Introduction
The antiestrogen tamoxifen has for several decades
and until recently been recommended as first-line endocrine
treatment for women with primary estrogen receptor α (ER)-positive
breast cancer. Although many patients respond well to the
treatment, resistance to tamoxifen is inevitable in advanced
disease and therefore a major clinical challenge. Upon progression
on tamoxifen, the pure antiestrogen fulvestrant (ICI 182,780 or
faslodex) is a treatment option (1). However, as for tamoxifen, resistance
to fulvestrant will eventually occur in patients with advanced
disease. Although the molecular mechanisms leading to antiestrogen
resistance are still not fully clarified, it is well recognized
that breast cancer cell growth, upon acquired antiestrogen
resistance, can switch from being ER-driven to be mediated by
members of the human epidermal growth factor receptor (HER) family
(2–6).
The HER receptor family comprises four type-1
transmembrane receptor tyrosine kinases: EGFR/HER1, HER2, HER3 and
HER4 (7,8). Increased HER expression and HER
signaling is a frequent phenomenon in various human cancers
(9). In breast tumors, the
expression and phosphorylation of EGFR, HER2 and HER3 have been
associated with poor prognosis, in contrast to the expression of
HER4, which has been linked to a better disease outcome (10–12).
Several ligands have been identified for EGFR, HER3 and HER4,
whereas no ligands have been identified for HER2 (7). The HER ligands are all synthesized as
transmembrane precursors that are proteolytically cleaved and
released (shed) from the membrane by metalloproteinases (7,13).
In the absence of ligand, the HER receptors are monomeric. However,
upon ligand activation of the HER receptors, changes in the
receptor conformation leads to formation of homo- or heterodimers
and consequently activation of the receptors via tyrosine
phosphorylation (except for HER3, which lacks a functional kinase
domain) (7). Activated HER
receptors can induce multiple downstream signaling pathways, which
leads to increased cell proliferation and reduced cell death
(7). Accordingly, as the HER
receptors represent important therapeutic targets for cancer
treatment, several therapies have been developed. These include
antibodies such as trastuzumab (directed against HER2) and
cetuxumab (directed against EGFR) as well as small tyrosine kinase
inhibitors like gefitinib (targeting EGFR) or lapatinib (targeting
EGFR/HER2). Although the HER targeted therapies have shown
promising results in the adjuvant and advanced setting, still many
patients exhibit de novo or acquired resistance (14,15).
It has been suggested that targeting the HER receptors individually
leads to treatment resistance as increased production of the HER
ligands may circumvent the loss of the function of a single
receptor (16,17). Thus, preventing HER ligand shedding
with metalloproteinase inhibitors may be an alternative and more
efficient way to prevent HER-mediated signaling and cancer cell
growth than targeting the HER receptors individually.
To investigate the mechanisms behind antiestrogen
resistant breast cancer cell growth, we have established a model
system with MCF-7 breast cancer cell lines with acquired resistance
to fulvestrant (18). The
fulvestrant-resistant MCF-7 cell lines have increased protein
expression or phosphorylation of EGFR and HER3 and can be growth
inhibited by targeting the HER receptors or signaling molecules
downstream of the HER receptors (2,6,19).
Thus, the breast cancer cell lines can switch from ER- to
HER-driven cell growth upon acquiring resistance to fulvestrant. In
agreement with the high levels of phosphorylated HER3, the level of
the HER3 activating ligands heregulin 2α and −2β was also increased
in resistant cell lines. Collectively, our study corroborate the
importance of the HER system in signaling and growth of
fulvestrant-resistant breast cancer cells (2). The work presented here was carried
out to unravel the importance of the HER receptors, in particular
HER3, and HER ligand shedding for growth and signaling in
fulvestrant-resistant MCF-7 cell lines and to explore whether
inhibition of ligand shedding could be a new treatment strategy for
resistant cells.
Materials and methods
Cell lines, culture conditions and
reagents
The MCF-7 cell line was originally obtained from the
Human Cell Culture Bank (Mason Research Institute, Rockville, MD,
USA). MCF-7 cells were maintained in growth medium without phenol
red (DMEM/F12; Gibco) supplemented with 1% FCS, 2 mM glutamax
(Gibco, Invitrogen, CA, USA) and 6 ng/ml insulin (Sigma-Aldrich,
St. Louis, MO, USA). The fulvestrant-resistant cell lines,
MCF-7/164R-5 (164R-5) and
MCF-7/164R-7 (164R-7) were established as
previously described (18) and
maintained in MCF-7 growth medium supplemented with 0.1 μM
fulvestrant (Tocris, Avonmouth, Bristol, UK). For experiments,
2.5×105 units of penicillin and 31.25 μg/l
streptomycin (Gibco) were added to the growth medium. TAPI-2, BB-94
and GM6001 were obtained from Calbiochem (Merck Biosciences,
Nottingham, UK), CI-1033 (Canertinib) and gefitinib from Selleck
(Chemicals, Munich, Germany), Ab5 from Thermo Fisher Scientific
(Fremont, CA, USA) and heregulin 1β, TGFα and EGF from R&D
Systems (Minneapolis, MN, USA). Stock solutions of 12 mM TAPI-2 was
dissolved in water, whereas stock solutions of 1 mM gefitinib, 10
mM CI-1033, 10 mM BB-94 and 10 mM GM6001 were dissolved in
DMSO.
Cell growth assays
The cell lines were seeded in 24-well multidishes in
growth medium and allowed to adhere for two days. When experiments
were initiated (day 0), growth medium containing fulvestrant (0.1
μM), HER ligands (10 ng/ml), gefitinib, CI-1033, TAPI-2,
BB-94 or GM6001 were added at concentrations indicated in the
figure. The control cells were added similar amount of vehicle as
the treated cells. Growth medium was replaced on day three, and
cell number was determined on day five, using a crystal violet
colorimetric assay as previously described (20). Each experiment was performed in
quadruplicate and repeated at least twice.
Western blot analysis
Western analyses were performed with cells lysed in
RIPA buffer (100 mM NaCl, 20 mM Tris-HCl, 1% Triton X-100, 0.5%
sodium deoxycholate, 0.1% SDS and 1 mM EDTA) supplemented with 1 mM
DDT, 1 mM NaF, 10 mM β-glycerol phosphate, 100 μM
Na3VO4, 150 μM PMSF and 1 tablet/10 ml
RIPA complete mini protease inhibitor cocktail (Roche). To
investigate the effect of HER ligands, HER inhibitors or
metalloproteinase inhibitors on expression of total and
phosphorylated forms of HER receptors or downstream signaling
molecules, cells were grown in 6-well multidishes until 60–80%
confluence and then treated for 15 min (HER ligands) or 24 h (HER
or metalloproteinase inhibitors) before cell extracts were obtained
by cell lysis in RIPA as described above. Conditioned medium was
prepared from MCF-7, 164R-5 and 164R-7 cells
grown with or without 20 μM TAPI-2. The cells were seeded in
T75 flasks and pre-treated for 24 h with 20 μM TAPI-2 before
new medium with 20 μM TAPI-2 was added. After additional 24,
48 or 72 h, the medium from each flask was collected and
concentrated ten times by centrifugation using iCON concentrators
with a molecular weight cut-off of 20 kDa (Thermo Fisher
Scientific, Fremont, CA, USA). MCF-7 cell cultures were grown to
60–70% confluence under standard conditions and treated for 15 min
with medium to which concentrated conditioned medium from MCF-7,
164R-5 or 164R-7 was added. Cell extracts
were obtained by cell lysis in RIPA as described above.
Determination of protein concentration in all cell
lysates was done using Bio-Rad Protein Assay kit (Bio-Rad
Laboratories, Copenhagen, Denmark). Total protein (15 μg)
was separated on 4–15% Tris-HCl or 3–8% Tris-Acetate resolving
criterion gels (Bio-Rad Laboratories) and transferred to an
ethanol-activated Immobilon-P membrane (Millipore, Bedford, MA,
USA). To prevent non-specific binding, membranes were blocked in
TBS containing 5% dry-milk, 0.2% FCS and 0.1% Tween-20 for 2–3 h at
room temperature. Incubation with the primary antibody was
performed overnight at 4°C followed by 1-h incubation with
species-specific peroxidase-conjugated secondary antibodies (Dako,
Glostrup, Denmark). Protein bands were visualized by enhanced
chemiluminescence [ECL Plus detection system (GE Healthcare,
Hilleroed, Denmark)] and detected by a CCD camera (LAS-1000,
Fujifilm). In order to detect multiple proteins, the antibodies
were stripped from the membrane by incubation for 15 min in 62.5 mM
Tris-HCl, 100 mM β-mercaptoethanol and 2% (w/v) SDS, pH 6.7, and
washed before incubated with another antibody. The following
antibodies were purchased from Cell Signaling Technology (Beverly,
MA, USA): phosphorylated HER3 (Tyr1289, 1:500, 4791),
phosphorylated Akt (Ser473, 1:500, 9271), phosphorylated
Erk (Thr202/Tyr204, 1:1000; 4377), Akt
(1:2000, 9272) and Erk (1:2000, 9102). HER3 (1:2000, M7297) was
purchased from Dako and Hsp70 (1:500.000, MS-482) from Thermo
Fisher Scientific. All western analyses were performed at least
twice and representative blots are shown.
Gene silencing with small interfering
RNA
HER3-targeting SMART pool siRNA (L-003127) and
scrambled sequence (non-targeting) control pool siRNA (D-001810)
were obtained from Dharmacon (Lafayette, CO, USA). Transfection of
cells with 300 nM HER3 or scramble siRNA in 100 μl
nucleofector solution (Cell Line Nucleofector kit V) was performed
using the Nucleofector device from Amaxa (Lonza, Cologne, Germany)
according to the manufacturer’s instructions. Transfected cells
were seeded in 6- and 24-well multidishes in standard growth medium
to measure protein expression and cell growth, respectively. Medium
was replaced at day one and the cell number in 24-well plates were
determined on day one, three and six using a crystal violet
colorimetric assay as previously described (20). Three days after transfection, cells
grown in 6-well multidishes were harvested in RIPA buffer and
subjected to western analysis as described above.
Statistics
Three independent growth experiments were performed
with quadruplicate measures. Two-sample unequal variance t-test
followed by Bonferroni’s correction was used to calculate whether
the observed differences in growth were statistically significant.
For all experiments, a P-value <0.05 was considered significant.
Representative experiments with mean and standard deviation (SD)
are shown.
Results
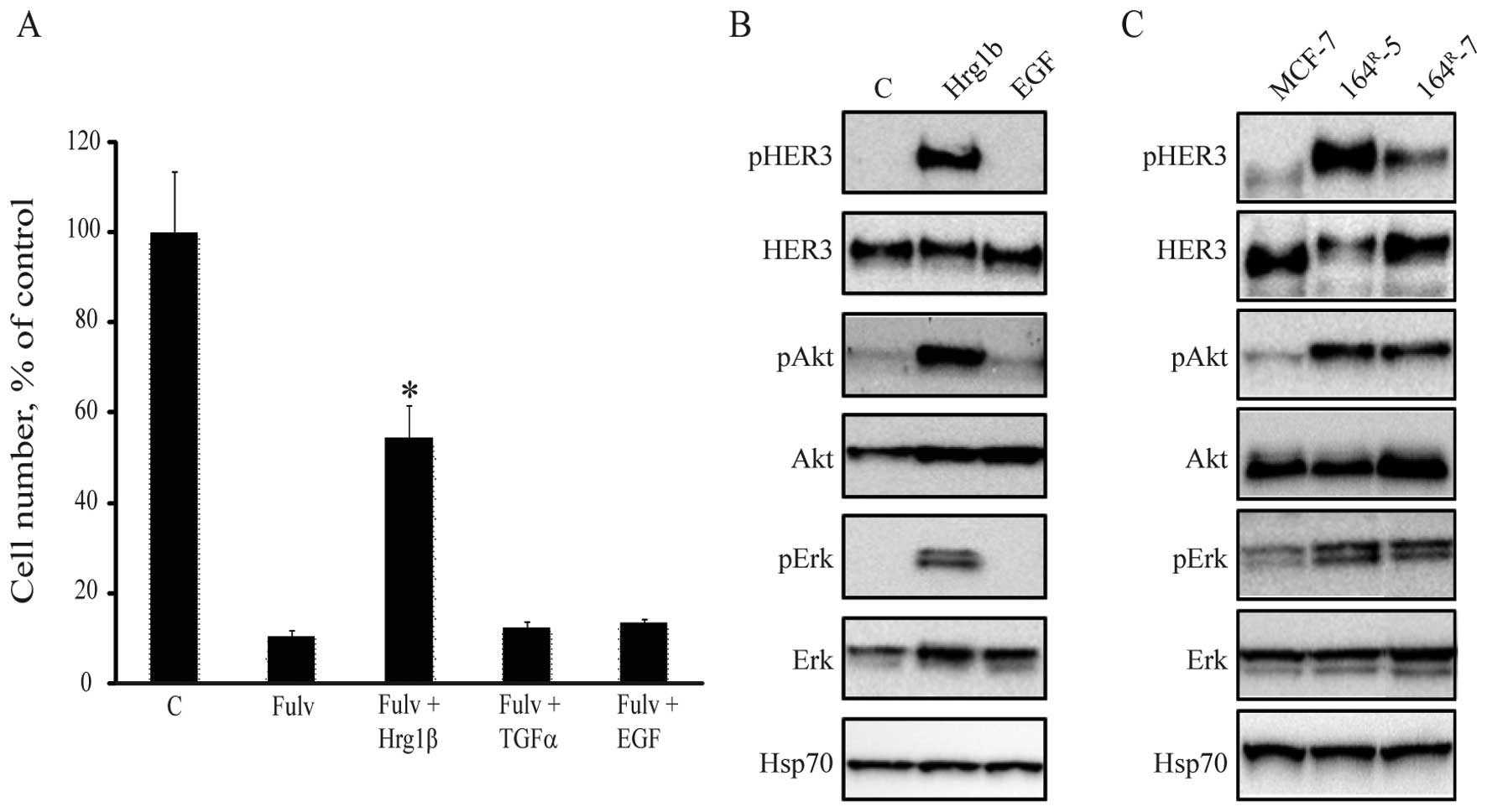
HER ligands partially rescue
fulvestrant-mediated growth inhibition
To clarify the importance of HER signaling in
response to treatment with fulvestrant, we investigated whether
different HER ligands could rescue fulvestrant-inhibited growth of
MCF-7 cells. Five days treatment with fulvestrant alone inhibited
cell growth of MCF-7 cells to 10% of the untreated control
(Fig. 1A). However, when heregulin
1β, which has the same function as heregulin 2β e.g. activation of
HER3 (2), was added,
fulvestrant-mediated inhibition of MCF-7 cell growth was partially
rescued. No effect on cell growth was observed when the EGFR
ligands EGF or TGFα were added to MCF-7 cells (Fig. 1A). The role of the HER ligands on
intracellular signaling in MCF-7 cells was investigated by western
blot analysis showing that heregulin 1β could induce HER3, Akt and
Erk phosphorylation in MCF-7 cells (Fig. 1B). Thus, the HER3 ligand heregulin
1β can partly rescue fulvestrant-inhibited cell growth via
activation of HER3 and downstream signaling molecules.
We have previously shown that our
fulvestrant-resistant MCF-7 cell lines produce increased levels of
the heregulins (2). In line with
this, the fulvestrant-resistant cell lines, 164R-5 and
164R-7, express increased levels of phosphorylated HER3,
Akt and Erk compared with their parental MCF-7 cells (Fig. 1C).
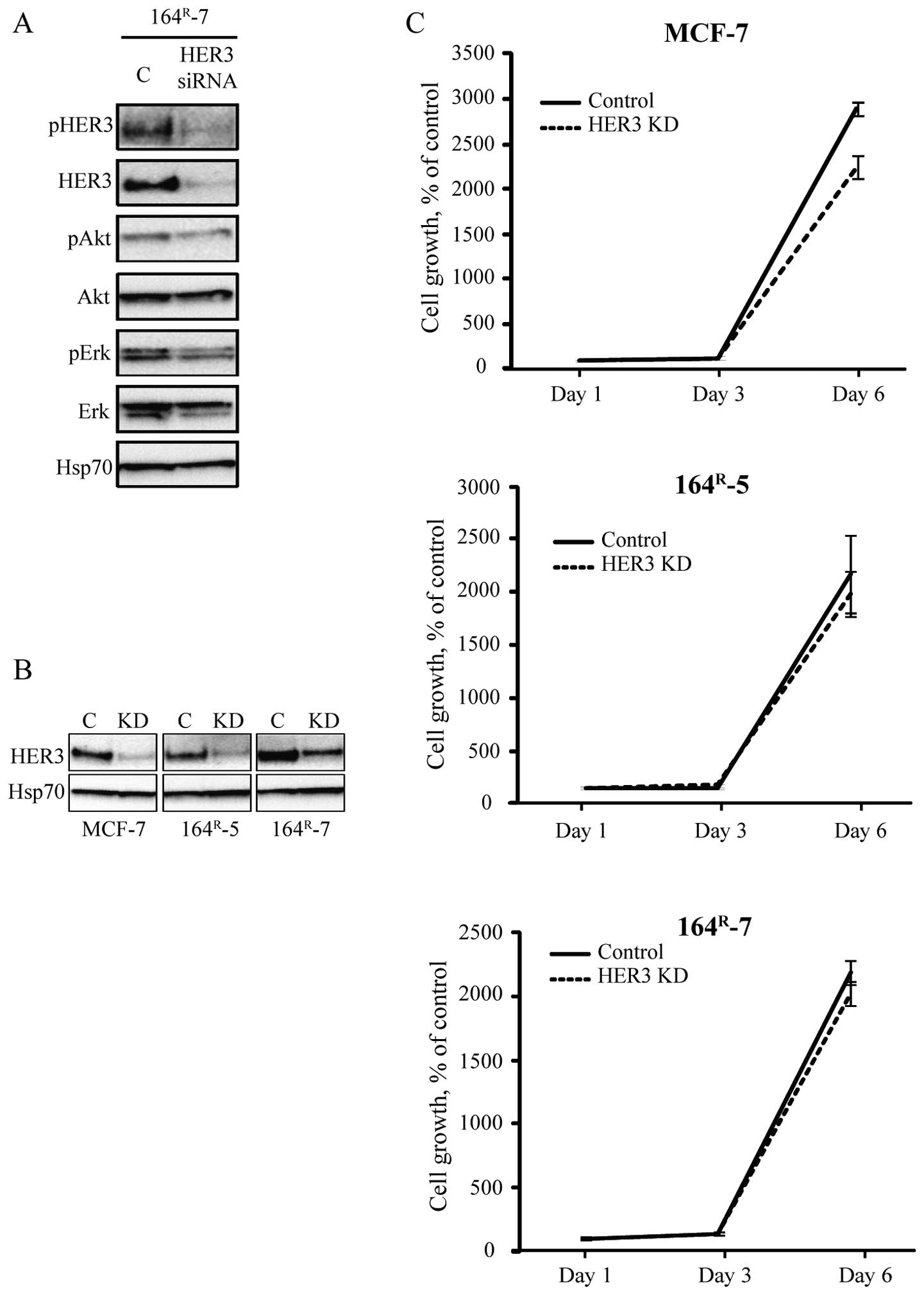
The knock-down of HER3 in parental and
fulvestrant-resistant cell lines has no effect on cell growth
To further explore the role of HER3 for
fulvestrant-resistant cell growth, siRNA-mediated knock-down of
HER3 was performed. HER3 knock-down in 164R-7
consequently depleted the cells of HER3 as well as reduced the
level of phosphorylated Akt and Erk (Fig. 2A). Next, growth of MCF-7 and the
fulvestrant-resistant 164R-5 and 164R-7 cells
was investigated upon transfection with control (scramble) or HER3
siRNA (Fig. 2B). As shown in
Fig. 2C, siRNA-mediated knock-down
of HER3 had no effect on growth up to six days after transfection
of the fulvestrant-resistant cell lines. In contrast, growth of the
parental MCF-7 cells was inhibited after the siRNA-mediated
knock-down of HER3 (Fig. 2C).
Thus, although signaling via HER3 is upregulated in the resistant
cells, knock-down of HER3 had no effect on resistant cell
growth.
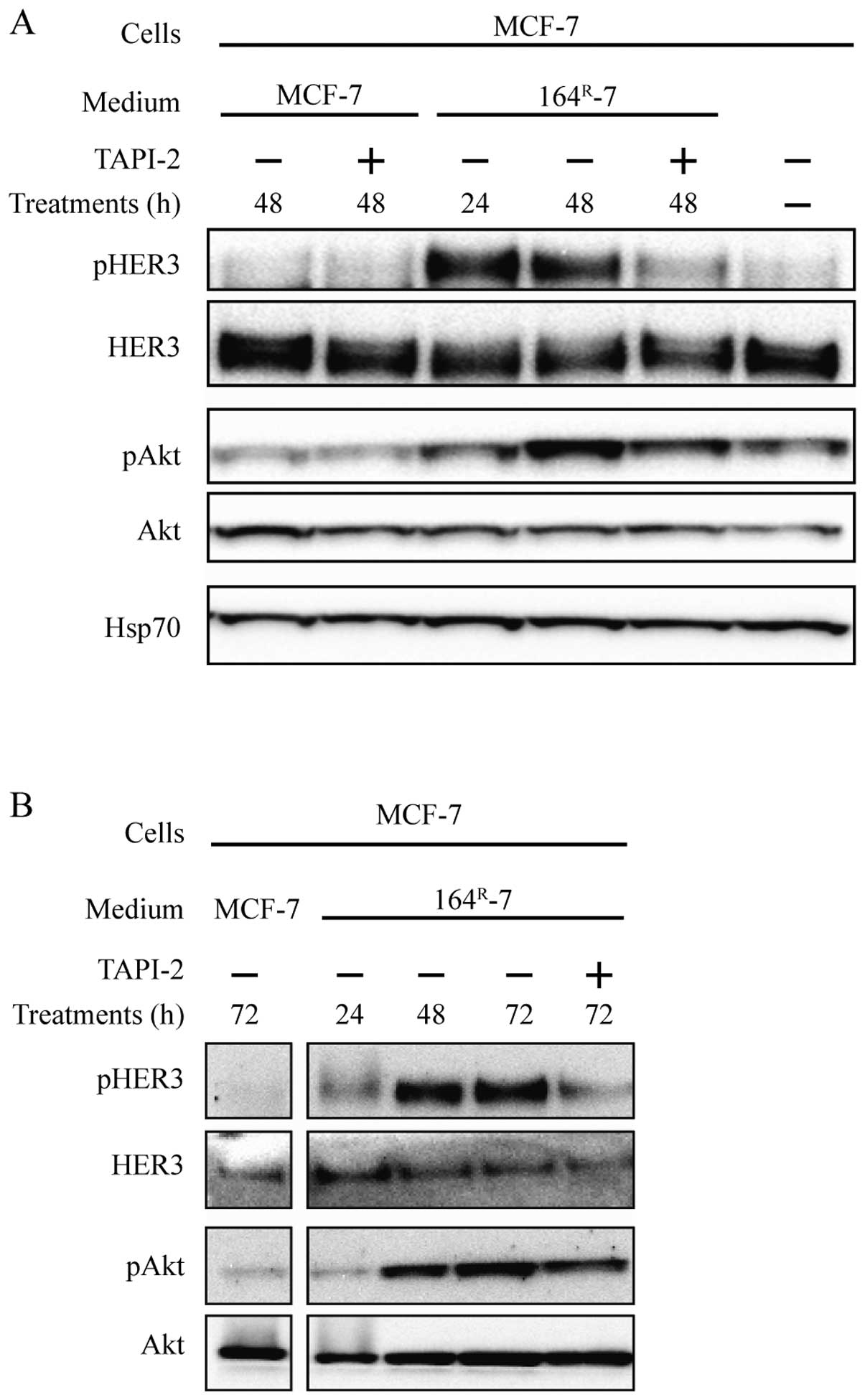
Metalloproteinase inhibitor TAPI-2
prevents release of HER3 activating ligands
Signaling in our fulvestrant-resistant cells is, at
least partly, due to endogenous synthesis of ligands which activate
HER3 (2). As the knock-down of
HER3 does not seem to be important for resistant cancer cell
growth, we investigated if inhibition of ligand shedding, and thus
prevention of HER3 activation, could abolish growth and signaling
in resistant cell lines. HER3 ligands are synthesized as
membrane-bound proteins, which are shed by metalloproteinases and
thus able to activate HER3. Initially, in order to reveal whether
metalloproteinase-mediated ligand shedding takes place in
fulvestrant-resistant cells, conditioned medium was collected from
the fulvestrant-resistant 164R-5 or 164R-7
cells grown with or without TAPI-2, a potent TACE inhibitor with
some activity towards other ADAMs and matrix metalloproteinases.
The medium was then concentrated and added to MCF-7 cells to
explore the effect on HER3 and Akt phosphorylation.
Medium conditioned by 164R-7 cells for 24
or 48 h induced phosphorylation of HER3 and Akt in MCF-7 cells
(Fig. 3A). When MCF-7 cells were
incubated with conditioned medium from 164R-7 cells
treated with TAPI-2 for 48 h, no phosphorylation of HER3 was seen
and downstream signaling to Akt was reduced (Fig. 3A). Similar results were obtained
with conditioned medium from 164R-5 (Fig. 3B). Treatment of MCF-7 cells with
conditioned medium from 164R-5 or 164R-7
cells treated with TAPI-2 had no effect on the expression of total
HER3 and total Akt (Fig. 3A and
B). Moreover, there were no differences in the expression and
phosphorylation levels of HER3 and Akt when MCF-7 cells were
incubated with medium conditioned by MCF-7 cells grown in the
presence or absence of TAPI-2 for 48 h (Fig. 3A). Collectively, these data support
that the fulvestrant-resistant cell lines 164R-5 and
164R-7, in contrast to MCF-7 cells, produce endogenous
HER3 ligands, which require cleavage by TACE, or another TAPI-2
sensitive metalloproteinase to be shed into the medium.
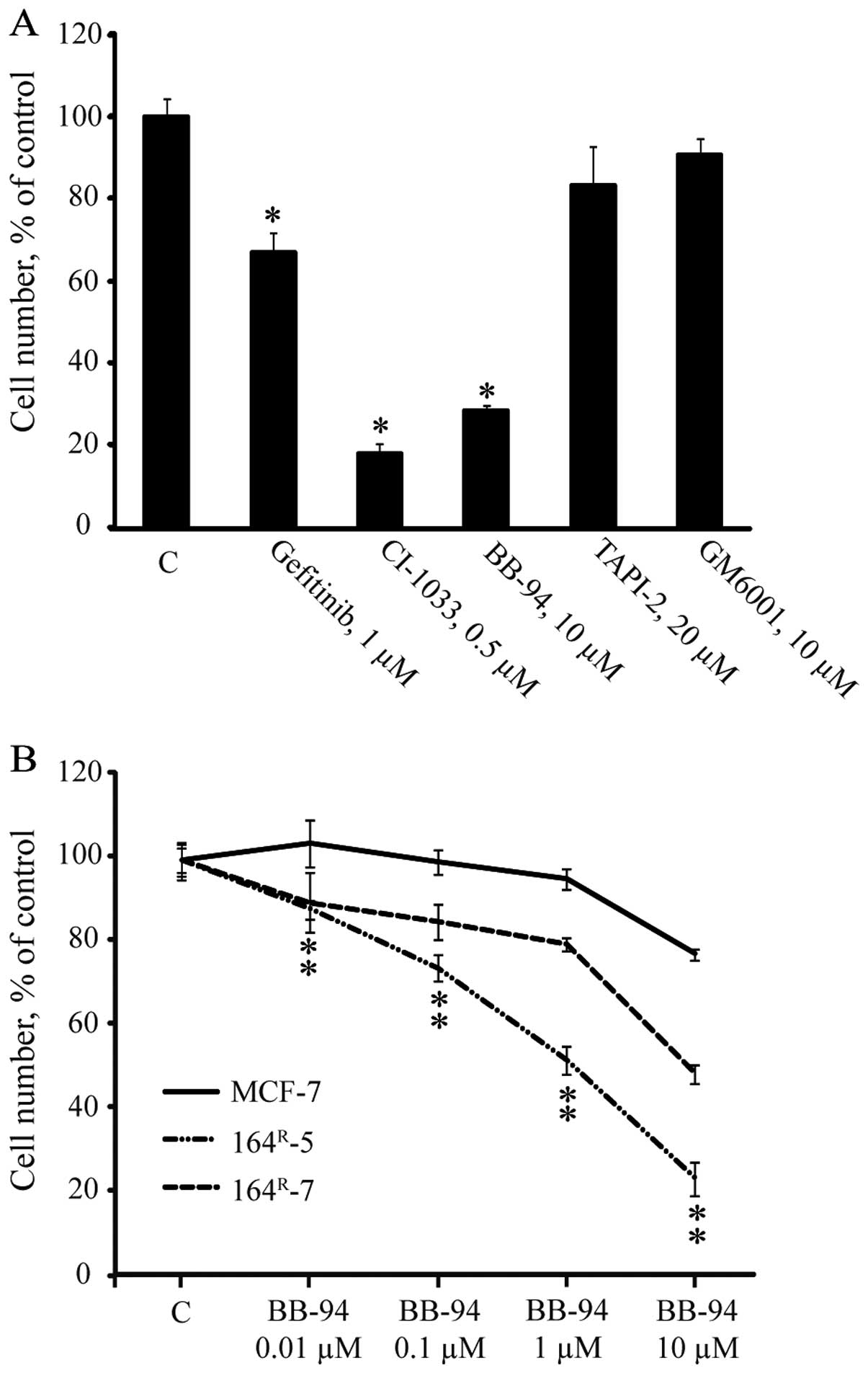
Metalloproteinase inhibitor BB-94 is able
to prevent growth and signaling of fulvestrant-resistant breast
cancer cell lines
To further investigate the importance of HER
receptors and HER ligand shedding for cell growth and signaling in
the resistant cell lines, 164R-7 was treated for five
days with gefitinib (preferentially targeting EGFR), CI-1033
(pan-HER inhibitor), or the metalloproteinase inhibitors TAPI-2,
BB-94 or GM6001 (Fig. 4A).
Compared to growth of the untreated control, 1 μM gefitinib
or 0.5 μM CI-1033 significantly inhibited growth of
164R-7 by 30 and 80%, respectively. Moreover, the
broad-spectrum metalloproteinase inhibitor BB-94 significantly
inhibited resistant growth by 70%, whereas the more selective
metalloproteinase inhibitors TAPI-2 and GM6001 had no inhibitory
effect on growth of 164R-7 (Fig. 4A). When the five-day dose-response
growth experiments with BB-94 were performed, increasing
concentrations (0.01–10 μM) of BB-94 resulted in a
dose-dependent growth inhibition up to 50% and 80% compared to the
untreated control for 164R-7 and 164R-5,
respectively, whereas growth of the parental MCF-7 cells in the
presence of increasing concentrations of BB-94 was <20%
inhibited compared to the untreated control (Fig. 4B). Thus, BB-94 preferentially
inhibited growth of fulvestrant-resistant MCF-7 cell lines.
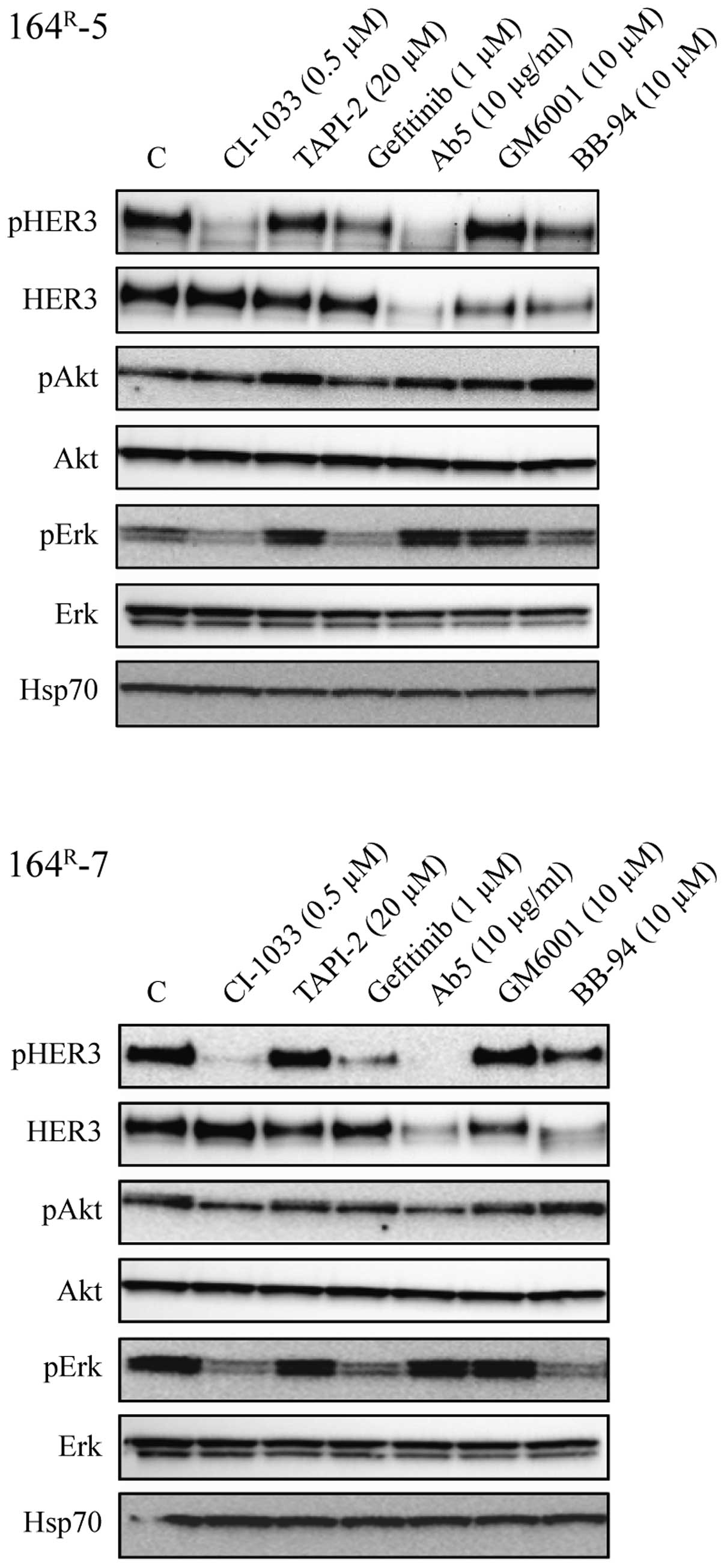
We then tested the effect of CI-1033, gefitinib, Ab5
(neutralizing antibody against HER3), TAPI-2, BB-94 and GM6001 on
signaling in 164R-5 and 164R-7 cells.
Treatment of the fulvestrant-resistant cells for 24 h with CI-1033
(0.5 μM) strongly inhibited phosphorylation of HER3 and Erk
whereas phosphorylation of Akt was only slightly reduced (Fig. 5). Treatment with gefitinib (1
μM) reduced the level of phosphorylated HER3 and strongly
reduced Erk phosphorylation, indicating that EGFR signals via Erk.
Treatment with Ab5 (10 μg/ml) prevented phosphorylation of
HER3, had no effect on phosphorylation of Erk and only little
effect on phosphorylation of Akt. Ab5 also resulted in degradation
of HER3. BB-94 (10 μM) inhibited phosphorylation of HER3 and
Erk, induced degradation of HER3 but had no effect on
phosphorylation of Akt. In contrast, TAPI-2 (20 μM) and
GM6001 (10 μM) had no effect on phosphorylation of HER3, Akt
or Erk.
Collectively, the experiments in Figs. 4 and 5 show that treatment with CI-1033, which
exerted the most severe growth inhibition, completely blocked HER3
phosphorylation and resulted in severe reduction of phosphorylated
Erk. Treatment with gefitinib resulted in reduced phosphorylation
of Erk, but did not totally reduce phosphorylation of HER3 and had
only a modest effect on cell growth. As the broad-spectrum
metalloproteinase inhibitor BB-94 severely reduced phosphorylation
of Erk, reduced expression and phosphorylation of HER3 and exerted
strong growth inhibition while TAPI-2 and GM6001 had no effect on
phosphorylation of HER3 and Erk, our data indicate that BB-94, in
contrast to TAPI-2 and GM6001, targets signaling of both via EGFR
and HER3.
Discussion
Several experimental studies have shown that breast
cancer cell lines can switch from ER- to HER-driven cell growth
upon acquiring antiestrogen resistance (2–4,6,21,22).
This is in agreement with the clinical observation that response to
endocrine treatment is reduced in ER-positive breast cancer
patients with HER2-positive tumors compared to the response in
HER2-negative tumors (23).
Moreover, it is in line with the clinical benefit for patients with
ER/HER2-positive breast cancer treated with a combination of
endocrine therapy and the EGFR/HER2 tyrosine kinase inhibitor
lapatinib, as compared to treatment with endocrine therapy alone
(24). In this study, we used
MCF-7 and MCF-7-derived breast cancer cell lines, which have
progressed from antiestrogen sensitive to anti estrogen resistant
cells during long-term treatment with the pure antiestrogen
fulvestrant. The expression of ER is maintained in the resistant
cells but at a reduced level. However, when grown in presence of
fulvestrant, ER is degraded and the resistant cells primarily use
the HER system for growth (2,6).
Previous studies have shown that the fulvestrant-resistant cell
lines secrete increased levels of the HER3/4 ligands heregulin 2α
and 2β, and in agreement with this also express significantly
increased level of activated HER3. The level of total EGFR,
phosphorylated EGFR and phosphorylated Erk is also increased in the
resistant cell lines, whereas HER4 level is severely reduced
(2). The aim of the current study
was to unravel the importance of HER receptors, in particular HER3,
and HER ligand shedding for growth of the fulvestrant-resistant
MCF-7 cell lines and to explore whether inhibition of ligand
shedding could be a new treatment strategy for resistant cells.
Initially, we tested whether prevention of ER
signaling and consequent cell growth arrest induced by fulvestrant
in the ER-positive MCF-7 cell line could be abrogated by activation
of the HER system via addition of exogenous HER ligands. We found
that MCF-7 cells were able to grow in the presence of fulvestrant
when the HER system was activated by the HER3/HER4 ligand heregulin
1β but not by TGFα or EGF. Western blot analysis showed that
exogenous heregulin 1β induced phosphorylation of HER3, Akt and Erk
in MCF-7 cells, supporting that activation of signaling via HER3
can abrogate fulvestrant induced growth inhibition.
To further explore the importance of HER3 for
resistant cell growth, siRNA mediated HER3 knock-down was performed
in two fulvestrant-resistant cell lines, 164R-5 and
164R-7. The HER3 knock-down resulted in close to total
abrogation of expression of HER3 and consequently also
phosphorylation of HER3, as well as reduced levels of
phosphorylated Akt and Erk, but cell growth was not significantly
inhibited. This demonstrates that Akt and Erk are downstream
mediators of HER3 signaling induced by endogenous ligands in the
resistant cell lines, in agreement with our finding that exogenous
heregulin 1β was able to activate HER3, Akt and Erk in parental
MCF-7 cells. The lack of significant growth inhibition by HER3
knock-down in fulvestrant-resistant cell lines is in agreement with
the finding that treatment with Ab5, a neutralizing antibody to
HER3, only resulted in a small growth inhibitory effect in two of
three tested fulvestrant-resistant cell lines (2). Ab5 exerted significant downregulation
of total and phosphorylated HER3, but had no effect on the level of
phosphorylated Akt or Erk. This may explain the lack of growth
inhibition. We presume that upon knock-down of HER3, other HER
receptors in the resistant cells may compensate for the lack of
HER3. EGFR has previously been shown to play a role for
fulvestrant-resistant cell growth (2,6,22),
but targeting EGFR alone with the kinase inhibitor gefitinib at 1
μM concentration had only a modest growth inhibition in the
order of 20–30%. This is again in agreement with our previous
finding that the EGFR neutralizing antibody cetuximab exerted only
a small inhibition of growth of the resistant cell lines (6). Similarly, targeting HER2 alone with
increasing concentrations of trastuzumab in fulvestrant-resistant
breast cancer cells had no effect on cell growth (25). Thus, targeting the HER receptors
individually had no inhibitory effect on resistant cell growth, in
contrast to targeting all HER receptors with the pan-HER inhibitor
CI-1033, which completely blocked growth and signaling via HER3 and
Erk in the resistant cells.
It has been shown that the effect of targeting the
HER receptors individually is compromised by increased expression
of HER ligands, which may circumvent the loss of the function of a
single receptor. For instance, the expression of the EGFR ligand
TGFα was increased upon trastuzumab treatment (16) and heregulin could partly bypass the
growth inhibitory effect of EGFR inhibitors (17). We therefore investigated the
importance of HER ligand shedding for signaling and growth of
breast cancer cells. In agreement with the lack of HER3
phosphorylation in MCF-7 cells, conditioned medium from MCF-7 cells
did not contain factors able to activate HER3 in MCF-7. In
contrast, HER3 and Akt were phosphorylated in MCF-7 cells treated
with conditioned medium from fulvestrant-resistant
164R-5 and 164R-7 cells, whereas conditioned
medium from antiestrogen resistant cells treated with TAPI-2, an
inhibitor of TACE and MMPs, showed no HER3 activation. This clearly
demonstrates that resistant cells produce HER3 activating factors,
which require cleavage by a metalloproteinase to be released from
the cells and supports that shedding of HER ligands could be
mediated by TACE as previously suggested (13).
When further investigating the effect of TAPI-2 as
well as another metalloproteinase inhibitor GM6001 on resistant
cell growth, we unexpectedly found that these metalloproteinase
inhibitors had no effect on growth of the fulvestrant-resistant
cell line neither on expression of activated HER3, Akt or Erk. The
maintained HER3 activation upon TAPI-2 treatment of resistant
cells, which was shown here to prevent shedding of HER3-activating
ligands, indicates that a juxtacrine mechanism may be responsible
for HER3 activation.
We have previously seen that the expression and
phosphorylation levels of EGFR is increased in our resistant cells
compared to MCF-7, but although increased, it is not possible to
measure phosphorylated EGFR by western blotting in either parental
or the resistant cell lines (2).
The fact that gefitinib and cetuxumab (unpublished data) strongly
inhibited Erk signaling in the resistant cells, supports that EGFR
signals via Erk and the lack of effect of TAPI-2 and GM6001 on
phosphorylation of Erk suggests that EGFR activation is not
inhibited by these metalloproteinase inhibitors. Instead, signaling
via EGFR, either as homodimer or heterodimer with HER2, appears to
be sufficient to maintain resistant cell growth even though HER3
activation is abrogated. In contrast to TAPI-2 and GM6001, the
broad-spectrum metalloproteinase inhibitor BB-94 exerted nearly
complete growth inhibition of the 164R-5 cells, about
50% reduction of growth of 164R-7 cells and only 20%
reduction of growth of parental cells. The growth inhibition was
concomitant with reduced expression of activated HER3 and Erk.
These findings support that BB-94, in contrast to TAPI-2 and
GM6001, blocks activation of EGFR. HER3 may still bind ligands by a
juxtacrine mechanism, but since EGFR is presumably not activated,
as indicated by reduced Erk phosphorylation, HER3 will not be able
to form heterodimers with EGFR, and this may explain the reduced
level of phosphorylated HER3 upon treatment with BB-94. Previously,
we found that the level of heregulin 2α and -2β is increased in
164R-5 and 164R-7 compared to the level in
MCF-7 and that TGFα, EGF, HB-EGF and betacellulin are expressed in
the resistant cells but at similar levels as in MCF-7 (2). These HER ligands have been shown to
be shed by TACE (TGFα, HB-EGF and heregulins) or ADAM10 (EGF,
betacellulin and HB-EGF) (13,26).
However, as neither TAPI-2 nor the broader metalloproteinase
inhibitor GM6001 reduced growth and signaling in the
fulvestrant-resistant cell lines, our data suggest that HER
receptor activation in fulvestrant-resistant cell lines can occur
independent of ligand shedding. Yet, the fact that the more
broad-spectrum metalloproteinase inhibitor BB-94 caused a marked
decrease in both resistant cell growth and levels of HER3 and Erk
phosphorylation strongly indicates some other metalloproteinase
implication not targeted by TAPI-2 and GM6001.
In this study, we investigated the importance of the
HER receptors and in particular HER3 as well as HER ligand shedding
for growth of our fulvestrant-resistant cell lines. Compared with
their parental MCF-7 cell lines, the resistant cells have severely
reduced HER4 expression, unchanged level of HER2, increased level
of EGFR, phosphorylated HER3, Erk and Akt and increased level of
heregulin 2α and -2β (2). We found
that neither HER3 nor EGFR alone was a key driver of growth of the
resistant cells. Inhibition of growth required concomitant
inhibition of HER3 activation and signaling via Erk and this was
achieved with the pan-HER inhibitor CI-1033. We also found that
targeting ligand shedding with a broad-spectrum metalloproteinase
inhibitor prevented growth and inhibited activation of HER3 and Erk
in the resistant cells. The less potent metalloproteinase inhibitor
TAPI-2, which blocked HER3 ligand shedding, did not prevent HER3
nor Erk activation, indicating that other mechanisms including
juxtacrine HER activation may occur. Thus, ligand shedding is
presumably not a target for treatment, and collectively, our
combined data underscore the complexity of the resistance
mechanisms and the requirement of targeting signaling from HER
receptors by multiple strategies.
Abbreviations:
|
ADAM
|
a disintegrin and
metalloproteinase
|
|
ER
|
estrogen receptor α
|
|
HER
|
human epidermal growth factor
receptor
|
|
MMP
|
matrix metalloproteinase
|
|
TACE
|
tumor necrosis factor α converting
enzyme
|
Acknowledgements
We thank Birgit Reiter, Jette Elm
Nielsen and Inger Heiberg for excellent technical assistance. The
study was supported by grants from Danish Cancer Society and the
Danish Medical Research Council (271-07-0704).
References
|
1.
|
Valachis A, Mauri D, Polyzos NP, Mavroudis
D, Georgoulias V and Casazza G: Fulvestrant in the treatment of
advanced breast cancer: a systematic review and meta-analysis of
randomized controlled trials. Crit Rev Oncol Hematol. 73:220–227.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Frogne T, Benjaminsen RV, Sonne-Hansen K,
et al: Activation of ErbB3, EGFR and Erk is essential for growth of
human breast cancer cell lines with acquired resistance to
fulvestrant. Breast Cancer Res Treat. 114:263–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
McClelland RA, Barrow D, Madden TA, et al:
Enhanced epidermal growth factor receptor signaling in MCF7 breast
cancer cells after long-term culture in the presence of the pure
antiestrogen ICI 182,780 (Faslodex). Endocrinology. 142:2776–2788.
2001.PubMed/NCBI
|
|
4.
|
Sommer A, Hoffmann J, Lichtner RB,
Schneider MR and Parczyk K: Studies on the development of
resistance to the pure antiestrogen Faslodex in three human breast
cancer cell lines. J Steroid Biochem Mol Biol. 85:33–47. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Massarweh S, Osborne CK, Creighton CJ, et
al: Tamoxifen resistance in breast tumors is driven by growth
factor receptor signaling with repression of classic estrogen
receptor genomic function. Cancer Res. 68:826–833. 2008. View Article : Google Scholar
|
|
6.
|
Sonne-Hansen K, Norrie IC, Emdal KB, et
al: Breast cancer cells can switch between estrogen receptor alpha
and ErbB signaling and combined treatment against both signaling
pathways postpones development of resistance. Breast Cancer Res
Treat. 121:601–613. 2010. View Article : Google Scholar
|
|
7.
|
Zahnow CA: ErbB receptors and their
ligands in the breast. Expert Rev Mol Med. 8:1–21. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Gullick WJ: c-erbB-4/HER4: friend or foe?
J Pathol. 200:279–281. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Witton CJ, Reeves JR, Going JJ, Cooke TG
and Bartlett JM: Expression of the HER1-4 family of receptor
tyrosine kinases in breast cancer. J Pathol. 200:290–297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tovey S, Dunne B, Witton CJ, Forsyth A,
Cooke TG and Bartlett JM: Can molecular markers predict when to
implement treatment with aromatase inhibitors in invasive breast
cancer? Clin Cancer Res. 11:4835–4842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Frogne T, Laenkholm AV, Lyng MB, Henriksen
KL and Lykkesfeldt AE: Determination of HER2 phosphorylation at
tyrosine 1221/1222 improves prediction of poor survival for breast
cancer patients with hormone receptor-positive tumors. Breast
Cancer Res. 11:R112009. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Blobel CP: ADAMs: key components in EGFR
signalling and development. Nat Rev Mol Cell Biol. 6:32–43. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Piccart-Gebhart MJ, Procter M,
Leyland-Jones B, et al: Trastuzumab after adjuvant chemotherapy in
HER2-positive breast cancer. N Engl J Med. 353:1659–1672. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Slamon DJ, Leyland-Jones B, Shak S, et al:
Use of chemotherapy plus a monoclonal antibody against HER2 for
metastatic breast cancer that overexpresses HER2. N Engl J Med.
344:783–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Valabrega G, Montemurro F, Sarotto I, et
al: TGFalpha expression impairs Trastuzumab-induced HER2
downregulation. Oncogene. 24:3002–3010. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Motoyama AB, Hynes NE and Lane HA: The
efficacy of ErbB receptor-targeted anticancer therapeutics is
influenced by the availability of epidermal growth factor-related
peptides. Cancer Res. 62:3151–8315. 2002.PubMed/NCBI
|
|
18.
|
Lykkesfeldt AE, Larsen SS and Briand P:
Human breast cancer cell lines resistant to pure anti-estrogens are
sensitive to tamoxifen treatment. Int J Cancer. 61:529–354. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Frogne T, Jepsen JS, Larsen SS, Fog CK,
Brockdorff BL and Lykkesfeldt AE: Antiestrogen-resistant human
breast cancer cells require activated protein kinase B/Akt for
growth. Endocr Relat Cancer. 12:599–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Lundholt BK, Briand P and Lykkesfeldt AE:
Growth inhibition and growth stimulation by estradiol of estrogen
receptor transfected human breast epithelial cell lines involve
different pathways. Breast Cancer Res Treat. 67:199–214. 2001.
View Article : Google Scholar
|
|
21.
|
Pancholi S, Lykkesfeldt A, Hilmi C, et al:
ERBB2 influences the subcellular localization of the estrogen
receptor in tamoxifen-resistant MCF-7 cells leading to the
activation of AKT and p90RSK. Endocr Relat Cancer. 15:985–1002.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Nicholson RI, Hutcheson IR, Harper ME, et
al: Modulation of epidermal growth factor receptor in
endocrine-resistant, oestrogen receptor-positive breast cancer.
Endocr Relat Cancer. 8:175–182. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Rasmussen BB, Regan MM, Lykkesfeldt AE, et
al: Adjuvant letrozole versus tamoxifen according to
centrally-assessed ERBB2 status for postmenopausal women with
endocrine-responsive early breast cancer: supplementary results
from the BIG 1–98 randomised trial. Lancet Oncol. 9:23–28.
2008.
|
|
24.
|
Schwartzberg LS, Franco SX, Florance A,
O'Rourke L, Maltzman J and Johnston S: Lapatinib plus letrozole as
first-line therapy for HER-2+ hormone receptor-positive
metastatic breast cancer. Oncologist. 15:122–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Larsen SS, Egeblad M, Jaattela M and
Lykkesfeldt AE: Acquired antiestrogen resistance in MCF-7 human
breast cancer sublines is not accomplished by altered expression of
receptors in the ErbB-family. Breast Cancer Res Treat. 58:41–56.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Sahin U, Weskamp G, Kelly K, et al:
Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six
EGFR ligands. J Cell Biol. 164:769–779. 2004. View Article : Google Scholar : PubMed/NCBI
|