells
used for cell viability and toxicity assays were treated with or
without doxorubicin for an additional 24 h prior to end-point
assays.
Western blot analysis
For protein analysis, cells were lysed in mammalian
protein extraction reagent (MPER) (Thermo Scientific, Waltham, MA,
USA) according to the product suggestions. Protein was quantified
using the Coomassie Blue Protein Assay Reagent (Bio-Rad, Hercules,
CA, USA), and lysates were separated on a 12% Mini Protean TGX gel
and electrophoretically transferred on to an ImmunBlot PVDF
membrane (Bio-Rad). Blots were blocked and incubated with primary
antibodies from Abcam [anti-Prdx1 (ab59538), anti-Prdx2 (ab15572),
anti-Prdx3 (ab16751), anti-Prdx4 (ab59542), anti-Prdx5 (ab16944)
anti-Prdx6 (ab16947); Cambridge, MA, USA]. An antibody for GAPDH
(Sigma, St. Louis, MO, USA) was used as a loading control. Blots
were subsequently processed with the appropriate secondary antibody
and chemiluminescent CDP-Star Reagent, and imaged with X-OMAT film
(Kodak). Bands were quantified using Image J software.
Measurement of cytotoxicity
Cytotoxicity was determined by the indirect
measurement of lactate dehydrogenase (LDH) activity using the
Cytotox 96 Assay (Promega, Madison, WI, USA). Cells were
transfected as described above and cultured for 48 h. To measure
released LDH, cell medium was removed and 40 μl was assayed
using an equal volume of substrate mix and processed according to
manufacturer's recommendations. Absorbance was measured at 490 nm
and absorbance of media blanks (with no cells) was subtracted from
each value.
Measurement of apoptosis
A membrane permeability/dead cell apoptosis kit
(Invitrogen) and Hoechst 33342 (Life Technologies) were used to
detect cell death. Cells were transfected according to the previous
methods and treated with or without 0.5 μM doxorubicin for
24 h. Cells were stained with 1 μl/ml Yo-Pro and 1
μg/ml Hoechst dye and photographed using phase contrast and
fluorescence microscopy. The field of view was quantified as a
percent of cells staining positive for Hoechst and Yo-Pro.
Statistical analysis
The means of individual treatment groups in each
quantitative experiment were statistically compared using a
two-tailed Student's t-test, assuming equal variances.
Results
We first compared the non-cancerous MCF-10A cell
line with the MCF-7 breast adenocarcinoma for sensitivity to
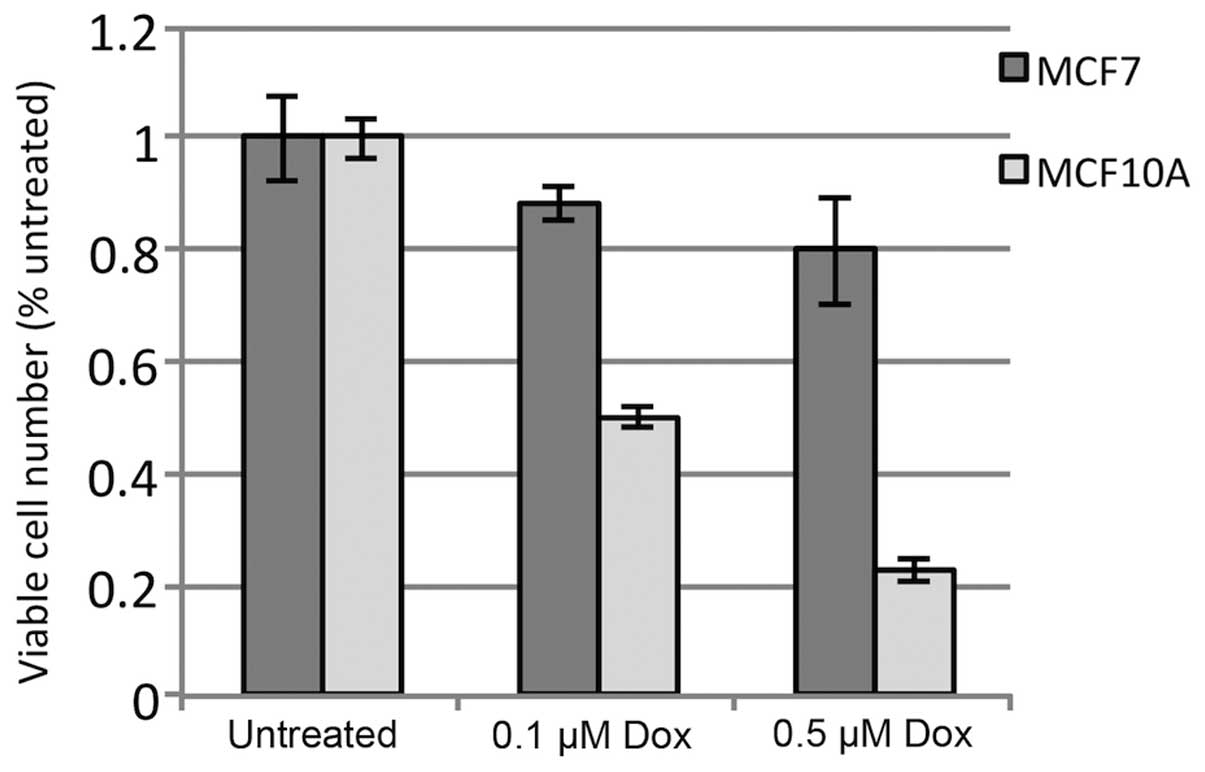
doxorubicin-induced toxicity. As shown in Fig. 1, MCF-10A cells exhibited an
approximately 50% reduction in viable cells after 24 h with 0.1
μM doxorubicin, as compared to untreated cells, and a nearly
80% reduction with 0.5 μM doxorubicin. In contrast, MCF-7
cells exhibited only a 10 and 20% reduction in viability with the
same treatments, respectively. The data show significant tolerance
of the MCF-7 cell line to this drug treatment. Analysis of
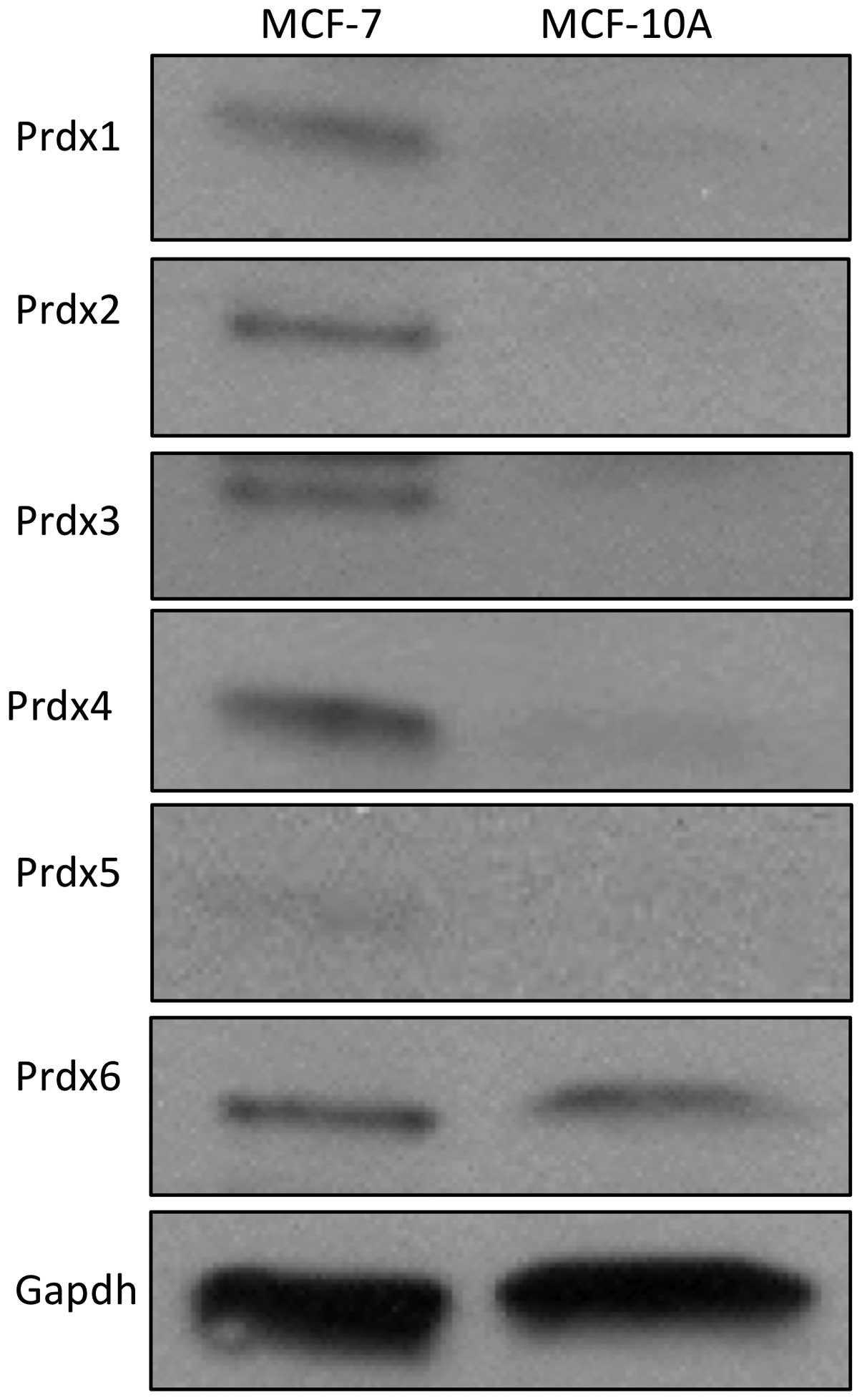
peroxiredoxin protein expression in these two lines revealed that
expression of five of the six Prdx proteins (Prdx1-5) are markedly
elevated in MCF-7 cells, as compared to MCF-10A cells (Fig. 2). Together, these data show a
correlation between doxorubicin resistance and peroxiredoxin
expression in MCF-7 cells.
In order to address the potential role of Prdxs in
MCF-7 cell survival, we used transient siRNA transfection
experiments to suppress individual Prdx proteins in these cells.
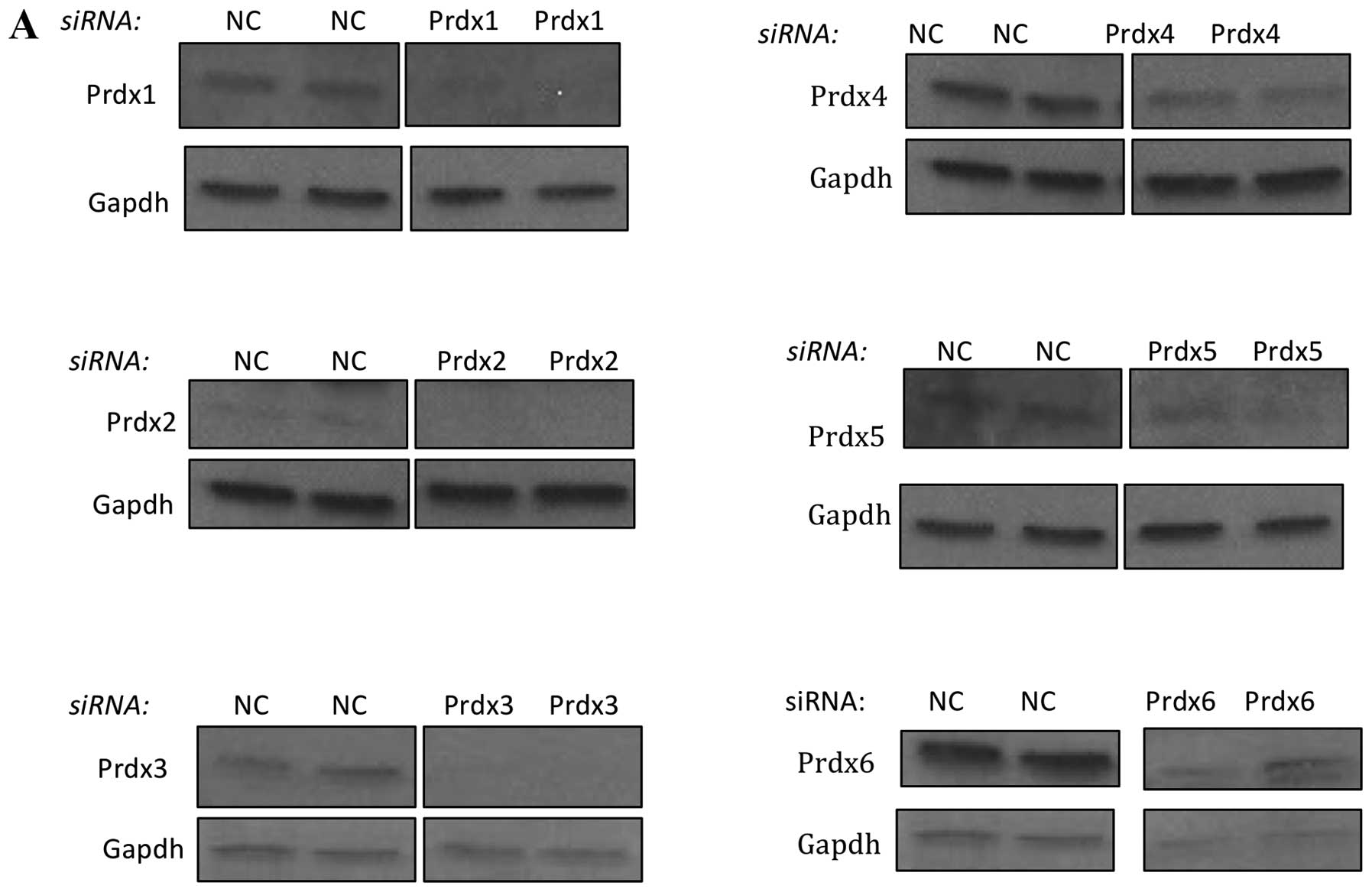
Cells were transfected with 33 nM siRNA and Prdx levels measured
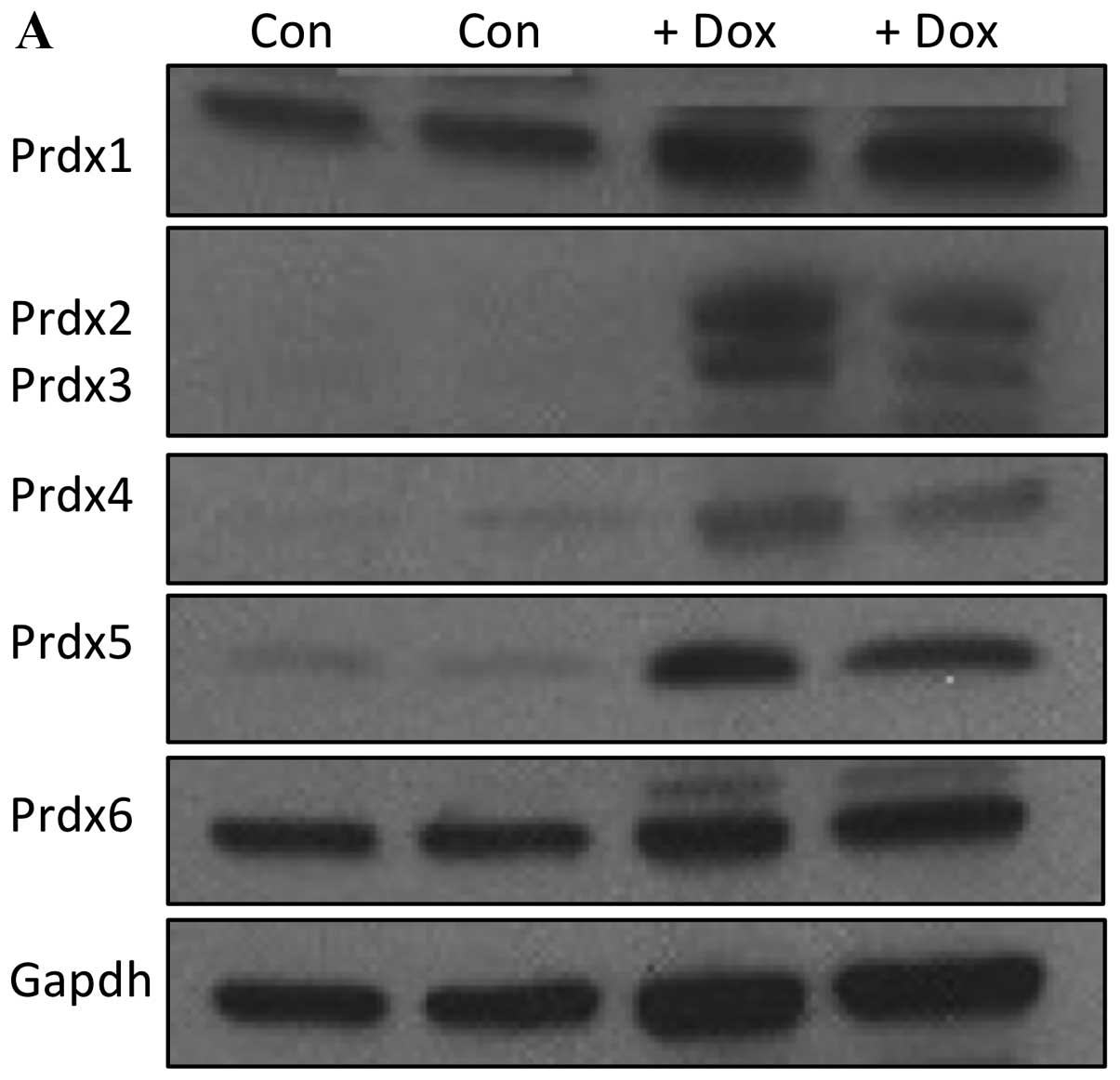
after 48 h by western blot analysis. As shown in Fig. 3A, we were able to greatly reduce
the expression of all six Prdx proteins by this method.
Quantification of these levels is shown in Fig. 3B, which demonstrates a range
between 70 and 90% protein suppression relative to cells
transfected with a negative control siRNA.
Before examining the effect of Prdx suppression on
doxorubicin sensitivity, we determined whether Prdx suppression in
these cells affected their morphology or viability. Seventy-two
hours after transfection (and 24 h after suppression was measured
by western blot analysis) the cells were examined by phase contrast
microscopy and analyzed for cytotoxicity using the released LDH
assay. First, we found no difference in either morphology or
cytotoxicity between untransfected cells and those transfected with
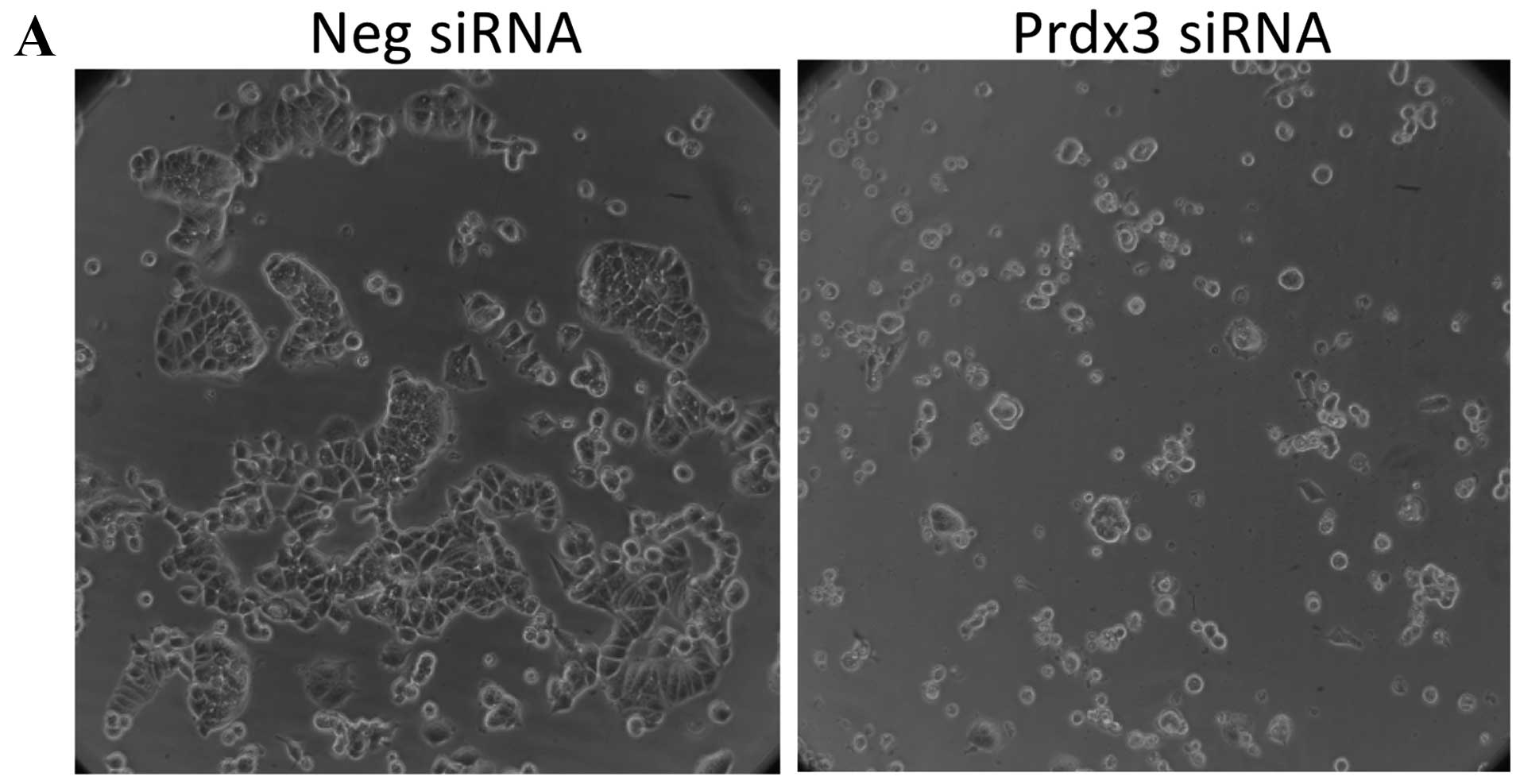
negative control siRNA (data not shown). The only morphological
change observed in transfected cells was with Prdx3. While
negative-control transfected cells appear to have a normal
cobblestone-like appearance (Fig.
4A), Prdx3-transfected cells are significantly smaller and
rounder (Fig. 4B). In addition,
there were fewer cells in all Prdx3-transfected replicate wells.
Likewise, cytotoxicity was significantly increased in
Prdx3-transfected cells in the absence of doxorubicin treatment,
relative to cells transfected with negative control siRNA (Fig. 4B). No other transfection condition
showed an effect. Together, these data show that Prdx3 suppression
renders MCF-7 cells more susceptible to death in the absence of
doxorubicin, suggesting an important role for this protein in the
general viability of these cells.
To examine the effect of Prdx suppression on
doxorubicin sensitivity, 48 h after transfection cells were treated
with 0.5 μM doxorubicin for 24 h, viable cell number was
measured using the MTT assay, which was originally used to compare
doxorubicin sensitivity in the cancerous and non-cancerous cell
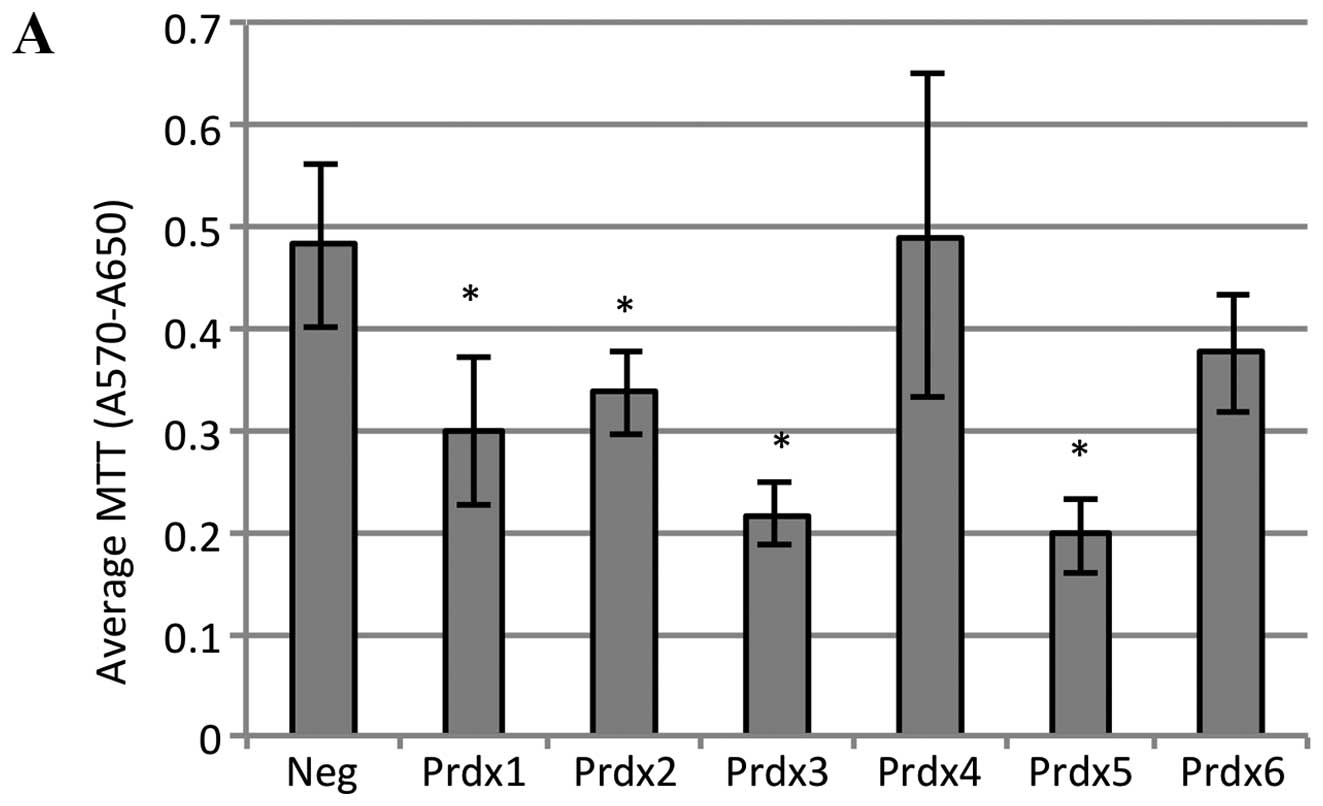
lines. As shown in Fig. 5A,
doxorubicin treatment led to a significant reduction in MTT
absorbance in cells transfected with either Prdx1, Prdx2, Prdx3 or
Prdx5. The magnitude of this decrease was about 40% for Prdx1 and
Prdx2, and over 50% for Prdx3 and Prdx5. These data suggest that
reduction in the levels of these Prdx proteins in MCF-7 cells
inhibits growth and/or induces death in response to doxorubicin
treatment. We attempted to address this by measuring cell death
using the Hoechst/Yo-Pro cell staining method. Representative phase
contrast and fluorescent images are shown for doxorubicin-treated
cells transfected with negative control siRNA, or one of the Prdxs
that showed an MTT reduction. A reduced cell number in all
Prdx-transfected cells, compared to the negative control, was
observed. These data are consistent with the MTT data. Analysis of
the stained cells shows a marked increase in the percentage of dead
cells in all Prdx-transfected conditions. These data strongly
suggest that suppression of Prdx1, Prdx2, Prdx3 and Prdx5 in MCF-7
cells increases the susceptibility of MCF-7 cells to
doxorubicin-induced cell death.
Since our data suggested a role for Prdxs in
doxorubicin resistance in MCF-7 cells, we asked whether long-term
treatment of these cells and selection of a highly resistant
subline would lead to a concomitant change in Prdx levels. This
experiment has important clinical significance since many breast
cancers develop resistance with prolonged chemotherapy treatment.
Cells were cultured in the presence of 0.1 μM doxorubicin
for 14 days and Prdx expression analyzed. As shown in Fig. 6A, 14 days of treatment led to a
marked increase in the expression of Prdxs 2, 3, 4 and 5.
Quantification of these levels are represented in Fig. 6B, revealing a nearly 10X increase
in Prdx2 expression, and an approximately 4-fold increase in levels
of Prdx3, Prdx4 and Prdx5. It is clear from these data that
culturing MCF-7 cells for 2 weeks in doxorubicin leads to a robust
induction of several Prdx proteins.
Discussion
In the present study, we showed a correlation
between doxorubicin-resistance and peroxiredoxin levels between
MCF-7 and MCF-10A cells, demonstrating significantly higher
resistance and Prdx expression in the cancer line. Using transient
transfections of MCF-7 cells with siRNA, we obtained marked
reduction in Prdx levels for all six proteins, leading to moderate
toxicity in Prdx3-suppressed cells. Subsequent treatment of
siRNA-transfected cells with doxorubicin resulted in a reduction in
viable cell number with suppression of either Prdx1, Prdx2, Prdx3
and Prdx5. We went on to show that this cell loss was, at least in
part, due to apoptotic death. Finally, we demonstrated that 2-week
treatment of MCF-7 cells with doxorubicin leads to a marked
induction of several Prdx proteins. Together, these data support
our hypothesis that Prdxs play a protective role in MCF-7 cells and
that doxorubicin-treatment leads to selection of drug-resistant
cells that possess elevated Prdx levels.
We and others previously reported the overexpression
of Prdxs in MCF-7 cells (22–24),
which is consistent with elevated Prdx levels found in breast
cancer tissue from patients. However, the mechanism by which these
cells upregulate Prdxs is not understood. The Prdx family is
inducible by oxidative stress in several systems, and ROS-induced
modifications include regulation at both the transcriptional and
post-transcriptional levels (4). A
previous study from our lab showed that Prdxs1-5 are elevated at
the mRNA level in these cells, compared to MCF-10A cells,
suggesting a transcriptional mechanism (24), but the signal transduction events
and transcription factors mediating higher basal levels are not
known. However, there is evidence that Nrf2 coordinately regulates
the Prdx gene family in macrophages (28), so we are currently investigating
this as a possible mechanism in MCF-7 cells.
Prdx suppression in many cells, including several
cancer cell types, is known to increase cell death. Our results
suggest a similar protective role for Prdx3 in MCF-7 cells, in the
absence of any added oxidative stress. Prdx3 is a mitochondrial
peroxiredoxin that is transcriptionally regulated by c-myc and is
required for proliferation, transformation, and apoptosis in
ovarian cancer cells (29).
Recently, a similar function was reported for Prdx3 in cervical
cancer cells (30). From these and
other studies, the importance of Prdx3 as a key protective protein
in cancer is well established. Our results are the first to
demonstrate this same function for Prdx3 in breast cancer cells,
suggesting that this protein may have a more ubiquitous survival
function in cancer.
We showed that MCF-7 breast cancer cells are
significantly more resistant to doxorubicin-induced toxicity at
both 0.1 and 0.5 μM concentrations than the non-cancerous
MCF-10A cells. Gajewski et al also demonstrated that MCF-10A
cells exposed to 0.1 μM doxorubicin (a clinically relevant
dosage) underwent growth arrest and apoptosis, and also developed
elevated levels of ROS (31).
However, our demonstration that siRNA-mediated Prdx suppression
markedly increases doxorubicin-induced apoptosis is a novel
finding, and one that is consistent with the known ROS-inducing
action of doxorubicin as well as the increased susceptibility of
Prdx-suppressed MCF-7 cells to ROS-induced apoptosis. For example,
Wang et al showed that Prdx2 suppression in MCF-7 cells
increased sensitivity to radiation-induced cell death (25), and a recent follow-up study
demonstrated that this occurred by alterations in cellular thiol
status and intracellular Ca2+ homeostasis (32). Likewise, Prdx1 suppression in MCF-7
cells leads to apoptosis induced by β-lapachone, an anticancer
agent that produces large amounts of ROS induced apoptosis
(33). In addition, Bae et
al showed that transgenic overexpression of Prdx1 and Prdx2 in
MCF-10A cells increases their resistance to peroxide-induced cell
death (22). Together, there is
strong evidence for Prdxs as key protective players against
ROS-induced death of breast cancer cells.
Our results further showed a functional relationship
between Prdx expression and doxorubicin resistance using a
prolonged doxorubicin treatment. The marked induction of several
Prdx proteins after a 2-week culture with 0.1 μM doxorubicin
suggests higher levels in drug-resistant cells. While it is not
clear that this Prdx induction is essential for clonal selection of
resistant cells, this observation coupled with the data from our
transfection experiments strongly suggests an important role for
these proteins in cell survival. Interestingly, short-term (4 or 24
h) treatment of MCF-7 cells with 0.1 or 0.5 μM doxorubicin
does not alter Prdx levels (data not shown), suggesting that the
changes in gene expression are likely associated with the selection
of resistant cells over time.
In conclusion, our data are the first to report an
effect of doxorubicin treatment on Prdx expression in breast cancer
cells, as well as a protective role for the peroxiredoxin protein
family in breast cancer cell resistance to doxorubicin. Since the
innate and acquired resistance of many breast tumors to doxorubicin
is of critical concern for patients, a better understanding of the
mechanisms governing this likely multifactorial phenomenon is
essential. While we do not yet understand the precise role of each
individual Prdx in the basal antioxidant defense system in these
cells, Prdxs may, in fact, play an essential role in the survival
of breast cancer cells in vivo. Based on the abundance and
obvious importance of this family of antioxidants in normal and
cancer cell biology, and the critical role of oxidative stress in
chemotherapy success, this area of research warrants further
investigation and is likely to provide an important new avenue for
new therapeutic interventions for the treatment of breast
cancer.
Acknowledgements
This study was supported in part by a
grant from the Connecticut Breast Health Initiative (CTBHI).
References
|
1.
|
Fujii J and Ikeda Y: Advances in our
understanding of peroxiredoxin, a multifunctional, mammalian redox
protein. Redox Rep. 7:123–130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Hofmann B, Hecht HJ and Flohe L:
Peroxiredoxins. Biol Chem. 383:347–364. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Wood ZA, Schröder E, Harris JR and Poole
LB: Structure, mechanism and regulation of peroxiredoxins. Trends
Biochem Sci. 28:32–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Immenschuh S and Baumgart-Vogt E:
Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid
Redox Signal. 7:768–777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Rhee SG, Chae HZ and Kim K:
Peroxiredoxins: A historical overview and speculative preview of
novel mechanisms and emerging concepts in cell signaling. Free Rad
Biol Med. 38:1543–1552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Halliwell B and Gutteridge J: Free
Radicals in Biology and Medicine. 3rd edition. Oxford University
Press; New York, NY: 1999
|
|
7.
|
Halliwell B: Oxidative stress and cancer:
have we moved forward? Biochem J. 401:1–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Valko M, Rhodes CJ, Moncol J, Izakovic M
and Mazur M: Free radicals, metals and antioxidants in oxidative
stress-induced cancer. Chem Biol Interact. 160:1–40. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Butterfield LH, Merino A, Golub SH and
Shau H: From cytoprotection to tumor suppression: the
multifactorial role of peroxiredoxins. Antioxid Redox Signal.
1:385–402. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Noh DY, Ahn SJ, Lee RA, Kim SW, Park IA
and Chae HZ: Overexpression of peroxiredoxin in human breast
cancer. Anticancer Res. 21:2085–2090. 2001.
|
|
11.
|
Kinnula VL, Lehtonen S, Sormunen R,
Kaarteenaho-Wiik R, Kang SW, Rhee SG and Soini Y: Overexpression of
peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J
Pathol. 196:316–323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Karihtala P, Mantyniemi A, Kang SW,
Kinnula VL and Soini Y: Peroxiredoxins in breast carcinoma. Clin
Cancer Res. 15:3418–3424. 2003.
|
|
13.
|
Lehtonen ST, Svensk AM, Soini Y, Paakko P,
Hirvikoski P, Kang SW and Saily M: Peroxiredoxins, a novel protein
family in lung cancer. Int J Cancer. 111:514–521. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Quan C, Cha EJ, Lee HL, Han KH, Lee KM and
Kim WJ: Enhanced expression of peroxiredoxin I and VI correlates
with development recurrence and progression of human bladder
cancer. J Urolol. 175:1512–1516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kinnula VL, Paakko P and Soini Y:
Antioxidant enzymes and redox regulating thiol proteins in
malignancies of human lung. FEBS Lett. 569:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Li DQ, Wang L, Fei F, Hou YF, Luo JM,
Wei-Chen, Zeng R, Wu J, Lu JS, Di GH, Ou ZL and Xia QC:
Identification of breast cancer metastasis-associated proteins in
an isogenic tumor metastasis model using two-dimensional gel
electrophoresis and liquid chromatography-ion trap-mass
spectrometry. Proteomics. 6:3352–3368. 2006. View Article : Google Scholar
|
|
17.
|
Neumann CA and Fang Q: Are peroxiredoxins
tumor suppressors? Curr Opin Pharm. 7:375–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di
GH, Jin W, Ou ZL and Shen ZZ: Identification of the functional role
of peroxiredoxin 6 in the progression of breast cancer. Breast
Cancer Res. 9:R762007. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ishii T, Warabi E and Yanagawa T: Novel
roles of peroxiredoxins in inflammation, cancer and innate
immunity. J Clin Biochem Nutr. 50:91–105. 2012.PubMed/NCBI
|
|
20.
|
Chahed K, Kabbage M, Hamrita B, Guillier
CL, Trimeche M, Remadi S, Ehret-Sabatier L and Chouchane L:
Detection of protein alterations in male breast cancer using two
dimensional gel electrophoresis and mass spectrometry: The
involvement of several pathways in tumorigenesis. Clin Chim Acta.
388:106–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Coley HM: Mechanisms and strategies to
overcome chemotherapy resistance in metastatic breast cancer.
Cancer Treat Rev. 34:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Bae J, Ahn S, Han W and Noh D:
Peroxiredoxin I and II inhibit H2O2-induced
cell death in MCF-7 Cell Lines. J Cell Biochem. 101:1038–1045.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Goncalves K, Sullivan K and Phelan SA:
Differential expression and function of peroxiredoxin 1 and
peroxiredoxin 6 in cancerous MCF-7 and noncancerous MCF-10A breast
epithelial cells. Cancer Invest. 30:38–47. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Tehan L, Taparra K and Phelan SA:
Peroxiredoxin overexpression in MCF-7 breast cancer cells and
regulation by cell proliferation and oxidative stress. Cancer
Invest. 31:374–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wang T, Tamae D, LeBon T, Shively JE, Yen
Y and Li JJ: The role of peroxiredoxin II in radiation-resistant
MCF-7 breast cancer cells. Cancer Res. 65:10338–10346. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hickman JA: Apoptosis induced by
anticancer drugs. Cancer Metastasis Rev. 11:121–139. 1992.
View Article : Google Scholar
|
|
27.
|
Osbild S, Brault L, Battaglia E and Bagrel
D: Resistance to cisplatin and adriamycin is associated with the
inhibition of glutathione efflux in MCF-7-derived cells. Anticancer
Res. 26:3595–3600. 2006.PubMed/NCBI
|
|
28.
|
Ishii T, Itoh K, Takahashi S, Sato H,
Yanagawa T, Katoh Y, Bannai S and Yamamoto M: Transcription factor
Nrf2 coordinately regulates a group of oxidative stress-inducible
genes in macrophages. J Biol Chem. 275:16023–16029. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Wonsey DR, Zeller KI and Dang CV: The
c-Myc target gene PRDX3 is required for mitochondrial homeostasis
and neoplastic transformation. Proc Natl Acad Sci USA.
99:6649–6654. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Li L, Zhang YG and Chen CL: Anti-apoptotic
role of peroxiredoxin III in cervical cancer cells. FEBS Open Bio.
3:51–54. 2012. View Article : Google Scholar
|
|
31.
|
Gajewski E, Gaur S, Akman SA, Matsumoto L,
van Balgooy JNA and Doroshowa JH: Oxidative DNA base damage in
MCF-10A breast epithelial cells at clinically achievable
concentrations of doxorubicin. Biochem Pharmacol. 73:1947–1956.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Diaz AJG, Tamae D, Yen Y, Li JJ and Wang
T: Enhanced radiation response in radioresistant MCF-7 cells by
targeting peroxiredoxin II. Breast Cancer (Dove Med Press).
5:87–101. 2013.PubMed/NCBI
|
|
33.
|
He T, Banach-Latapy A, Vernis L, Dardalhon
M, Chanet R and Huang ME: Peroxiredoxin 1 knockdown potentiates
β-lapachone cytotoxicity through modulation of reactive oxygen
species and mitogen-activated protein kinase signals.
Carcinogenesis. 34:760–769. 2013.
|