Introduction
Infection with Helicobacter pylori is the
strongest risk factor for the development of chronic gastritis,
gastric ulcer and gastric carcinoma, which is the second most
common cause of cancer-related death worldwide (1,2).
Some H. pylori strains possess a cytotoxin-associated gene
(cag) pathogenicity island (cag PAI) that is present in about half
of the Western strains and most of the Eastern strains (3). One constituent of the cag PAI is
cagA, which encodes a 120–140-kDa CagA protein. The CagA protein is
directly injected into gastric epithelial cells by the type IV
secretory system of H. pylori. Once inside host cells, CagA
is tyrosine phosphorylated on conserved carboxyl-terminal
Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs by Src family kinases (4,5). The
phosphorylated and unphosphorylated CagA targets multiple enzymes
to activate downstream signal pathways, such as the
Ras/mitogen-activated protein kinase (MEK)/extracellular
signal-regulated kinase (ERK) pathway, nuclear factor κB (NF-κB)
pathway, Src kinase and the PI3 kinase pathway (6–9).
Eventually, these events are important for cell function and are
also implicated in carcinogenesis since they regulate fundamental
cellular responses such as cell proliferation, cell morphology,
cell motility and apoptosis. According to these observations,
bacterial oncoprotein CagA participates in gastric epithelial cell
injury caused by cagA-positive H. pylori infection.
Epidemiological studies reveal that strains of H. pylori
carrying the virulence factor CagA protein are associated with an
increased risk of gastric mucosal inflammation as well as severe
atrophic gastritis and gastric carcinoma compared to strains of
H. pylori lacking CagA (10,11).
However, details of the exact molecular mechanisms by which CagA
promotes carcinogenesis are fragmentary.
In order to illuminate the pathogenesis of H.
pylori-induced gastric diseases, our group has developed a
cagA gene knock out isogenic mutant strain. In our previous
study, an in vitro model was established to characterize
proteins differentially expressed in cagA-positive H.
pylori-infected gastric epithelial cells versus the cagA
mutant strain. We found that α-enolase (ENO1) expression level was
increased in cagA-positive H. pylori-infected cells
as compared to mutant strain by two-dimensional electrophoresis
(2-DE) and matrix-assisted laser desorption/ionization-time of
flight mass spectrometry (MALDI-TOF MS) analysis (12). ENO1 is an isoenzyme of enolase, a
key protein in the glycolytic pathway. Overexpression of ENO1 is
considered to take part in unlimited cellular proliferation of
cancer. ENO1 has also been described as a stress protein induced by
hypoxia (13,14).
As mentioned above, H. pylori infection could
affect the expression level of gastric carcinogenic factors, such
as Myc, cyclin D1 and COX-2 (15–18).
However, up to now, the relationship between H. pylori
infection and ENO1 expression has not been reported. To figure out
the effect of CagA on ENO1 may help us to understand the
carcinogenic mechanism of H. pylori infection. In the
present study, we designed CagA transfection in gastric epithelial
cells and tried to investigate whether main virulence factor CagA
of H. pylori could increase the expression of ENO1 in
vitro. Furthermore, we used signal inhibitors to examine which
pathway was involved in the CagA-mediated regulation of ENO1
expression.
Materials and methods
Materials
Bacterial strains and culture
conditions
Hp27 (cagA+) isolated from a patient with
chronic atrophy gastritis in Zhengzhou, China, was grown on
Brucella agar plates containing 10% sheep blood supplemented with
10 mg/l vancomycin, 2,500 U/l polymyxin B, 2 mg/l amphotericin and
5 mg/l trimethoprim under microaerophilic conditions at 37°C for 3
days. Hp27 is the East Asia type strain, which has EPIYA-ABD type
CagA.
Cell lines and culture conditions
The AGS gastric cancer cells were purchased from the
Type Culture Collection of the Chinese Academy of Sciences,
Shanghai, China. Cells were cultured in F12 medium (Gibco Life
Technologies) with 10% FBS (Gibco Life Technologies) at 37°C in 5%
CO2.
Methods
Construction of pcDNA3.1 (+)
expression vectors containing cagA and of site-directed
mutations
Full-length cagA DNA (QD306710) was amplified
by high-fidelity PCR from the DNA of H. pylori strain
Hp27 using primers containing EcoRI/XhoI. The
resultant PCR fragment was digested with EcoRI and
XhoI, cloned into pcDNA3.1 (+) vector giving rise to
wild-type cagA/pcDNA3.1 (+) plasmid (WT-cagA).
Hp27 has three copies of tyrosine phosphorylation site
sequences which is the ABD type EPIYA.
We generated the CagA site-directed mutagenesis in
which the tyrosine residues present in the B or D copies of the
EPIYA sequences were replaced by cysteine by using splice overlap
extension PCR. Specific primers were designed for B or D copies
respectively. The first step involves synthesis of individual
fragments containing mutant sites. Then the mutations were
connected by overlap extension PCR. After the results were
validated by gene sequencing, the mutated cagA gene was
cloned into pcDNA3.1 (+) vector. The phosphorylation-resistant
plasmids were named PRB-cagA and PRD-cagA,
respectively. The specific primers used for the gene site directed
mutations are listed as follows: P1, 5′-CATGAATT
CGCCACCATGACTAACGAAAC-3′; P2, 5′-CGCCTCGA GTTAAGATTTCTGGAAAC-3′;
B1, 5′-GCCCTGAAGAGC CCATTTGCGCTCAAGTTGCTAAAAAG-3′; B2, 5′-CTTT
TTAGCAACTTGAGCGCAAATGGGCTCTTCAGGGC-3′; D1,
5′-CTAGTCCTGAACCCATTTGCGCTACAATTGATT TTGATG-3′; D2,
5′-CATCAAAATCAATTGTAGCGCAAAT GGGTTCAGGACTAG-3′.
Transient transfection
Lipofectamine 2000 reagent (Invitrogen) was used to
transfect the plasmid WT-cagA, PR-cagA or blank
vector into 5×105 AGS cells in 6-well plates. For
transient transfection, 5 μg of the plasmid was transfected into
cells with 10 μl transfection reagent according to the
manufacturer’s protocol and all experiments were repeated three
times. After the transfection, cells were harvested and lysed with
RIPA buffer supplemented with protease inhibitors before freezing
at −80°C. To investigate associated signal pathways involved in
CagA-mediated ENO1 upregulation in the epithelial cells, we used
the following reagents: BAY 11-7082 (5 μM), LY294002 (50 μM), PP1
(10 μM), U0126 (10 μM) (all from Cell Signaling Technology). A
respective reagent was used to pre-incubate cells for 1 h and then
the plasmid was transfected into cells.
Quantitative detection of eno1
mRNA
The quantitative RT-PCR was designed to detect eno1
mRNA level in which GAPDH gene was used as the reference
gene. Briefly, total RNA of AGS cells that were infected with
different recombinant plasmids for desired time were extracted by
using TRIzol reagent (Invitrogen) and then reverse transcribed to
cDNA with random primers by using PrimeScript RT reagents kit
(Takara). QRT-PCR was performed on an ABI PRISM 7500 Real-Time PCR
system (Applied Biosystems) using SYBR® Premix Ex Taq™
(Takara) according to the manufacturer’s protocols. Relative gene
expression values were obtained by normalization to the reference
gene GAPDH using the −2ΔΔCt method, where
−2ΔΔCt=ΔCt sample−ΔCt calibrator as described. The
following primer pairs were used for PCR amplification:
eno1-forward, 5′-TGTACCGCCACATCGC TGA-3′;
eno1-reverse, 5′-TGAAGTTT-GCTGCACCG CTG-3′.
GAPDH-forward, 5′-ACCACAGTCCATGCCATCAC-3′;
GAPDH-reverse, 5′-TCCACCACCCTGTTGCTGTA-3′.
Western blot analysis
Cells were lysed with RIPA buffer (pH 7.4, 50 mM
Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na-deoxycholate,
1 mM PMSF, 1 mM orthovanadate, 1 μM leupeptin, 1 μM pepstatin) for
30 min at 4°C. Cell lysates were centrifuged at 12,000 g for 20 min
at 4°C. After measuring the concentration of the protein using a
BCA reagent kit (China), the lysates were boiled at 100°C for 5 min
in the presence of 2% β-mercaptoethanol. Equal amounts of protein
were separated by SDS-PAGE (12% SDS-acrylamide gels), and
transferred to polyvinylidene difluoride membrane (Millipore,
Bedford, MA, USA). After blocking with 1% bovine serum albumin in
TBST buffer (pH 7.6, 20 mM Tris-HCl, 137 mM NaCl, 0.1% Tween-20),
membranes were incubated overnight with either mouse anti-CagA
antibody (1:1,000, Abcam, USA), rabbit anti-ENO1 antibody (1:1,000,
Abcam) or mouse anti-GAPDH antibody (1:3,000, Abcam) in TBST-milk.
Blots were then incubated with the corresponding secondary
horseradish peroxidase-conjugated antibody (1:5,000, Abcam, USA)
for 1 h at room temperature. After washing the membranes, the
reaction was visualized using the ECL Detection kit (GE Healthcare,
Buckinghamshire, UK) and by exposing the blots to Fuji medical
X-ray film. ImageJ software was used to analyse the western blot
results.
Statistical analysis
SPSS17.0 software (SPSS Inc., USA) was adopted for
statistical analysis. Statistical significance of the measured
values was analyzed using the ANOVA. P<0.05 was considered
statistically significant.
Results
Identification of recombinant vector
The recombinant vector WT-cagA and PR-cagA were
detected as recombined properly with PCR and digestion experiments.
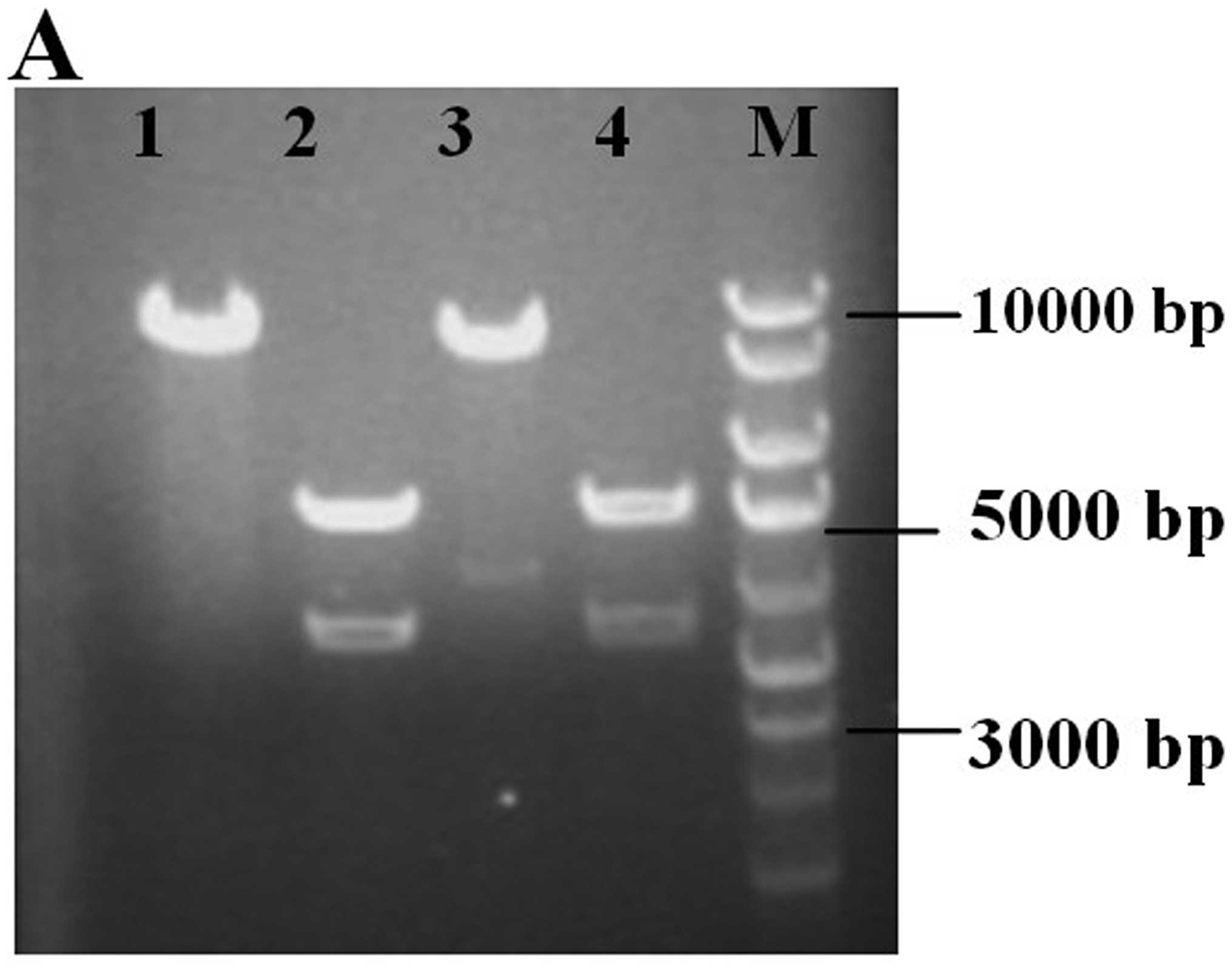
Fig. 1A shows the cagA
fragment and vector pcDNA3.1 analyzed by EcoRI and
XhoI double digestion. By PCR amplification from recombinant
vector, 3510 bp of cagA was produced and further verified by
sequencing.
Mutation analysis was performed by direct
sequencing. The DNA sequencing results revealed that the codon TAC
which codes the tyrosine residues present in EPIYA sequences were
replaced by cysteine (TGC). The CagA phosphorylation-resistant
expression vector was constructed successfully (Fig. 1B).
Detection of CagA mRNA and protein
expression in cell transfection
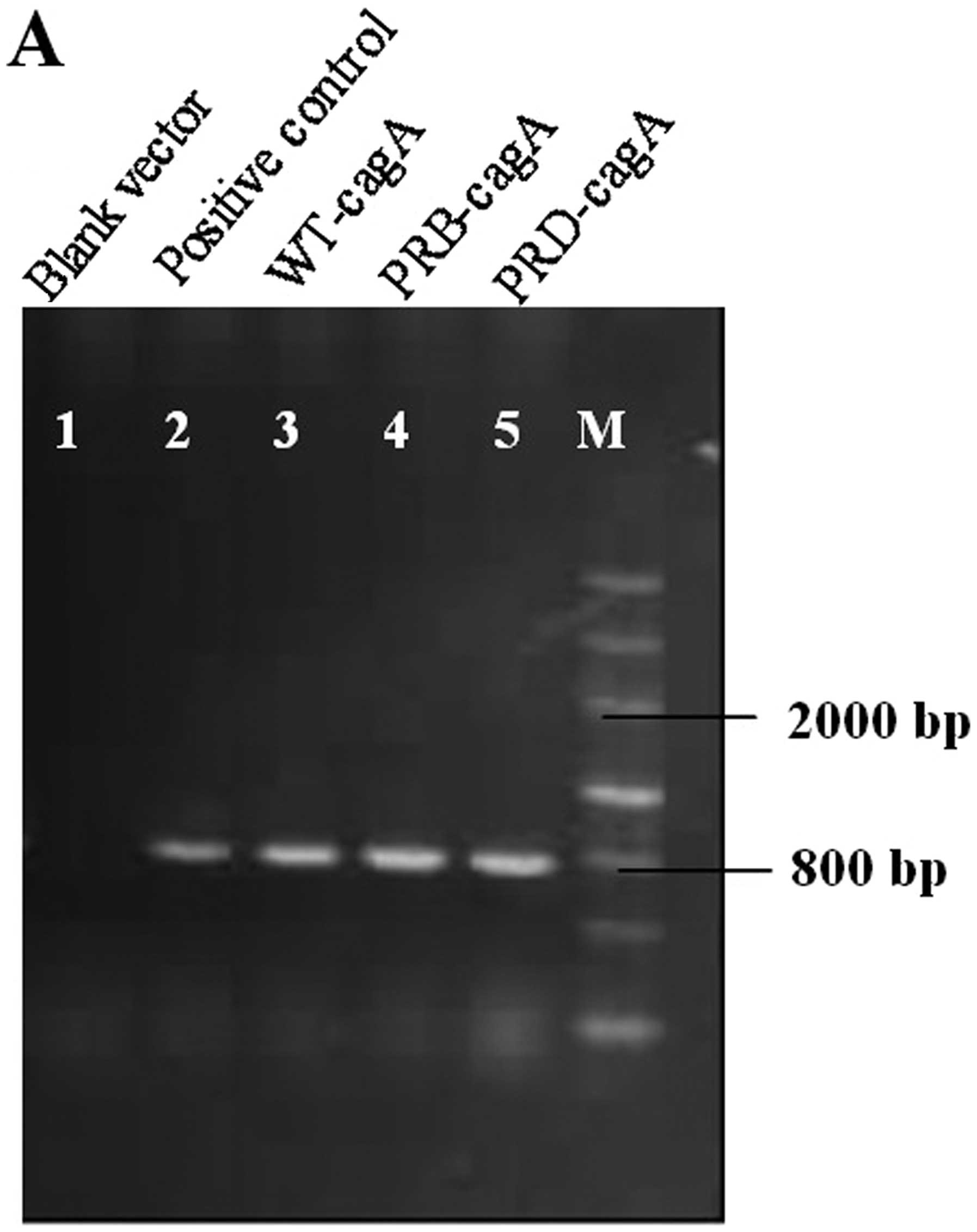
To confirm whether CagA protein was involved in cell
transfection, the RT-PCR and western blot analysis were used to
detect cagA mRNA and protein in the transformed cells. We
designed the specific primers for cagA of ~780 bp in length.
In the PCR results, we found a cagA cDNA band, 780 bp in
length (Fig. 2A), confirming to
the results of DNA sequencing, while it was absent in the negative
control. Similarly, western blot results indicated that CagA
protein band was clearly visible in the cagA-transformed cell group
with molecular weight of 121 kDa, but not in the control group
(Fig. 2B).
CagA-transformed cells promote the
increase in eno1 mRNA expression
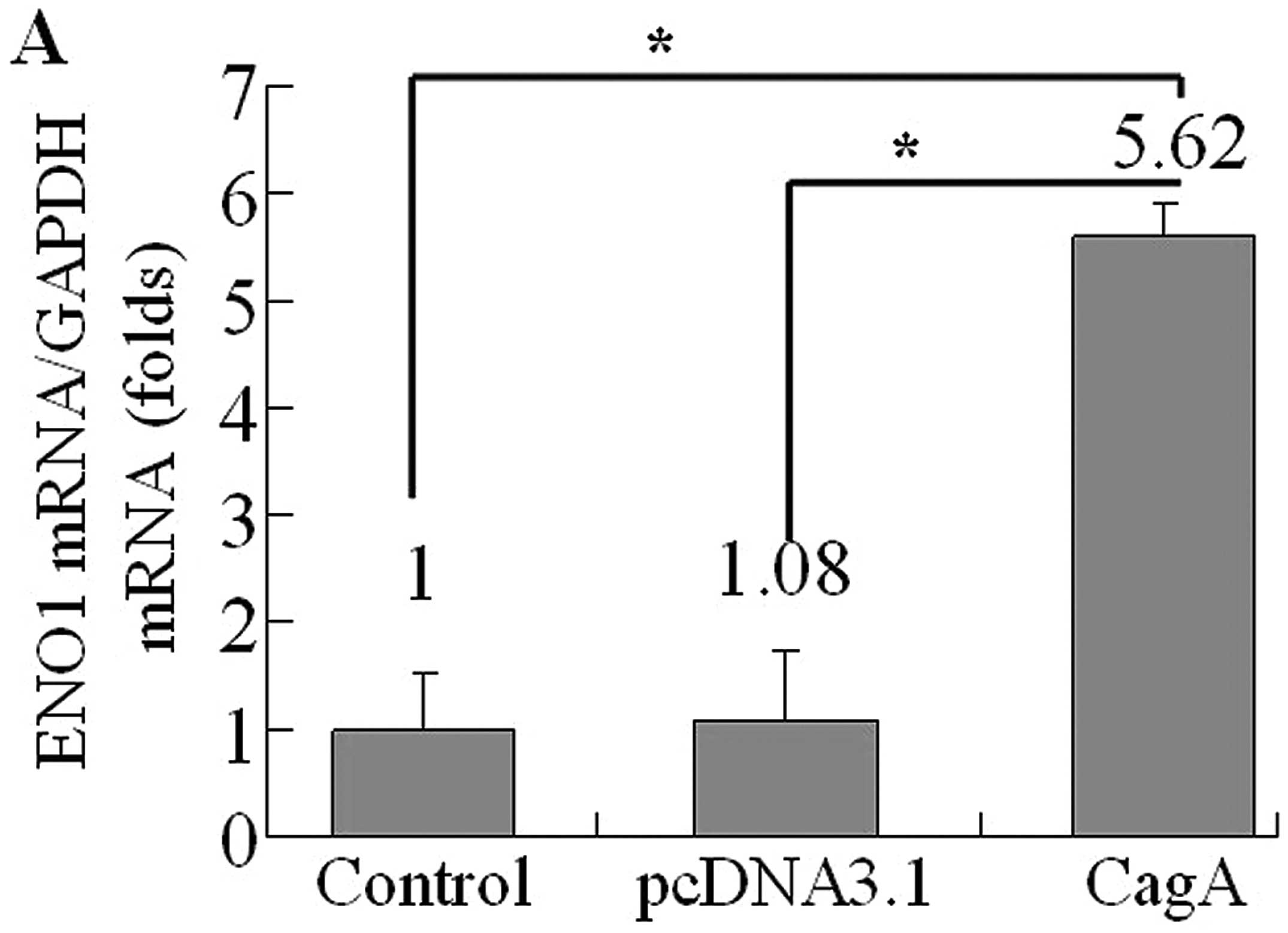
In order to evaluate the effect of CagA on eno1
expression level, AGS cells were transfected with WT-cagA or
pcDNA3.1, respectively. Then we determined eno1 mRNA levels by
quantitative RT-PCR analysis at 36 h after transfection. The
results showed that eno1 expression at RNA level was dramatically
increased in WT-cagA overexpressing cells compared to non-induced
control cells and blank vector transformed cells (Fig. 3A). Next, we investigated whether
the upregulation is dependent on the CagA tyrosine phosphorylation.
We used the PR-cagA plasmids to transtect AGS cells. The PR-cagA
plasmid encodes a CagA protein with a mutation in the EPIYA B motif
or D motif, required for CagA tyrosine phosphorylation. Thus the
PR-CagA mutant protein cannot be phosphorylated nor can it transmit
the signal pathway. After 36 h transfection of PR-cagA plasmid into
AGS cells, eno1 mRNA levels were unchanged in PR-cagA
overexpressing cells (Fig 3B).
ENO1 protein expression was upregulation
by HP WT-CagA in AGS cells
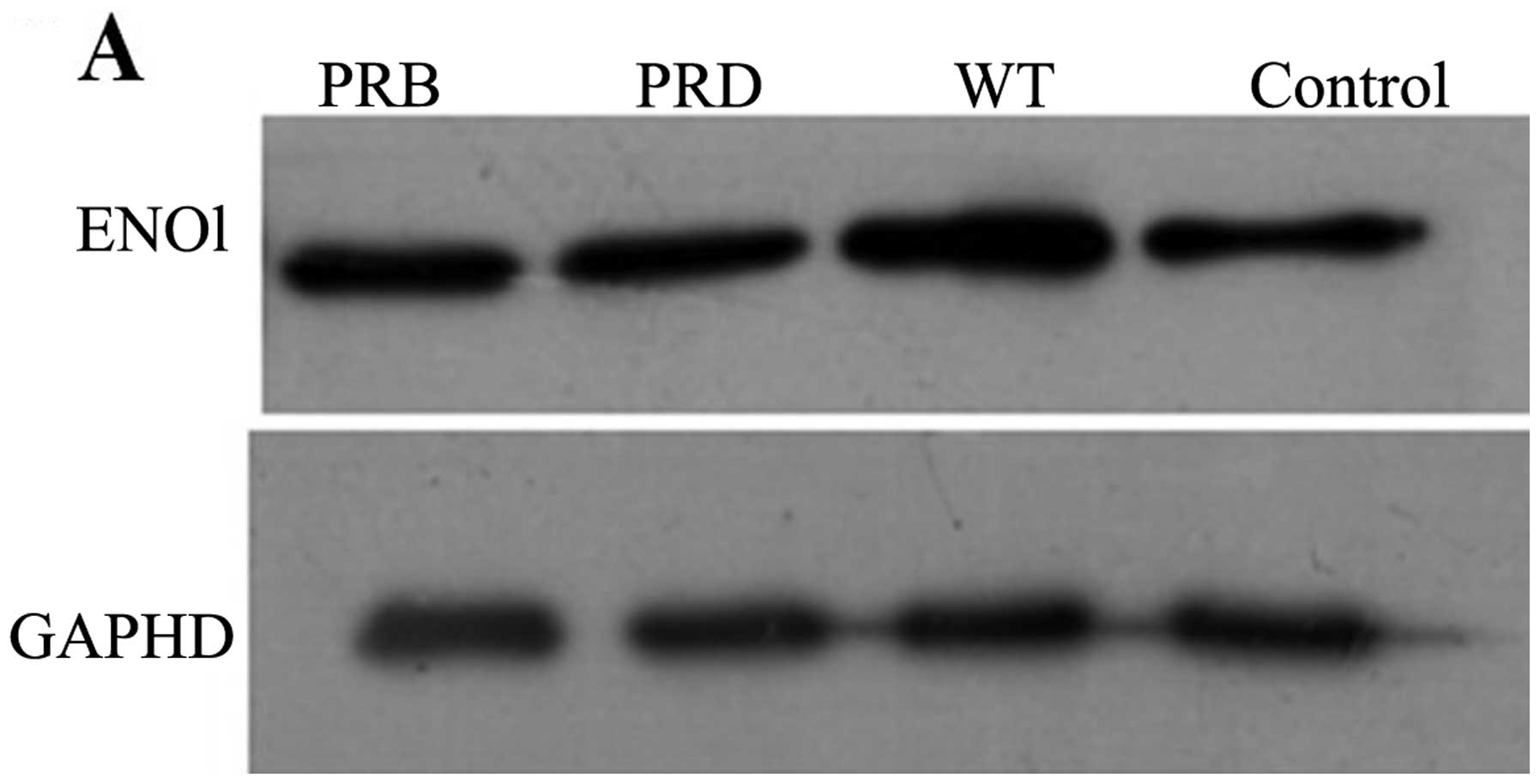
As eno1 mRNA level upregulation was demonstrated to
be associated with CagA, we next evaluated the relationship between
CagA and ENO1 at the protein level. The WT-cagA or PR-cagA plasmid
was transfected in AGS cells for 36 h. Total cellular protein
analysed by western blot analysis indicated that the ENO1
expression level in AGS cells expressing WT-CagA was significantly
greater than that in normal cells. In contrast, there was no
increase in ENO1 expression level in AGS cells transfected with
vector expressing PR-CagA compared with control cells. Taken
together, bacterial oncoprotein CagA can upregulate the expression
of ENO1 and this response is mediated specifically by tyrosine
phosphorylate, but not unphosphorylated CagA (Fig. 4).
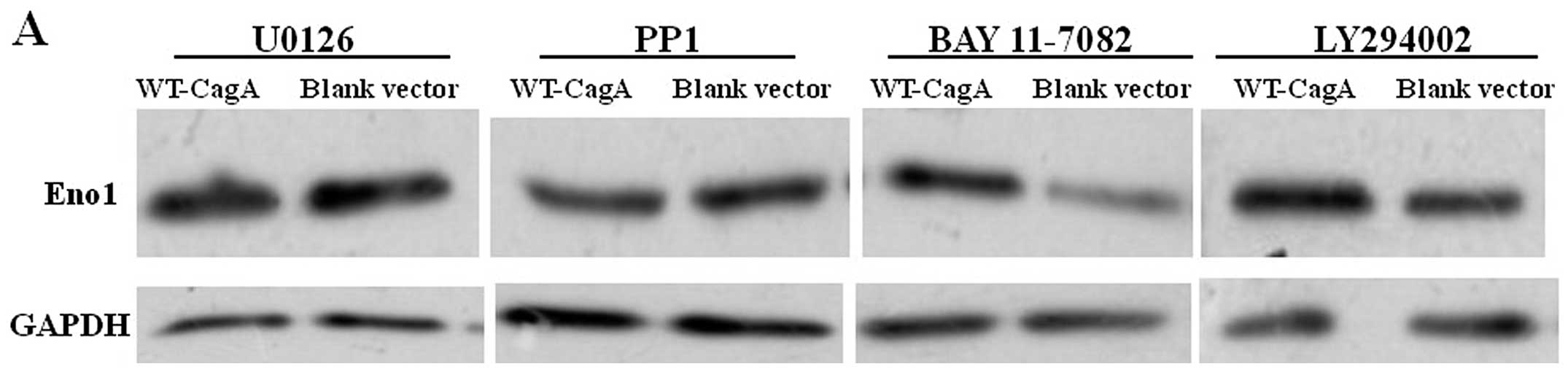
Signal inhibitor effects on ENO1
expression in AGS cells transfected with WT-cagA plasmid
We next wanted to determine the pathway(s) through
which CagA affected ENO1. For this purpose, we used the MAP kinase
inhibitor U0126 to block MEK1 and MEK2 kinase activity, thereby
inhibiting ERK1/2 phosphorylation, BAY 11-7082 to inhibit the NF-κB
pathway, PP1 to inhibit the Scr-family tyrosine kinase activity and
LY294002 to block PI3 kinase activity. Each signal inhibitor was
incubated with gastric cells for 1 h prior to plasmid transfection.
After 36-h transfection, western blot analysis was used to detect
the ENO1 expression level. Fig. 5
shows that ENO1 upregulation by CagA was attenuated by U0126 and
PP1, ENO1 upregulation was maintained using BAY 11-7082 and
LY294002. In total, the results indicated that MEK/ERK and
Scr-family tyrosine kinase pathways participated in the
upregulation of ENO1 by CagA.
Discussion
Infection by H. pylori is one of the major
causes of chronic active gastritis and peptic ulcer and is closely
related to the occurrence and progression of gastric cancer
(19). CagA is one of the most
important virulence factors encoded by H. pylori. H.
pylori colonizing the gastric epithelial surface inject CagA
into host cells via its type IV secretion system. CagA can interact
with various host cellular proteins to trigger distinct signaling
pathways in a tyrosine phosphorylation-dependent and -independent
manner (9,20–22).
The phosphorylation capability of a
glutamate-proline-isoleucine-tyrosine-alanine (EPIYA) motif in CagA
is one of the major markers that correlates with H. pylori
pathogenicity and the phosphorylation status is a key determinant
of CagA-mediated signaling and transcriptional outcomes. The
C-termini of the CagA proteins of different H. pylori
strains exhibit considerable variations, which mainly include
different numbers of EPIYA tandem repeats as well as different
amino acid sequences flanking the motif (23). Higashi et al reported that
most Asian H. pylori isolates express a highly pathogenic
CagA protein, harboring the EPIYA-D tyrosine phosphorylation motif,
which exhibits high affinity for binding to SHP-2 (24,25).
In our study, Hp27 is the East Asia type strain, which has
EPIYA-ABD type CagA, thus we design the site-directed mutations at
B and D motif (12).
In the former work of our laboratory (12), we had discovered ENO1 expression
level was increased in wild-type H. pylori
(cagA+)-infected cells as compared to cagA
mutant H. pylori (cagA−). ENO1, a key
glycolytic enzyme, belonging to a novel class of surface proteins.
ENO1 serves as a plasminogen receptor on the surface of a variety
of hematopoietic, epithelial and endothelial cells. It is involved
in various pathological events such as tissue remodeling and the
spread of transformed tumor cells induced by plasminogen activation
(26). Some evidence indicates a
relation between ENO1 and the progression of tumors, such as
neuroendocrine tumors, neuroblastoma, lung cancer and
hepatocellular carcinoma (27). In
cancer cells, the rate-determining enzyme for glycolysis is
converted to an isoenzyme different from that in normal cells and
the capability of glycolysis is increased because of increased cell
proliferation. In response to upregulated ENO1 expression, the
fibrinolytic system is inordinately accelerated. Consequently,
increased local fibrinolysis may contribute to cancer cell invasion
and metastasis. More recently, oncogenic AKT and Myc have been
shown to stimulate anaerobic glycolysis directly and ENO1 is one of
the Myc driven target genes (28,29).
ENO1 has also been described as a stress protein induced by
hypoxia. In large tumors, oxygen is relatively decreased in the
central region. Thus, it is reasonable that ENO1 is upregulated in
large tumors and it has been suggested to be a diagnostic marker
for certain tumors.
However, up to now, the relationship between gastric
carcinogenic pathogen H. pylori infection and gastric cancer
oncoprotein ENO1 overexpression has not been reported. Therefore,
we explored the relationship between H. pylori CagA, ENO1
overexpression and signaling pathways leading to gastric
cancer.
After translocation into epithelial cells, H.
pylori CagA protein was able to activate some major pathways,
including the Src kinase, MEK/ERK pathway, PI3K/Akt pathway and
NF-κB pathway. Kim et al reported that CagA transfection
activated the Src kinase, and Zhu et al proved that CagA
transfection and phosphorylated CagA protein activated the ERK1/2
pathway (30,31). It was previously shown that signal
inhibitors could be used to elucidate which pathway was involved in
the H. pylori infection-induced cyclooxygenase 2
upregulation (32) or the H.
pylori CagA inhibition of Runx3 expression (33). To seach for the signal pathways
involved in ENO1 regulation, we used four specific signal protein
inhibitors to block corresponding signal transduction. We
identified that Src and MEK/ERK pathways were involved in ENO1
upregulation by CagA. Taken together, CagA protein was
tyrosine-phosphorylated by Src kinase, and thereafter activated the
MEK/ERK pathway to upregulate ENO1 expression in AGS cells.
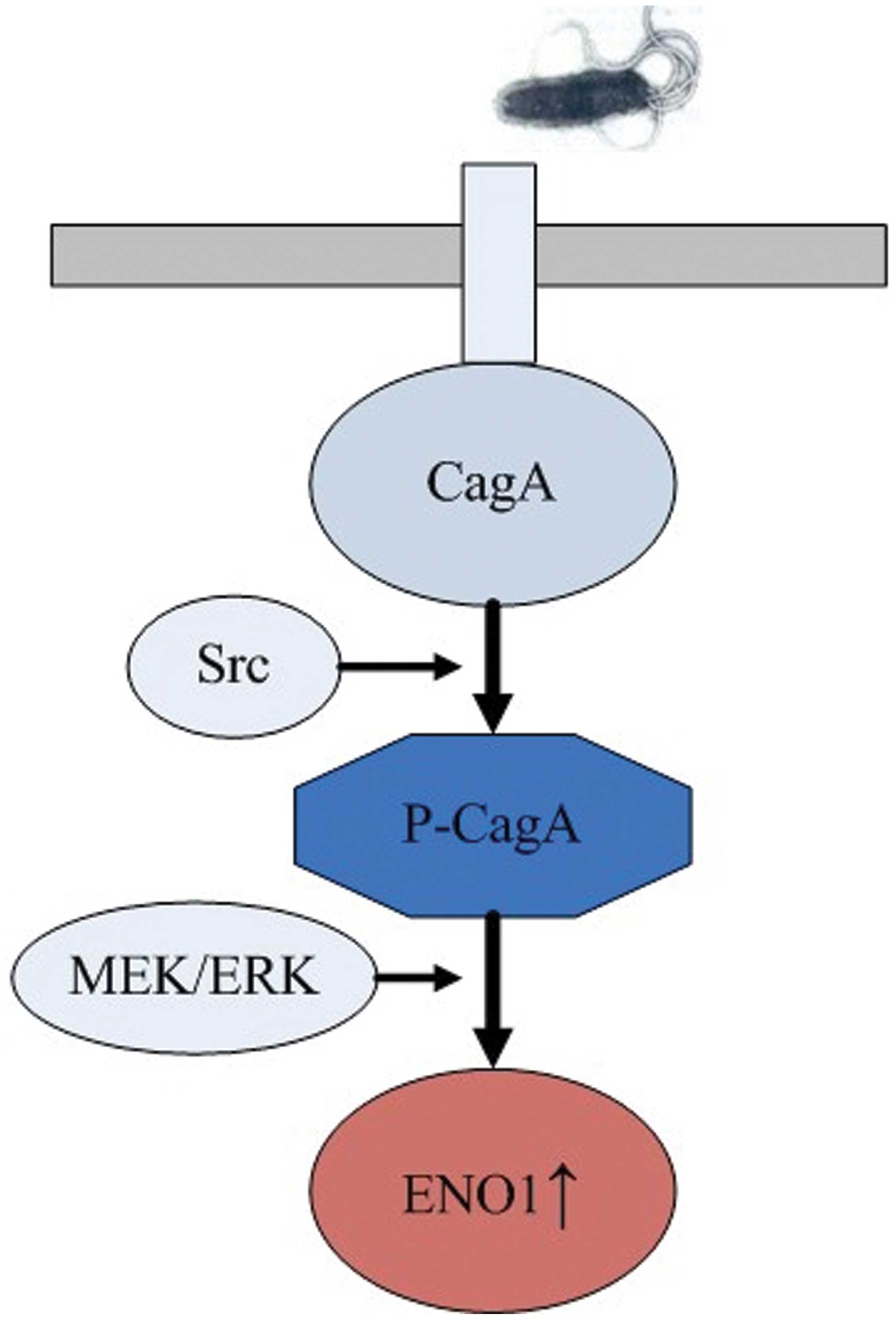
The combination of data from our study and others
led us to propose the regulation mechanisms of H. pylori
CagA on the expression of ENO1 (Fig.
6).
After translocating into gastric epithelial cells
through the TFSS, bacterial oncoprotein CagA was
tyrosine-phosphorylated by Src kinase. P-CagA activated the MEK/ERK
pathway and upregulated ENO1 expression. Upregulation of ENO1
expression by H. pylori provided a further explanation for
the gastric carcinogenic effects of H. pylori infection.
In conclusion, this study identifies the H.
pylori effector protein CagA as potentially enhancing the risk
for gastric cancer by increasing ENO1 expression in epithelial
cells. This is the first time that the mechanism by which H.
pylori infection and bacterial oncoprotein CagA upregulated
ENO1 expression in gastric cell lines has been elucidated. CagA
protein activated the Src and MEK/ERK signal pathways, resulting in
the elevation of expression of ENO1 protein in AGS cells. These
findings may be useful in identifying the mechanism by which
gastric tumors are caused by H. pylori infection in
humans.
Acknowledgements
We thank members of the Henan Key Laboratory of
Molecular Medicine for helpful discussions and critical reading of
the manuscript.
References
|
1
|
Cavaleiro-Pinto M, Peleteiro B, Lunet N
and Barros H: Helicobacter pylori infection and gastric
cardia cancer: systematic review and meta-analysis. Cancer Causes
Control. 22:375–387. 2011. View Article : Google Scholar
|
|
2
|
Peek RM and Crabtree JE:
Helicobacter infection and gastric neoplasia. J Pathol.
208:233–248. 2006. View Article : Google Scholar
|
|
3
|
Zhu Y-L, Zheng S, Du Q, Qian K-D and Fang
P-C: Characterization of CagA variable region of Helicobacter
pylori isolates from Chinese patients. World J Gastroenterol.
11:880–884. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hatakeyama M: Oncogenic mechanisms of the
Helicobacter pylori CagA protein. Nat Rev Cancer. 4:688–694.
2004.
|
|
5
|
Backert S and Selbach M: Role of type IV
secretion in Helicobacter pylori pathogenesis. Cell
Microbiol. 10:1573–1581. 2008. View Article : Google Scholar
|
|
6
|
Saadat I, Higashi H, Obuse C, et al:
Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt
epithelial cell polarity. Nature. 447:330–333. 2007. View Article : Google Scholar
|
|
7
|
Brandt S, Kwok T, Hartig R, Konig W and
Backert S: NF-kappaB activation and potentiation of proinflammatory
responses by the Helicobacter pylori CagA protein. Proc Natl
Acad Sci USA. 102:9300–9305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao DP, Liu ZF, Ding J, et al:
Helicobacter pylori CagA upregulation of CIP2A is dependent
on the Src and MEK/ERK pathways. J Med Microbiol. 59:259–265. 2010.
View Article : Google Scholar
|
|
9
|
Churin Y, Al-Ghoul L, Kepp O, Meyer TF,
Birchmeier W and Naumann M: Helicobacter pylori CagA protein
targets the c-Met receptor and enhances the motogenic response. J
Cell Biol. 161:249–255. 2003. View Article : Google Scholar
|
|
10
|
Kuipers EJ, Perez-Perez GI, Meuwissen SG
and Blaser MJ: Helicobacter pylori and atrophic gastritis:
importance of the cagA status. J Natl Cancer Inst. 87:1777–1780.
1995. View Article : Google Scholar
|
|
11
|
Nomura AM, Lee J, Stemmermann GN, Nomura
RY, Perez-Perez GI and Blaser MJ: Helicobacter pylori CagA
seropositivity and gastric carcinoma risk in a Japanese American
population. J Infect Dis. 186:1138–1144. 2002. View Article : Google Scholar
|
|
12
|
Huang ZG, Duan GC, Fan QT, et al: Mutation
of cytotoxin-associated gene A affects expressions of antioxidant
proteins of Helicobacter pylori. World J Gastroenterol.
15:599–606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plow EF, Herren T, Redlitz A, Miles LA and
Hoover-Plow JL: The cell biology of the plasminogen system. FASEB
J. 9:939–945. 1995.PubMed/NCBI
|
|
14
|
Jiang BH, Agani F, Passaniti A and Semenza
GL: V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1)
and transcription of genes encoding vascular endothelial growth
factor and enolase 1: involvement of HIF-1 in tumor progression.
Cancer Res. 57:5328–5335. 1997.PubMed/NCBI
|
|
15
|
Zhu Y, Shu X, Chen J, et al: Effect of
Helicobacter pylori eradication on oncogenes and cell
proliferation. Eur J Clin Invest. 38:628–633. 2008.
|
|
16
|
Hirata Y, Maeda S, Mitsuno W, et al:
Helicobacter pylori activates the cyclin D1 gene through
mitogen-activated protein kinase pathway in gastric cancer cells.
Infect Immun. 69:3965–3971. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu C-Y, Wang C-J, Tseng C-C, et al:
Helicobacter pylori promote gastric cancer cells invasion
through a NF-kappaB and COX-2-mediated pathway. World J
Gastroenterol. 11:31972005. View Article : Google Scholar
|
|
18
|
Lee KS, Kalantzis A, Jackson CB, et al:
Helicobacter pylori CagA triggers expression of the
bactericidal lectin REG3γ via gastric STAT3 activation. PloS One.
7:e307862012. View Article : Google Scholar
|
|
19
|
Atherton JC and Blaser MJ: Coadaptation of
Helicobacter pylori and humans: ancient history, modern
implications. J Clin Invest. 119:2475–2487. 2009.PubMed/NCBI
|
|
20
|
Selbach M, Moese S, Hauck CR, Meyer TF and
Backert S: Src is the kinase of the Helicobacter pylori CagA
protein in vitro and in vivo. J Biol Chem. 277:6775–6778.
2002.PubMed/NCBI
|
|
21
|
Poppe M, Feller SM, Romer G and Wessler S:
Phosphorylation of Helicobacter pylori CagA by c-Abl leads
to cell motility. Oncogene. 26:3462–3472. 2007.PubMed/NCBI
|
|
22
|
Amieva MR, Vogelmann R, Covacci A,
Tompkins LS, Nelson WJ and Falkow S: Disruption of the epithelial
apical-junctional complex by Helicobacter pylori CagA.
Science. 300:1430–1434. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wroblewski LE, Peek RM and Wilson KT:
Helicobacter pylori and gastric cancer: factors that
modulate disease risk. Clin Microbiol Rev. 23:713–739. 2010.
View Article : Google Scholar
|
|
24
|
Higashi H, Tsutsumi R, Muto S, et al:
SHP-2 tyrosine phosphatase as an intracellular target of
Helicobacter pylori CagA protein. Science. 295:683–686.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Higashi H, Tsutsumi R, Fujita A, et al:
Biological activity of the Helicobacter pylori virulence
factor CagA is determined by variation in the tyrosine
phosphorylation sites. Proc Natl Acad Sci USA. 99:14428–14433.
2002.
|
|
26
|
Pancholi V: Multifunctional alpha-enolase:
its role in diseases. Cell Mol Life Sci. 58:902–920. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takashima M, Kuramitsu Y, Yokoyama Y, et
al: Overexpression of alpha enolase in hepatitis C virus-related
hepatocellular carcinoma: association with tumor progression as
determined by proteomic analysis. Proteomics. 5:1686–1692. 2005.
View Article : Google Scholar
|
|
28
|
Feo S, Arcuri D, Piddini E, Passantino R
and Giallongo A: ENO1 gene product binds to the c-myc promoter and
acts as a transcriptional repressor: relationship with Myc
promoter-binding protein 1 (MBP-1). FEBS Lett. 473:47–52. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim J-W, Zeller KI, Wang Y, et al:
Evaluation of myc E-box phylogenetic footprints in glycolytic genes
by chromatin immunoprecipitation assays. Mol Cell Biol.
24:5923–5936. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim SY, Lee YC, Kim HK and Blaser MJ:
Helicobacter pylori CagA transfection of gastric epithelial
cells induces interleukin-8. Cell Microbiol. 8:97–106. 2006.
View Article : Google Scholar
|
|
31
|
Zhu Y, Zhong X, Zheng S, Du Q and Xu W:
Transformed immortalized gastric epithelial cells by virulence
factor CagA of Helicobacter pylori through Erk
mitogen-activated protein kinase pathway. Oncogene. 24:3886–3895.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Q, Liu N, Shen B, et al:
Helicobacter pylori enhances cyclooxygenase 2 expression via
p38MAPK/ATF-2 signaling pathway in MKN45 cells. Cancer Lett.
278:97–103. 2009. View Article : Google Scholar
|
|
33
|
Liu Z, Xu X, Chen L, et al:
Helicobacter pylori CagA inhibits the expression of Runx3
via Src/MEK/ERK and p38 MAPK pathways in gastric epithelial cells.
J Cell Biochem. 113:1080–1086. 2012. View Article : Google Scholar
|