Introduction
Gastric cancer (GC) is currently the fourth most
common malignancy and the second leading cause of cancer-related
deaths worldwide (1,2). It is estimated that more than 900,000
new cases were diagnosed and over 700,000 people died from GC in
2011 (3,4). Due to the absence of specific
symptoms, patients are usually not diagnosed until reaching an
advanced stage or metastasis (5).
Surgical resection is the most effective treatment modality for GC,
yet up to half of the patients experience recurrence after curative
surgery with a 5-year survival rate of only 20–30% (1,6).
Despite efforts of multiple fields, current interventional
strategies have shown little success in increasing the disease-free
survival rate of GC patients due to a limited understanding of the
disease causing mechanisms (6).
Therefore, it is imperative to conduct an in-depth study
characterizing GC pathogenesis to identify novel molecular makers
and therapeutic targets.
Protein-coding sequences cover only a small fraction
(~2%) of the human genome, with up to 70% of the genome transcribed
as ncRNAs (7,8). Over the past decade, much attention
has focused on miRNA, a class of short ncRNAs ranging in length
from 21–25 nucleotides, and their involvement in proliferation,
development, differentiation, apoptosis, tumorigenesis and other
biological processes (9–11). Recently, a large number of lncRNAs
have been found in mouse, fly and human systems (12). Unlike miRNAs, lncRNAs code
transcripts longer than 200 nucleotide, with mRNA-like properties
including 5′ capping, splicing and polyadenylation, yet lack a
significant ORF (8,13). LncRNAs can regulate gene expression
through cis- or trans-acting pathways (14), and have been shown to play
important roles in a broad range of biological processes such as
genomic imprinting, nuclear-cytoplasmic trafficking, dosage
compensation, immune responses, developmental patterning and
differentiation (15–17). Although thousands of lncRNAs have
been identified, the exact function of most lncRNAs remains unknown
requiring characterization to reveal the mechanisms by which they
regulate biological processes (12,18).
However, unlike protein-coding genes whose sequence motifs are
usually indicative of their function, the sequences of most lncRNAs
are poorly conserved rendering a direct functionality prediction
difficult (14). Moreover,
evidence indicates that dysregulation of lncRNAs is associated with
a wide range of human diseases, possibly through the regulation or
interactions of lncRNAs with protein-coding genes (14,18,19).
These interactions could ultimately contribute to tumor
pathogenesis, making an understanding of these molecular mechanisms
imperative. Recently, Cao et al (20) identified a set of differentially
expressed lncRNAs in GC by analyzing publicly available human exon
array data sets from the GEO, yet the mechanistic properties of
this dysregulation remains unknown. In addition, integrated
analysis correlating changes in the expression patterns of lncRNAs
and mRNAs, as it relates to GC pathogenesis, requires
examination.
In this study, we analyzed lncRNA and mRNA
expression profiles in samples of GC tissues, in conjunction with
paired adjacent normal tissues, utilizing microarray technology.
Differentially expressed lncRNAs were subjected to bioinformatic
analysis for target gene prediction and findings integrated with
differentially expressed mRNA data to increase predictive accuracy.
GO and pathway analysis was performed to determine associated
functions of the predicted targets, ultimately leading to the
construction of a lncRNA-mRNA correlation network. Our results
demonstrate that the altered expression levels of lncRNAs and mRNA
may have implications in GC pathogenesis, and that integrative
analysis of lncRNAs and mRNA may provide a new foundation for
diagnosing and treating GC.
Materials and methods
Patient samples and ethics statement
Eight primary gastric adenocarcinoma samples with
paired adjacent normal tissue (≥5 cm away from the tumor margin)
samples were collected at Shenzhen People’s Hospital (Shenzhen,
China) following gastrectomy or partial gastrectomy. The diagnosis
of gastric adenocarcinoma was determined by histopathological
examination for the exclusion of precancer lesions such as gastric
epithelial dysplasia and intestinal metaplasia. The patient donors
did not received chemotherapy or radiotherapy prior to surgical
resection and the participant rights were explained prior to
consent form signing. All the clinical characteristics of the eight
patients with gastric cancer are shown in Table I. This investigation was carried
out according to the Helsinki Declaration guidelines on ethical
principles for medical research involving human subjects and
approved by the ethics committee of the Shenzhen People’s Hospital.
Written informed consent was obtained from all participants. All
tissue samples were snap-frozen and stored in liquid nitrogen until
needed for further testing.
 | Table IClinical characteristics of the eight
patients with gastric cancer. |
Table I
Clinical characteristics of the eight
patients with gastric cancer.
| Characteristic | Gastric cancer
patients |
|---|
| Age (mean ± SD),
years | 58.75±11.80 |
| Sex (no.) |
| Male | 6 |
| Female | 2 |
| Tumor location |
| Upper third | 2 |
| Middle third | 1 |
| Lower third | 5 |
| Histological type
(no.) |
|
Adenocarcinoma | 8 |
| Differentiation of
cancer tissue |
| High
differentiation | 1 |
| Moderate
differentiation | 5 |
| Poor
differentiation | 2 |
| Lauren
classification |
| Intestinal
type | 6 |
| Diffuse type | 2 |
| TNM stage |
| Stage I | 1 |
| Stage II | 3 |
| Stage III | 4 |
| Stage IV | 0 |
RNA extraction
Frozen tissue samples were homogenized by an
electric tissue homogenizer, total RNA extracted using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) and a RNeasy kit (Qiagen,
Hilden, Germany) according to the manufacturer’s protocol, to
include a DNase digestion step. RNA quantification and purity was
assessed using a NanoDrop ND-1000 spectrophotometer measuring
absorbance ratios of A260/A280 and
A260/A230, with RNA integrity evaluated by
standard denaturing agarose gel electrophoresis.
Analysis of lncRNA and mRNA expression
profiles
lncRNA and mRNA microarray. The Human 12x135k lncRNA
expression microarray (Roche NimbleGen, Inc.) provide global
profiling of long transcripts, containing 22,192 lncRNAs and 20,447
protein-coding mRNAs in human genome, each transcript is accurately
identified by specific exon or splice junction probes. To improve
statistical confidence, each transcript was targeted with 1–5
strategic probes, with ncRNAs shorter than 200 bp or highly similar
sequences excluded. To ensure hybridization quality, negative
probes and probes for reference genes were printed multiple times.
Human lncRNAs and mRNAs were collected from databases such as UCSC,
RNAdb, NCBI RefSeq, UCRs and related microarray detection
literature, with both represented on a single array to provide
consistent hybridization.
RNA labeling and array hybridization
After completing the RNA quality assessment, 5 μg of
total RNA served as a template to synthesize ds-cDNA using a cDNA
synthesis kit (Superscript Double-Stranded cDNA Synthesis kit,
Invitrogen). After incubation with 4 μg RNase A for 10 min at 37°C,
ds-cDNA was cleaned and labeled with Cy3 overnight using a
One-Color DNA labeling kit (Roche NimbleGen, Inc.) per the
manufacturer’s protocol. Cy3 labeled ds-cDNA in hybridization
buffer (NimbleGen hybridization component A) was hybridized for
16–20 h at 42°C in a hybridization chamber (Hybridization
System-NimbleGen Systems, Inc., Madison, WI, USA). Following
hybridization, slides were washed and scanned on a GenePix 4000B
microarray scanner (Axon Instruments, Union City, CA, USA).
Data analysis
Scanned microarray image files (TIFF format) were
analyzed using NimbleScan software (version 2.5, Roche NimbleGen,
Inc.) to extract raw data as pair files for grid alignment and
normalization. Spot intensities were obtained by subtracting local
background intensity from the total intensity, and intensities were
normalization via quantile normalization and Robust Multichip
Average (RMA) (NimbleScan software). The probe level
(*_norm_RMA.pair) files and mRNA level
(*_RMA.calls) files produced following normalization
were further analyzed via Agilent GeneSpring Software (version
11.0, Agilent) for further normalization adjustments, with
differentially expressed lncRNAs and mRNAs exhibiting a fold change
≥2-fold.
Quantitative PCR
Following total RNA extraction, samples were reverse
transcribed to generate cDNA with appropriate primers. qRT-PCR was
carried out in triplicate with a total reaction volume of 10 μl (5
μl of 2X RT2 real-time SYBR Green PCR Master Mix
(SuperArray Bioscience, Frederick, MD, USA), 0.5 μl of PCR forward
primer (10 μM), 0.5 μl of PCR reverse primer (10 μM), 2 μl of
double-distilled water and 2 μl of cDNA) and incubated (95°C for 10
min, then 40 cycles at 95°C for 10 sec, 60°C for 60 sec). After PCR
amplification, melt curve analysis was performed to confirm
reaction specificity; with Human U6 snRNA (for lncRNAs) and 18S
rRNA (for mRNA) used as internal controls. Expression fold changes
were calculated via the 2−ΔΔCt method (21). Differences in gene expression
levels between GC samples and their paired adjacent tissue sample
were analyzed using Student’s t-test, with a p-value <0.05
considered statistically significant. All primers used in qRT-PCR
and cDNA synthesis are shown in Tables II and III.
| Table IIPrimers used for cDNA synthesis and
real-time quantitative PCR of lncRNAs. |
Table II
Primers used for cDNA synthesis and
real-time quantitative PCR of lncRNAs.
| lncRNAs | Sense primer for
qRT-PCR (5′-3′) | Antisense primer
for qRT-PCR and cDNA synthesis (5′-3′) | Product (bp) |
|---|
| uc003iqu |
AGTCCCGAATCCCAAGACACT |
AAGCTCCATGAACCACCACC | 122 |
| uc003tfx |
TCAAGCAATTCTCCTGCCTCA |
CACGCCTGTAATCCTAGCACTTT | 189 |
| AK022971 |
ATCCCGATTGTTCCCTTTAGTC |
CTTTGGTACATGCACGGTTTCT | 250 |
| uc.341 |
ACAGCAAAGAGCGGAAGGAA |
TCGCGTCAAATACATATTGAACAG | 96 |
| HIV1230 |
TTCTTCCCTTTCTACTCACCTTTG |
TTCCACCTTCTGCCCTACTTG | 303 |
| BC011663 |
AGGTCTGCGTCTGGGAAGG |
AGCTCGGCGAAGAGGTGA | 143 |
| AK057054 |
CTGTGCTGGCTCCTCTACCTG |
TGGGACCTCCTGCCTCTACTT | 99 |
| M14574 |
CTGGGCATGTGGAGACAGAG |
CAGCCTAAGGGTGGGAAAAT | 89 |
| U6 snRNA |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT | 94 |
| Table IIIPrimers used for real-time
quantitative PCR of mRNAs. |
Table III
Primers used for real-time
quantitative PCR of mRNAs.
| mRNAs | Sense primer
(5′-3′) | Antisense primer
(5′-3′) | Product (bp) |
|---|
| CCAR1 |
TGGATGGACCAGACCCAGAA |
CAGGGCGATGGTAGCGAAT | 138 |
| HOXC10 |
GAACATCTGGAATCGCCTCA |
TCTCCTCTTTCGCTTCGTTATC | 205 |
| RRM2 |
GAGAGTAGGCGAGTATCAGAGGA |
CAGCCAAGTAAGGGCACATC | 109 |
| DSCR1 |
GGACCTGCTCCTTCCTGCTT |
GGTTTTCCTTCGATGGCAAA | 249 |
| ALAS1 |
AAACAGCCGAGTGCCAAAGTA |
GGCATGACTCCATCCCGAT | 261 |
| NPTXR |
CCTCGCTTTGGTCATTTGCT |
TGAACCTTGCCCTGGACTCT | 184 |
| RBMS2 |
TCTCCACCCGTATCCTTCG |
CATGACGCCCATGTCTGC | 257 |
| TRIM74 |
TCCACTGGTTCCTCCATTCA |
AGGCTGGTCTCGAACTCCC | 281 |
| 18srRNA |
CCTGGATACCGCAGCTAGGA |
GCGGCGCAATACGAATGCCCC | 112 |
Association analysis of the different
expression of lncRNAs and mRNAs
Target genes prediction for different
expression lncRNAs
To unveil potential associations of lncRNAs with
mRNAs, differentially expressed lncRNAs (fold change, ≥3.0) were
subjected to bioinformatic analysis for target gene prediction. Two
independent algorithms were applied to predict possible target
genes for both cis- or trans-acting lncRNAs independently.
In the cis-acting analysis, lncRNAs and their
potential paired target genes were obtained and visualized using
the UCSC genome browser and genome annotation. The algorithms
included an ORF-Predictor and BLASTP pipeline (e<1E-5) to
identify lncRNA genes in the human genome and searched for their
nearest known protein-coding gene according to distance
stratification. The genes located within 10 and 10–300 kb away from
lncRNAs were considered as cis10k and cis300k target genes,
respectively (22,23). Generally, an equidirectional
transcriptive target gene is for promoting expression in the
promoter region, otherwise it is for suppression. In some
conditions, a reversed direction is possible to promote expression
in the 3′-UTR region. The predictive algorithms for the cis-acting
analysis were performed as previously described (24–26).
For the trans-acting analysis, a RNAplex program
(http://www.bioinf.uni-leipzig.de/Software/RNAplex/)
was used to identify possible target genes for lncRNAs. RNAplex was
especially designed to quickly localize possible hybridization
sites for a query RNA in large RNA databases by applying a simpler
energy model in the first screening phase followed by a full energy
model to refine potential binding sites (27,28).
First, we performed BLAST (e<1E-5) between lncRNA and the known
protein-coding genes followed by RNAplex software to further
screening the trans-acting target genes (parameters were set as -e
-20). The RNAplex program for trans-acting analysis was performed
as previously described (22,25–28).
Combination of differentially expressed
mRNAs with target prediction
To increase the accuracy of target prediction,
differentially expressed mRNAs (fold change, ≥3.0) were integrated
with the predicted potential of lncRNA targets.
Gene Ontology (GO) analysis and pathway
analysis
GO (www.geneontology.org) is a functional analysis
utilizing GO categories to describe genes and gene product
attributes including molecular function, cellular components and
biological processes. To understand the potential roles of
differentially expressed lncRNAs, we applied GO categories to
analyze the biological functions for the correlated lncRNAs/gene
targets. Additionally, we used the KEGG database (http://www.genome.jp/kegg/) to identify significant
pathways for predicted target genes. GO term enrichment and the
biological pathways utilized significant p-values (recommended
p-value <0.05) relating to the target genes of differentially
expressed lncRNAs.
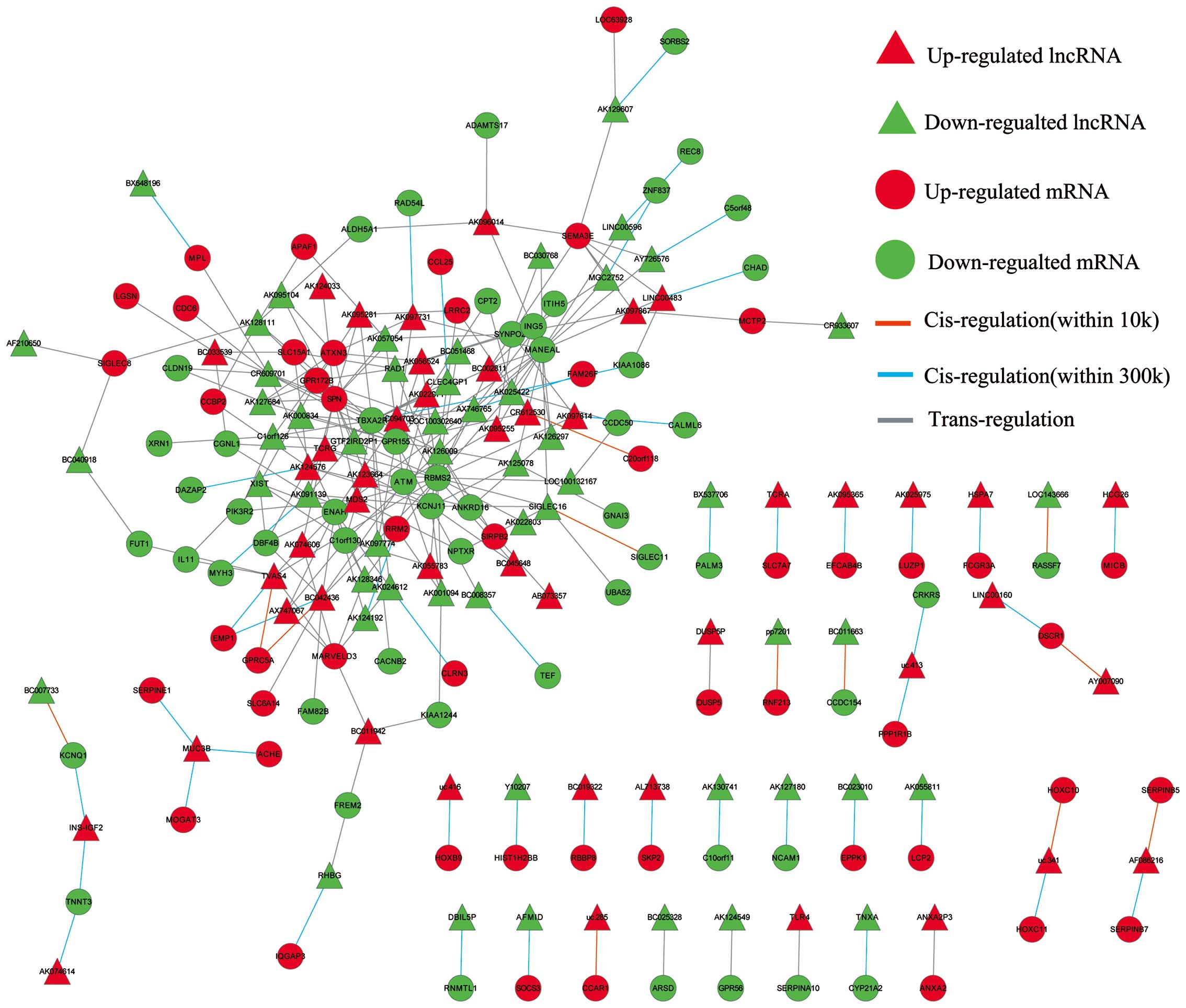
LncRNA-mRNA correlation network
Superimposing lncRNA target predictions with
differentially expressed lncRNAs and mRNA profiles in Cytoscape
(http://www.cytoscape.org) enabled the
construction of a lncRNA-mRNA correlation network. As shown in
Fig. 5, in this network, circular
nodes represent mRNA, triangular nodes represent lncRNA, while red
nodes represent an upregulation and green nodes a downregulation
relative to matched normal tissue. Direct connections were drawn
between lncRNAs and mRNAs using either an orange line
(cis-interaction within 10 kb), sapphire line (cis-interaction
within 10–300 kb) or grey line (trans-interaction).
Statistical analysis
Statistical analyses were accomplished using SPSS
software (version 13.0, SPSS Inc.), with all data expressed as
means ± standard deviation (SD). The Student’s t-test was used for
evaluating the statistical significance of difference in the means
between groups with a stringency of p<0.05.
Results
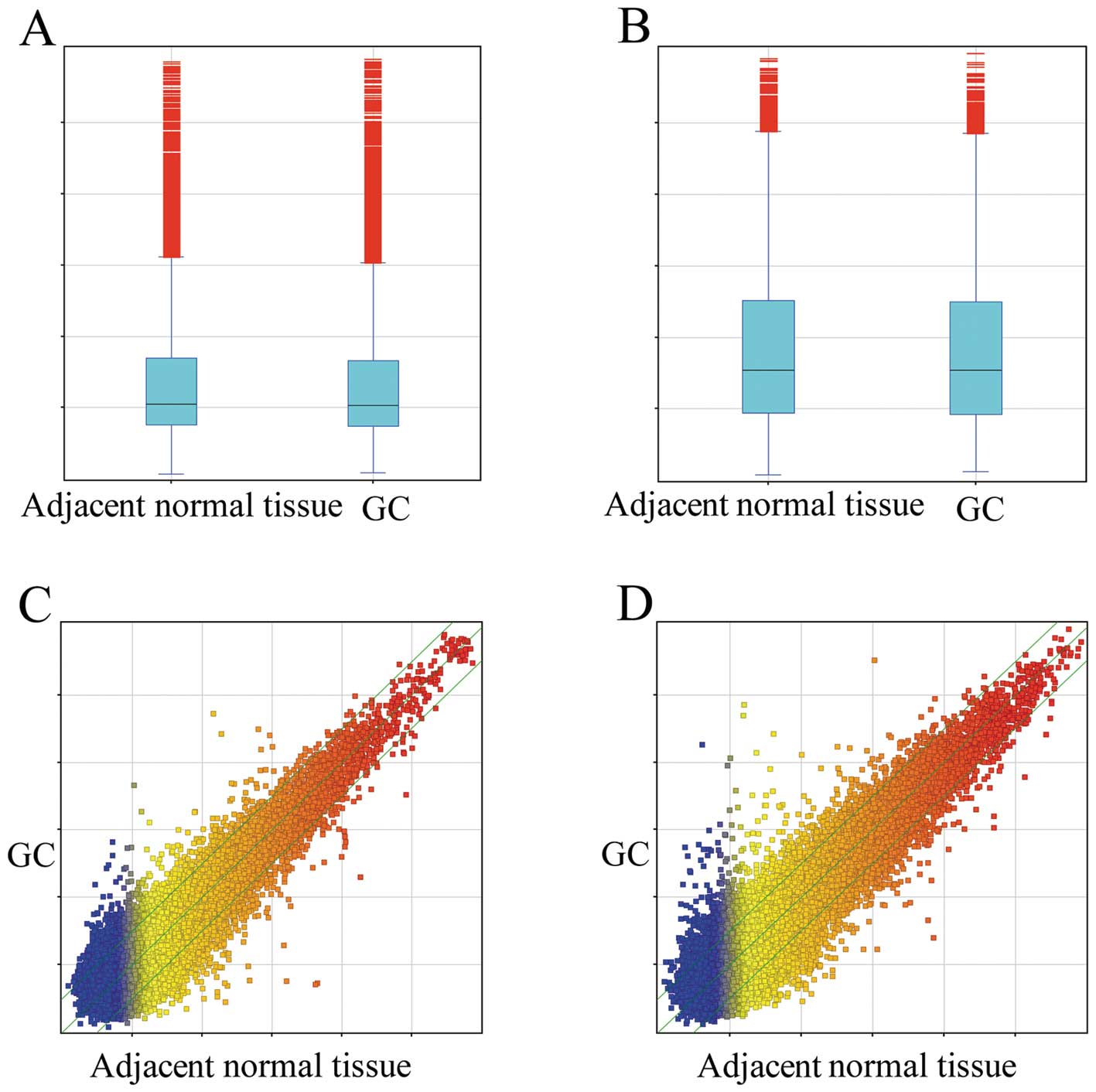
Aberrant lncRNA and mRNA expression in
GC
To investigate the possible biological functions of
lncRNAs in GC, we analyzed lncRNA and mRNA expression profiles in
GC samples and their matched adjacent normal tissue via microarray
technology. Among the 22,192 lncRNA transcripts examined, we found
2,621 (11.8%) lncRNAs were differentially expressed (fold change,
≥2.0) in GC samples relative to their matched counterparts
(Fig. 1A and C), with 1,215 being
upregulated, while 1,406 were downregulated. Uc010lho (fold change,
39.068) was the most significantly upregulated lncRNA, while
HIV1213 (fold change, 56.240) was the most significantly
downregulated lncRNA.
The mRNA expression profiling data showed 15.3%
(3,121/20,447) of mRNAs to be differentially expressed (fold
change, ≥2.0) in GC samples relative to their matched counterparts
(Fig. 1B and D), with 1,523
upregulated, while 1,598 were downregulated. Among these mRNAs,
MUC2 (fold change, 162.24048) showed the highest degree of
upregulation, while WDR20 (fold change, 29.62003) was the most
downregulated protein-coding gene.
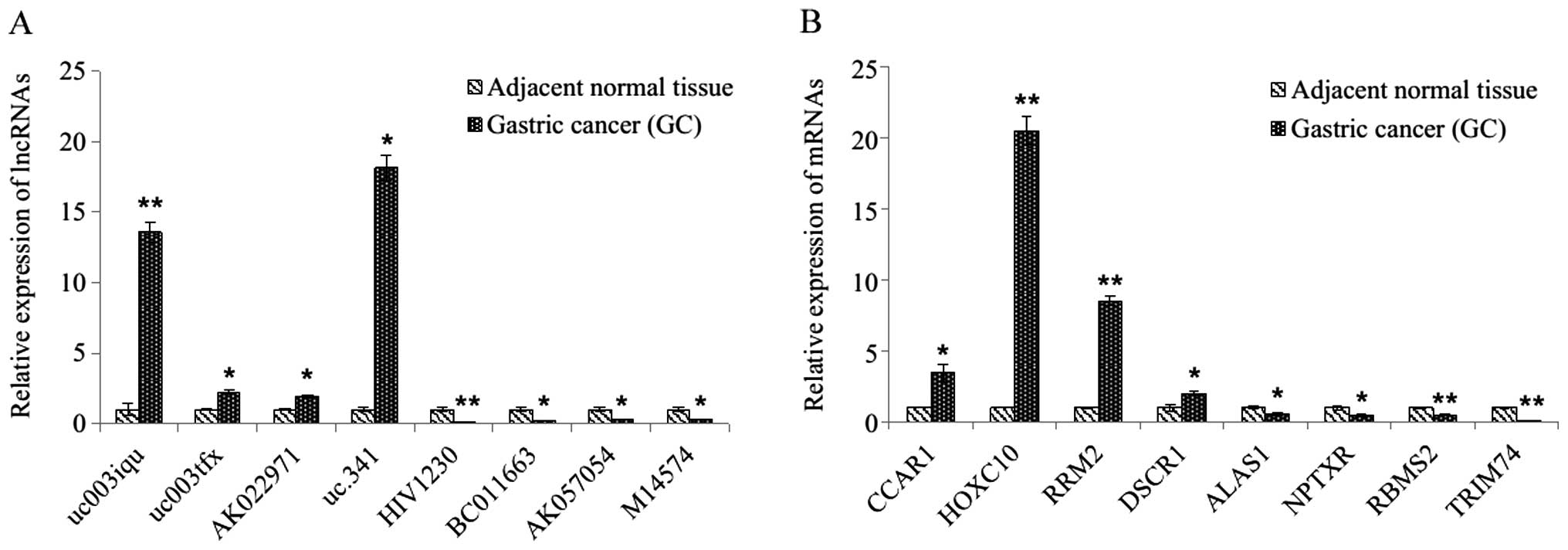
Real-time quantitative PCR
validation
To validate the microarray results, 8 differentially
expressed lncRNAs and 8 differentially expressed mRNAs were
randomly selected and analyzed via qRT-PCR. For the lncRNAs, the
results demonstrated that uc003iqu, uc003tfx, AK022971 and uc.341
were upregulated and that HIV1230, BC011663, AK057054 and M14574
were downregulated in the GC tissues relative to their matched
counterparts (all p<0.05, Fig.
2A). For the mRNAs, the expression of CCAR1, HOXC10, RRM2,
DSCR1, ALAS1, NPTXR, RBMS2 and TRIM74 showed statistically
significant differences between matched samples (p<0.05 for each
mRNAs, Fig. 2B), with these
results being consistent with the microarray data.
Integrating differentially expressed
mRNAs into the predicted targets of differentially expressed
lncRNAs
Increasing evidence has confirmed that lncRNAs serve
an important role by regulating the expression of protein-coding
genes. Therefore, we applied bioinformatic analysis to aid in
target gene prediction to explore the potential correlation between
lncRNA and mRNA expression profiles. First, we used bioinformatic
algorithms to predict possible target genes for lncRNAs (fold
change, ≥3.0). Second, differentially expressed mRNAs (fold change,
≥3.0) were integrated with predicted lncRNA targets. The result
showed that 60 differentially expressed protein-coding genes were
significantly associated with each of the 75 differentially
expressed lncRNAs to generate 221 lncRNA-mRNA target pairs. Among
them, 119 pairs were differentially expressed unidirectionally (up
or down), and 102 pairs were differentially expressed
bidirectionally. Within our data sets, we found that 3.6% (8/221)
of lncRNA-mRNA target pairs interacted in a cis-manner, while 96.4%
(213/221) pairs interacted in a trans-manner.
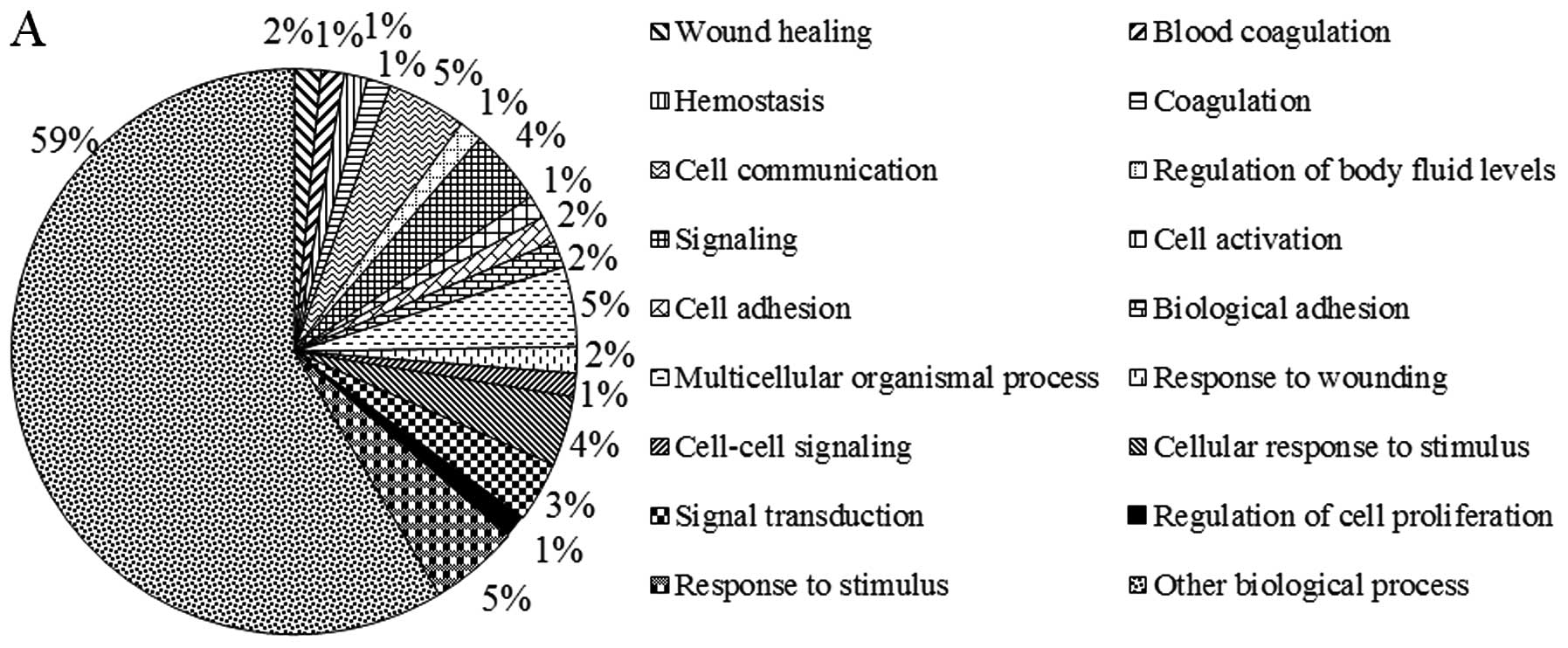
Ontological and pathway analysis of
target genes for differentially expressed lncRNAs
To investigate whether the differentially expressed
lncRNAs correlate with functional groupings, GO categories were
used to analyze biological process, cellular components and
molecular functioning for the correlated targets of specific
lncRNAs (Fig. 3). In the GO
biological processes classification, most of the lncRNA targets
were involved in response to stimuli, cell communication and
multicellular organismal processes. The majority of target genes
related to cellular components were located in membrane components,
the plasma membrane and cell periphery. The GO molecular function
classification showed a large proportion of target genes associated
with enzyme regulatory activity, peptidase regulatory activity and
enzyme inhibitor activity. Additionally, these target genes were
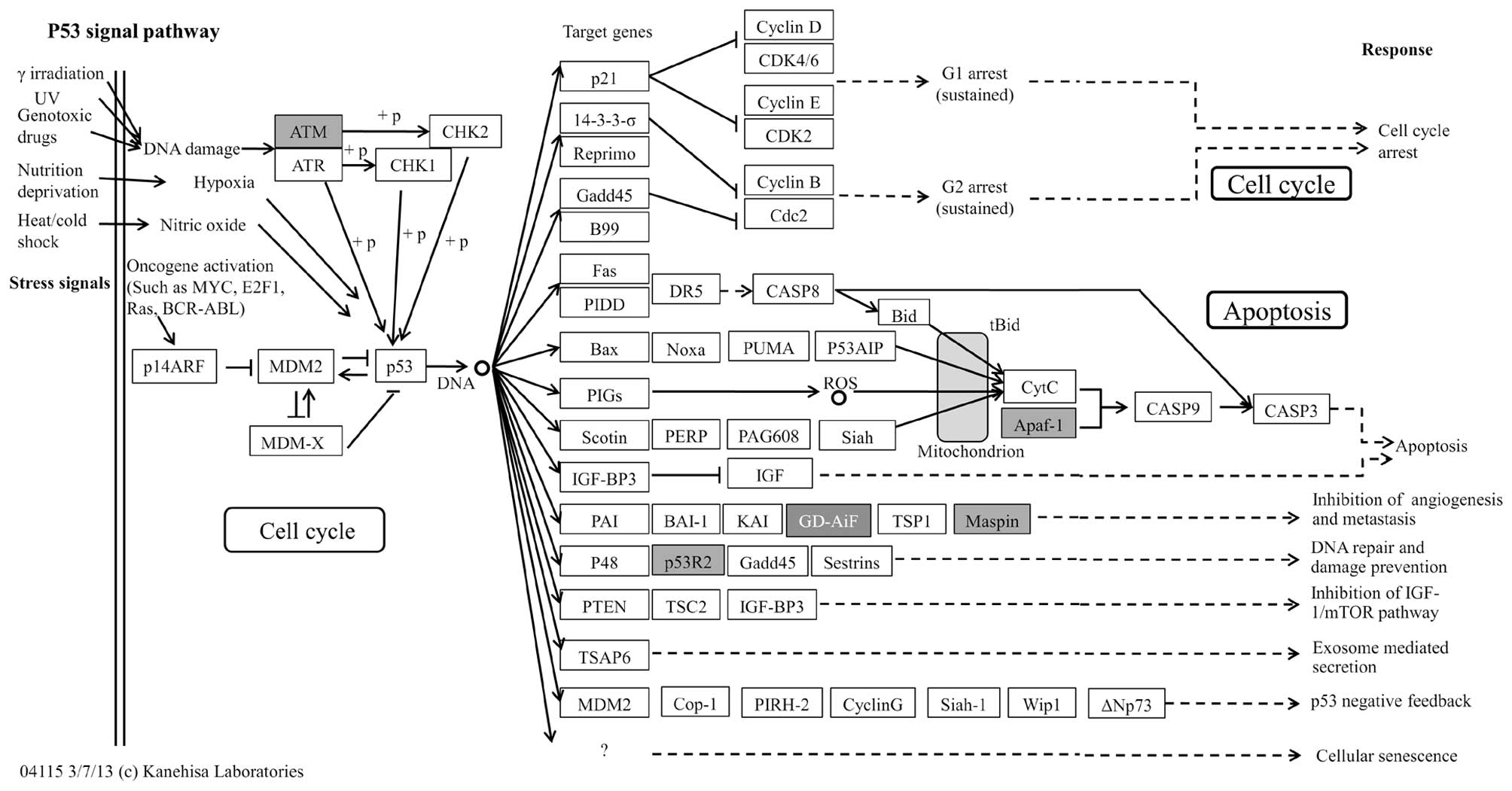
significantly enriched in 7 different pathways (Table IV), of which the p53 pathway
(29,30) was the most significant (Table IV and Fig. 4) followed by apoptosis (31) and Jak-STAT signaling pathways
(32,33) which have all been previously
implicated in GC.
| Table IVTarget gene-related pathways. |
Table IV
Target gene-related pathways.
| Pathway ID | Definition | Fisher-p-value | Selection
counts |
Enrichment_Score | Genes |
|---|
| hsa04115 | p53 signaling
pathway - Homo sapiens | 0.000140843 | 4 | 3.851266 |
APAF1//ATM//RRM2//SERPINB5 |
| hsa04210 | Apoptosis - Homo
sapiens | 0.005059445 | 3 | 2.295897 |
APAF1//ATM//PIK3R2 |
| hsa04670 | Leukocyte
transendothelial migration - Homo sapiens | 0.01136341 | 3 | 1.944491 |
CLDN19//GNAI3//PIK3R2 |
| hsa04930 | Type II diabetes
mellitus - Homo sapiens | 0.01567604 | 2 | 1.804764 | KCNJ11//PIK3R2 |
| hsa04630 | Jak-STAT signaling
pathway - Homo sapiens | 0.02429151 | 3 | 1.614545 |
IL11//MPL//PIK3R2 |
| hsa04914 |
Progesterone-mediated oocyte maturation -
Homo sapiens | 0.04628376 | 2 | 1.334571 | GNAI3//PIK3R2 |
| hsa05222 | Small cell lung
cancer - Homo sapiens | 0.04628376 | 2 | 1.334571 | APAF1//PIK3R2 |
Construction of lncRNA-mRNA correlation
network
After merging lncRNA target predictions with lncRNA
and mRNA expression profiles, the lncRNA-mRNA correlation network
was constructed to include differentially expressed lncRNAs and
their target genes (Fig. 5). The
correlation network contained 135 nodes and 221 connections between
the 75 lncRNAs and the 60 mRNAs. Further analysis of this network
indicated that ataxia telangiectasia mutated (ATM) was a predicted
target of six lncRNAs (AK001094, AK057054, BC042436, uc003iqu.1,
AK022971 and uc003tfx.1), ribonucleotide reductase M2 polypeptide
(RRM2) a predicted target of three lncRNAs (AK091139, AK001094 and
uc003dwf.3) and serpin peptidase inhibitor, clade B (ovalbumin),
member 5 (SERPINB5) a predicted target of AF086216. These target
genes participate in the p53 signaling pathway, and have been
previously implicated in the development of GC (34–36).
Furthermore, the result also showed that one lncRNA could target up
to 5 coding genes and that one coding gene was predicted to be a
target of up to 22 lncRNAs in this network.
Discussion
Gastric cancer is one of the most common and lethal
malignancies in humans and comprises 8% of all cancer cases and 10%
of cancer mortalities worldwide (3). Despite extensive efforts to elucidate
the genetic and epigenetic mechanisms involved in disease
development and progression, the pathogenesis of GC remains poorly
understood to date (37).
Numerous studies in recent decades have demonstrated
the importance of lncRNA in cancer pathogenesis, with aberrant
expression frequently linked to different kinds of human cancers.
Certain lncRNAs, behave like oncogenes or tumor-suppressors
displaying an important function in cancer initiation, progression,
metastasis and recurrence (38,39).
lncRNAs appear to serve a regulatory function by controlling gene
expression, making the understanding of their dysregulation and how
it relates to cancer pathology essential (19). Hence, constructing lncRNA
expression patterns in cancer tissues and isolating potential
target molecules is required to enhance cancer diagnostics and
therapeutics (38). Recently, a
study indicated that the lncRNA H19 was upregulated in GC
cells/tissues and that partial inactivation of p53 was linked to
the H19 promotion of cellular proliferation and inhibition of
apoptosis to display oncogenic functioning (40). However, the functional properties
of the vast majority of lncRNAs relating to GC pathogenesis are
still unknown. Therefore, we analyzed lncRNA and mRNA expression
profiles in GC tissue samples relative to matched normal samples to
reveal the potential role of lncRNAs during pathogenesis (Fig. 1). Microarray data revealed 2621
lncRNAs and 3121 mRNAs to be significantly differentially expressed
(fold change, ≥2.0), with a subset of these findings corroborated
via qRT-PCR (Fig. 2).
lncRNAs are gaining recognition as important
functional components in eukaryotic gene regulation, with cis- and
trans-regulatory mechanisms becoming more clearly characterized
(14,41,42).
In general, lncRNAs can remodel the chromatin status of surrounding
regions in a cis-fashion via co-transcription (competes for the
transcription-binding complex between lncRNAs and their neighboring
genes) by anchoring to RNA polymerase II or act as an artificial
miRNA sponge to competitively inhibit the ability of miRNAs to bind
their mRNA targets (43,44). In addition, cis-regulatory lncRNAs
may also regulate the transcription of nearby genes by facilitating
access to enhancers and promoters for transcriptional machinery
molecules (45). One classic
cis-regulatory example involving two recently discovered lncRNAs,
HOTTIP and HOTAIRM1, located at opposite ends of the HoxA cluster
and help to promote the expression of neighboring HoxA genes
(46).
Unlike cis-regulatory lncRNAs that act locally on
the genomic region from which they are transcribed,
trans-regulatory lncRNAs exert their effects by regulating the
expression of target genes at geographically distant locations,
even including different chromosomes (42,46).
For example, HOTAIR is a 2.2 kb lncRNA that originates from the
HOXC locus, but recruits Polycomb Repressive Complex 2 (PRC2) to
silence the HOXD locus of a different chromosome (42). For the trans-acting analysis,
possible target genes of differentially expressed lncRNAs were
identified using the RNAplex program which finds the optimal target
sites of a query RNA relative to an mRNA target by computing
secondary structures for their hybridization. The ability of
RNAplex to afford a high degree of run-time efficiency without
noticeable loss of specificity by performing a comparative target
search allows poorly conserved interactions to be discarded.
Subsequently, RNAplex is a powerful tool for the rapid and reliable
prediction of RNA-RNA interactions, which is well suited for
finding high-confidence ncRNA targets amongst a large genomic
dataset (27,28).
To increase the accuracy of target prediction,
differentially expressed mRNA data were integrated with predicted
lncRNA targets. To understand the potential functional roles of
lncRNAs, GO category and KEGG pathway annotation were utilized to
analyze the target gene pool. KEGG annotation showed the predicted
target genes to be significantly enriched in 7 different pathways
in GC tissue compared with matched normal gastric tissue. Among
these pathways, the p53 signaling pathway (Fig. 4) was the most significant and has
been previously implicated in GC pathogenesis (29,30).
Furthermore, construction of the lncRNA-mRNA correlation network
displaying differentially expressed lncRNAs and their target genes
(Fig. 5) implicated three target
genes ATM, RRM2 and SERPINB5 which have been previously implicated
in GC development and found to have involvement in p53 signaling in
the present study (34–36). These findings indicate a possible
role of the p53 signaling pathway in the dysregulation of lncRNAs
during GC pathogenesis. Moreover, we postulate that the 10
differentially expressed lncRNAs (AK001094, AK057054, BC042436,
uc003iqu.1, AK022971, uc003tfx.1, AK091139, AK001094, uc003dwf.3
and AF086216) and their predicted targets (ATM, RRM2 and SERPINB5)
relate to the p53 signaling pathway and may play a significant
collective role in GC pathogenesis.
In conclusion, this is the first study that
describes the global expression profiling of lncRNAs and mRNAs
relating to GC using microarray technology. In this study, we
observed a large number of aberrantly expressed lncRNAs and mRNAs
in GC samples when compared matched normal samples. Bioinformatic
analysis to include lncRNA target prediction, GO category
classification and KEGG pathway annotation enabled the uncovering
of possible associations between lncRNAs and protein-coding genes
to reveal potential functional roles of lncRNAs in GC pathogenesis.
While the regulatory roles of several lncRNAs related to p53
signaling, the exact regulatory mechanisms still require further
elucidation. Additionally, it is worth noting that each lncRNA-mRNA
target pair serves as a strong candidate for GC diagnosis and
therapeutics warranting further investigation.
Acknowledgements
This study was supported by the Science and
Technology Planning Project of Shenzhen (201102176). We are deeply
grateful to all donors who participated in this program.
Abbreviations:
|
lncRNAs
|
long non-coding RNAs
|
|
GC
|
gastric cancer
|
|
GO
|
Gene Ontology
|
|
ORF
|
open reading frames
|
|
GEO
|
Gene Expression Omnibus
|
|
KEGG pathway
|
Kyoto encydopedia of gene and
genomes
|
|
ds-cDNA
|
double-stranded cDNA
|
|
ncRNAs
|
non-coding RNAs
|
|
miRNA
|
microRNA
|
|
JAK-STAT signaling pathway
|
Janus kinase and signal transducer and
activator of transcription signaling pathway
|
References
|
1
|
Thiel A and Ristimäki A: Gastric cancer:
basic aspects. Helicobacter. 1:26–29. 2012. View Article : Google Scholar
|
|
2
|
Leja M, Wex T and Malfertheiner P: Markers
for gastric cancer premalignant lesions: where do we go? Digest
Dis. 30:268–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guggenheim DE and Shah MA: Gastric cancer
epidemiology and risk factors. J Surg Oncol. 107:230–236. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
5
|
Wagner AD, Grothe W, Haerting J, et al:
Chemotherapy in advanced gastric cancer: a systematic review and
meta-analysis based on aggregate data. J Clin Oncol. 24:2903–2909.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang J, Wang Q, Liu H, et al: MicroRNA
expression and its implication for the diagnosis and therapeutic
strategies of gastric cancer. Cancer Lett. 297:137–143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:38–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gutschner T and Diederichs S: The
hallmarks of cancer: a long non-coding RNA point of view. RNA Biol.
9:703–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Griffiths JS, Saini HK, van Dongen S, et
al: miRBase: tools for microRNA genomics. Nucleic Acids Res.
36:D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Storz G, Opdyke JA and Zhang A:
Controlling mRNA stability and translation with small, noncoding
RNAs. Curr Opin Microbiol. 7:140–144. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morey C and Avner P: Employment
opportunities for non-coding RNAs. FEBS Lett. 567:27–34. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao Q, Liu C, Yuan X, et al: Large-scale
prediction of long non-coding RNA functions in a coding-non-coding
gene co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Au PC, Zhu QH, Dennis ES, et al: Long
non-coding RNA-mediated mechanisms independent of the RNAi pathway
in animals and plants. RNA Biol. 8:404–414. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma H, Hao Y, Dong X, et al: Molecular
mechanisms and function prediction of long noncoding RNA. Sci World
J. 2012:5417862012.PubMed/NCBI
|
|
15
|
Sui W, Yan Q, Li H, et al: Genome-wide
analysis of long noncoding RNA expression in peripheral blood
mononuclear cells of uremia patients. J Nephrol. 26:731–738. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin Z, Guan D, Fan Q, et al: lncRNA
expression signatures in response to enterovirus 71 infection.
Biochem Biophys Res Commun. 430:629–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu G, Yao W, Wang J, et al: LncRNAs
expression signatures of renal clear cell carcinoma revealed by
microarray. PLoS One. 7:e423772012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen G, Wang Z, Wang D, et al:
LncRNADisease: a database for long-non-coding RNA-associated
diseases. Nucleic Acids Res. 41:D983–D986. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harries LW: Long non-coding RNAs and human
disease. Biochem Soc. 40:902–906. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao WJ, Wu HL, He BS, et al: Analysis of
long non-coding RNA expression profiles in gastric cancer. World J
Gastroenterol. 19:3658–3664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:452001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sui W, Lin H, Peng W, et al: Molecular
dysfunctions in acute rejection after renal transplantation
revealed by integrated analysis of transcription factor, microRNA
and long noncoding RNA. Genomics. 102:310–322. 2013. View Article : Google Scholar
|
|
23
|
Szafranski P, Dharmadhikari AV, Brosens E,
et al: Small noncoding differentially methylated copy-number
variants, including lncRNA genes, cause a lethal lung developmental
disorder. Genome Res. 23:23–33. 2013. View Article : Google Scholar
|
|
24
|
Jia H, Osak M, Bogu GK, et al: Genome-wide
computational identification and manual annotation of human long
noncoding RNA genes. RNA. 16:1478–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han L, Zhang K, Shi Z, et al: LncRNA
profile of glioblastoma reveals the potential role of lncRNAs in
contributing to glioblastoma pathogenesis. Int J Oncol.
40:2004–2012. 2012.PubMed/NCBI
|
|
26
|
Bu Q, Hu Z, Chen F, et al: Transcriptome
analysis of long non-coding RNAs of the nucleus accumbens in
cocaine-conditioned mice. J Neurochem. 123:790–799. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tafer H and Hofacker IL: RNAplex: a fast
tool for RNA-RNA interaction search. Bioinformatics. 24:2657–2663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tafer H, Amman F, Eggenhofer F, et al:
Fast accessibility-based prediction of RNA-RNA interactions.
Bioinformatics. 27:1934–1940. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ryu DS, Lee HS, Lee GS, et al: Effects of
the ethylacetate extract of Orostachys japonicus on induction of
apoptosis through the p53-mediated signaling pathway in human
gastric cancer cells. Biol Pharm Bull. 35:660–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J, Xie YS, Wang FL, et al:
Cytotoxicity of 5-Aza-2′-deoxycytidine against gastric cancer
involves DNA damage in an ATM-P53 dependent signaling pathway and
demethylation of P16(INK4A). Biomed Pharmacother. 67:78–87.
2013.
|
|
31
|
Ma GF, Chen SY, Sun ZR, et al: FoxP3
inhibits proliferation and induces apoptosis of gastric cancer
cells by activating the apoptotic signaling pathway. Biochem
Biophys Res Commun. 430:804–809. 2013. View Article : Google Scholar
|
|
32
|
Yu RX, Hu XM, Xu SQ, et al: Effects of
fucoxanthin on proliferation and apoptosis in human gastric
adenocarcinoma MGC-803 cells via JAK/STAT signal pathway. Eur J
Pharmacol. 657:10–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
To KF, Chan MW, Leung WK, et al:
Constitutional activation of IL-6-mediated JAK/STAT pathway through
hypermethylation of SOCS-1 in human gastric cancer cell line. Br J
Cancer. 91:1335–1341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim JW, Im SA, Kim MA, et al:
Ataxia-telangiectasia mutated (ATM) protein expression with
microsatellite instability in gastric cancer as prognostic marker.
Int J Cancer. 134:72–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morikawa T, Hino R, Uozaki H, et al:
Expression of ribonucleotide reductase M2 subunit in gastric cancer
and effects of RRM2 inhibition in vitro. Hum Pathol. 41:1742–1748.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Terashima M, Maesawa C, Oyama K, et al:
Gene expression profiles in human gastric cancer: expression of
maspin correlates with lymph node metastasis. Br J Cancer.
92:1130–1136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hudler P: Genetic aspects of gastric
cancer instability. Sci World J. 2012:7619092012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qiu MT, Hu JW, Yin R, et al: Long
noncoding RNA: an emerging paradigm of cancer research. Tumour
Biol. 34:613–620. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qi P and Du X: The long non-coding RNAs, a
new cancer diagnostic and therapeutic gold mine. Mod Pathol.
26:155–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang F, Bi J, Xue X, et al: Up-regulated
long non-coding RNA H19 contributes to proliferation of gastric
cancer cells. FEBS J. 279:3159–3165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim ED and Sung S: Long noncoding RNA:
unveiling hidden layer of gene regulatory networks. Trends Plant
Sci. 17:16–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
De LF and Dean C: Long non-coding RNAs and
chromatin regulation. Curr Opin Plant Biol. 14:168–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qu Z and Adelson DL: Identification and
comparative analysis of ncRNAs in human, mouse and zebrafish
indicate a conserved role in regulation of genes expressed in
brain. PLoS One. 7:e522752012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ponjavic J, Oliver PL, Lunter G, et al:
Genomic and transcriptional co-localization of protein-coding and
long non-coding RNA pairs in the developing brain. PLoS Genet.
5:e10006172009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|