Introduction
Tumorigenesis is the result of malfunction of many
genes, proteins and metabolites. Among these molecules, TGFβ and
EGF have prominent places as strong regulators of tumorigenesis.
TGFβ has a double role in tumorigenesis. In the early stage of
cancer, TGFβ has a tumor suppressor role which results in growth
inhibition, cell cycle arrest and apoptosis. In the advanced stage
of cancer, TGFβ promotes tumorigenesis. The cancer cells may lose
responsiveness to TGFβ and may acquire aberrant TGFβ signaling,
followed by promotion of survival, proliferation, EMT and increased
motility and invasiveness of the cells (1). Numerous signaling pathways converge
with the TGFβ pathway to modulate its effects; including signaling
induced by EGF (2). EGF has
predominately pro-mitogenic role in tumorigenesis (3). EGF promotes also cell survival,
angiogenesis and differentiation. Deregulation of EGF pathways by
overexpression or constitutive activation can promote
tumorigenesis, including angiogenesis and metastasis, and is
associated with a poor prognosis in many human malignancies
(4). TGFβ and EGF intracellular
signaling involves sharing intracellular signaling mechanisms. The
extensive cross-talk between TGFβ and EGF involves such proteins
and genes such as Smads, Erk1/2, p38 and PI3K.
Mammalian sterile-like 1 (MST1), a serine/threonine
kinase of the sterile 20-like superfamily, has been reported to be
a stress-activated protein participating in a wide range of
apoptotic responses (5,6). MST1 plays also an important role in
mammalian development, cell cycle progression and tumorigenesis
(7). Loss or reduction of MST1
expression has been observed in head and neck squamous cell
carcinoma (8), soft tissue sarcoma
(9), glioblastoma (10) and colorectal cancers (6), along with poorer cancer prognosis
(9). Other in vivo studies
have also indicated that the conditional ablation of MST1 resulted
in liver enlargement (11).
Despite the recent advance of our knowledge on the role of MST1 in
tumorigenesis, the involvement of MST1 in other signaling pathway
and regulation of invasiveness of endometrial cancer cells is
unexplored. We report here that MST1 modulates cross-talk between
TGFβ and EGF in regulation of cell proliferation, migration and
invasiveness.
Materials and methods
Cell culture
HEC-1-A cells were obtained from American Type
Culture Collection (Manassas, VA) and were cultured in McCoy’s 5A
Modified Medium supplemented with 10% fetal bovine serum, 1%
penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO). Human
recombinant TGFβ1 and EGF were obtained from PeproTech (Rocky Hill,
NJ).
Cell transfection
A day before transfection HEC-1-A cells were
cultured to 50% confluence for transfection. HEC-1-A cells were
transfected with pcDNA3 control vector, wild-type and Ser82Ala
mutant MST1-3XFLAG in pRC/CMV10 in 12-well plates using
GeneJuice® transfection reagent, as recommended by the
supplier (Novagen, Darmstadt, Germany). The expression constructs
were kindly provided by Dr Zengqiang Yuan. Medium was changed 6 h
after transfection and cells were incubated in complete medium for
48 h prior to treatments.
Cell proliferation assay
Cell proliferation in response to TGFβ1 and EGF
treatment was measured by using CellTiter 96® AQueous
One Solution Cell assay (Promega) according to the manufacturer’s
recommendations. Cells were grown in McCoy’s 5A Modified Medium
supplemented with 10% FBS and 1% penicillin/streptomycin with or
without TGFβ1 (5 ng/ml) and EGF (10 ng/ml) for 24 h.
Cell apoptosis assay
Cell apoptosis was determined by using Cell Death
Detection ELISAPlus (Roche, Germany). Briefly, cell lysates were
placed in a streptavidin-coated microplate. A mixture of
anti-histone-biotin and anti-DNA-peroxidase (anti-DNA-POD) was
added and incubated for 2 h at 25°C. After removal of unbound
antibodies by washing steps, POD was determined photometrically at
405 nm with ABTS as substrate.
Wound healing assay
Cells were grown in culture media containing 10% of
fetal bovine serum for 48 h, until the cells reached the
confluence. Monolayers of confluent cultures were scratched with a
20 μl-pipette tip, and images of the areas of scratches were taken
under a microscope. TGFβ1 (5 ng/ml) and EGF (10 ng/ml) were added,
and the cells were incubated for 24 h. After 24 h, images of the
areas of scratches were taken. Quantification was done by measuring
the open wound area, which is the fraction of open image area at a
later time-point compared to the initial time-point, given as a
percentage using TScrach software (12).
Migration assay
Cells were seeded on the membranes of wells of the
96-well plate ChemoTx® chemotaxis system (cat. no.
#116-8; Neuro Probe Inc.) in a culture medium containing growth
factors as indicated for treatments. After 24 h, the membrane was
washed twice in PBS and fixed with 70% ethanol. The non-migrated
cells were removed by cotton swab from the upper side of the
membrane. Membrane was stained with 0.5% crystal violet, and
subsequently visualized and quantified by using ImageJ software
(13).
Invasion assay
Membranes of the 96-well plate ChemoTx chemotaxis
system (cat. no. #116-8) were covered with 3% gelatin, and cells
were seeded on the membranes in a culture medium containing 5 ng/ml
TGFβ1 and 10 ng/ml EGF. After 24 h, the membrane was washed twice
in PBS and fixed with 70% ethanol. The non-invaded cells were
removed by cotton swab from the upper side of the membrane.
Membrane was stained with 0.5% crystal violet, and subsequently
visualized and quantified by using ImageJ software (13).
Immunoblot analysis
Cell lysates were resolved in 10% SDS polyacrylamide
mini-gels (Mini-protein Tetra Cell, Bio-Rad) and transferred onto
nitrocellulose membranes (Whatman, Protran, Dassel, Germany).
Membranes were blocked with 5% (W/V) BSA in TBS-T for 1 h, and
incubated with primary antibodies against target proteins at
dilution recommended by the manufacturer, and followed by
incubation with HRP-conjugated secondary antibody (GE Healthcare,
Uppsala, Sweden). The proteins were visualized using Western
Blotting Luminol Reagents (Santa Cruz Biotechnology Inc.). The
antibodies used were: MST1 (AP7922a; Abgent), Flag M5, Smad2/3
(sc-6032), pSmad2 (sc-135644), anti-ERK1/2 (sc-135900), pERK1/2
(sc-7383), Rb (sc-50), pRb (sc-12901), vimentin (sc-7557),
E-cadherin (sc-7870), FAK (sc-558), actin (sc-8432) all from Santa
Cruz Biotechnology Inc., and anti-FAK (phospho Y397) (ab4803;
Abcam).
Systemic analysis
Systemic analysis was performed with use of
Cytoscape. GO terms of MST1, TGFβ1 and EGF were uploaded to
generate a network, with inclusion of neighbors of the uploaded
proteins, and databases were explored by Cytoscape plug-in
MiMi.
Statistical analysis
Statistical significance of observed differences was
evaluated using Mann-Whitney test among unpaired groups and among
multiple groups by the Kruskal-Wallis test followed by Dunn’s
Multiple Comparison Test. Analysis were conducted using Graph Prism
6 software (GraphPad Software, San Diego, CA) and a P<0.05 was
considered significant.
Results
Expression of MST1 in HEC-1-A human
endometrial cells
We identified earlier MST1 as a protein deregulated
in endometrial cancer (14).
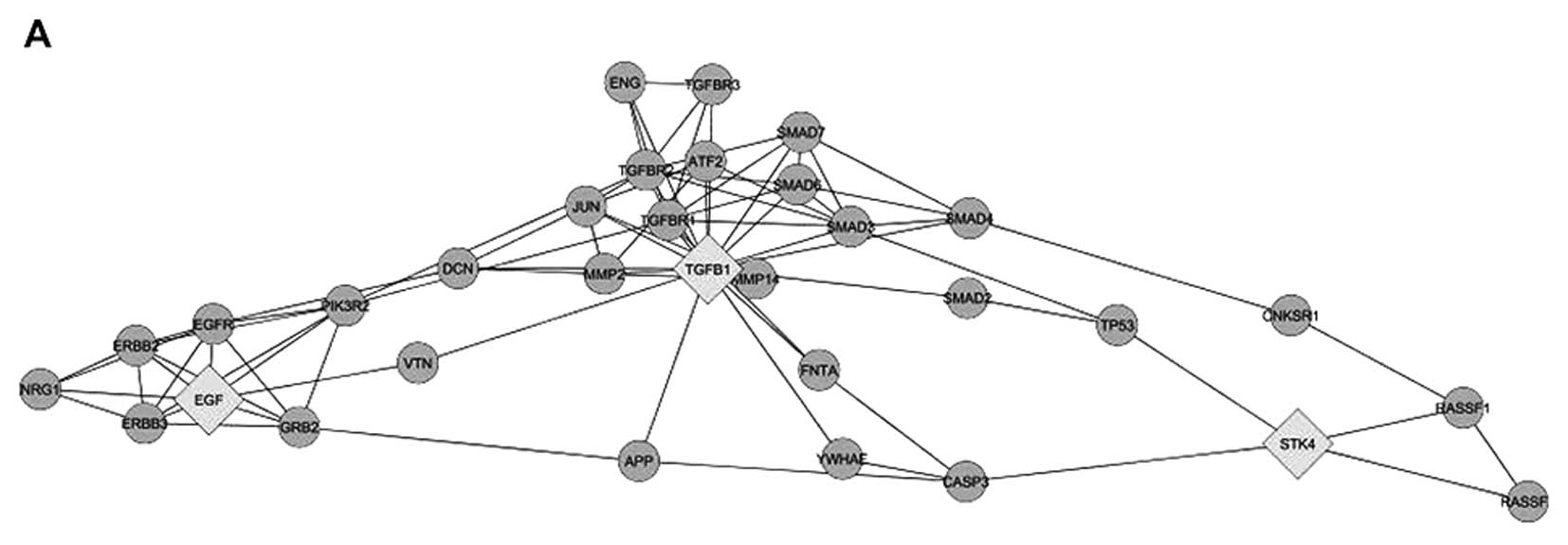
Network analysis suggested that MST1 may be involved in a
cross-talk between TGFβ and EGF (Fig.
1A). The involvement of MST1 may have an impact on cell
proliferation via regulators of Ras and on cell death via
regulation of caspase-3 and p53 (Fig.
1A). It has to be noted that molecular mechanisms of MST1
action are under extensive exploration, and therefore we may expect
further interactions between MST1, TGFβ and EGF. However, available
data are sufficient to suggest that MST1 may be involved in TGFβ
and EGF cross-talk. Therefore, to explore the role of MST1 in
responsiveness of cells to TGFβ1 and EGF, we used overexpression of
the wild-type and Ser82Ala mutant of MST1 in human endometrial
carcinoma HEC-1-A cells. The mutant MST1 was reported to have
strongly decreased phosphorylation and disrupted dimerization
(15). Both MST1 constructs were
expressed to similar levels, and their expression was not affected
by treatment of the cells with TGFβ1 and EGF alone or in
combination (Fig. 1B). This
indicates that TGFβ1 and EGF did not affect stability of the MST1
expressed in HEC-1-A cells. MST1-transfected cells showed no signs
of enhanced cell death, indicating that MST1 itself under the used
conditions did not induce cell death. Therefore, HEC-1-A cells
transfected with the wild-type and mutant MST1 were used for
further study.
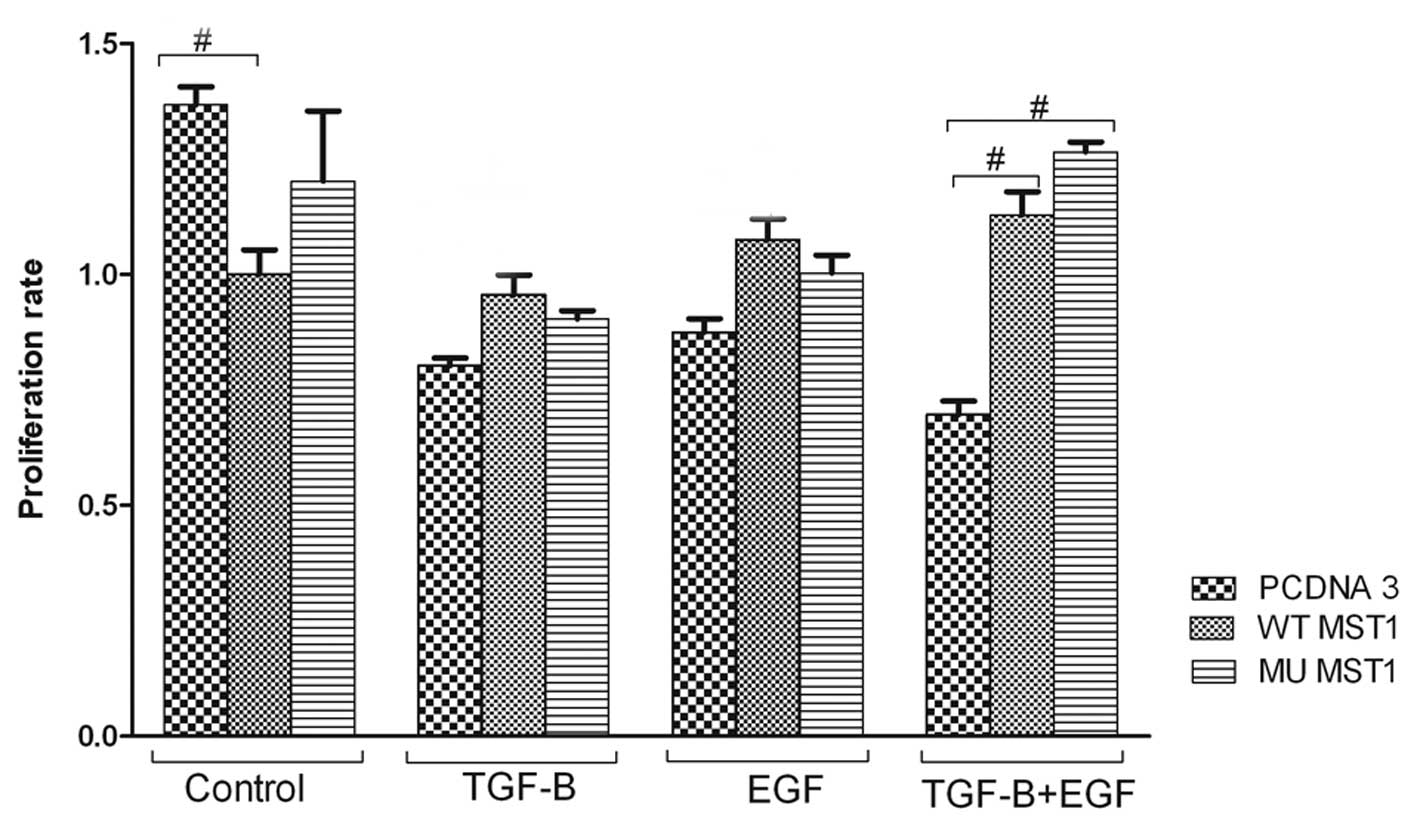
Impact of MST1 on cell proliferation
First, we explored whether MST1 may affect cellular
physiology and response to TGFβ1 and EGF in regulation of cell
proliferation, apoptosis, migration and invasiveness. To study cell
proliferation we performed MTT assay. TGFβ1 and/or EGF reduced the
proliferation rate of cells (Fig.
2). The inhibitory effect of EGF was unexpected, but it was
reproducible. The MST1 effect was rather marginal when the cells
were treated with TGFβ1 or EGF only. The wild-type MST1 inhibited
MTT activity in non-treated cells, but this effect was not observed
when cells were treated with TGFβ1 or EGF. The inhibitory effect of
combined treatment with TGFβ1 and EGF was prevented in cells
expressing WT or mutant MST1 (Fig.
2). An interesting observation is that the MST1 mutant had
similar impact as compared to the wild-type construct. These
results indicate that the mutation of Ser82 in MST1 is not
essential for the MST1 negative impact on TGFβ1 and EGF cross-talk
(Fig. 2).
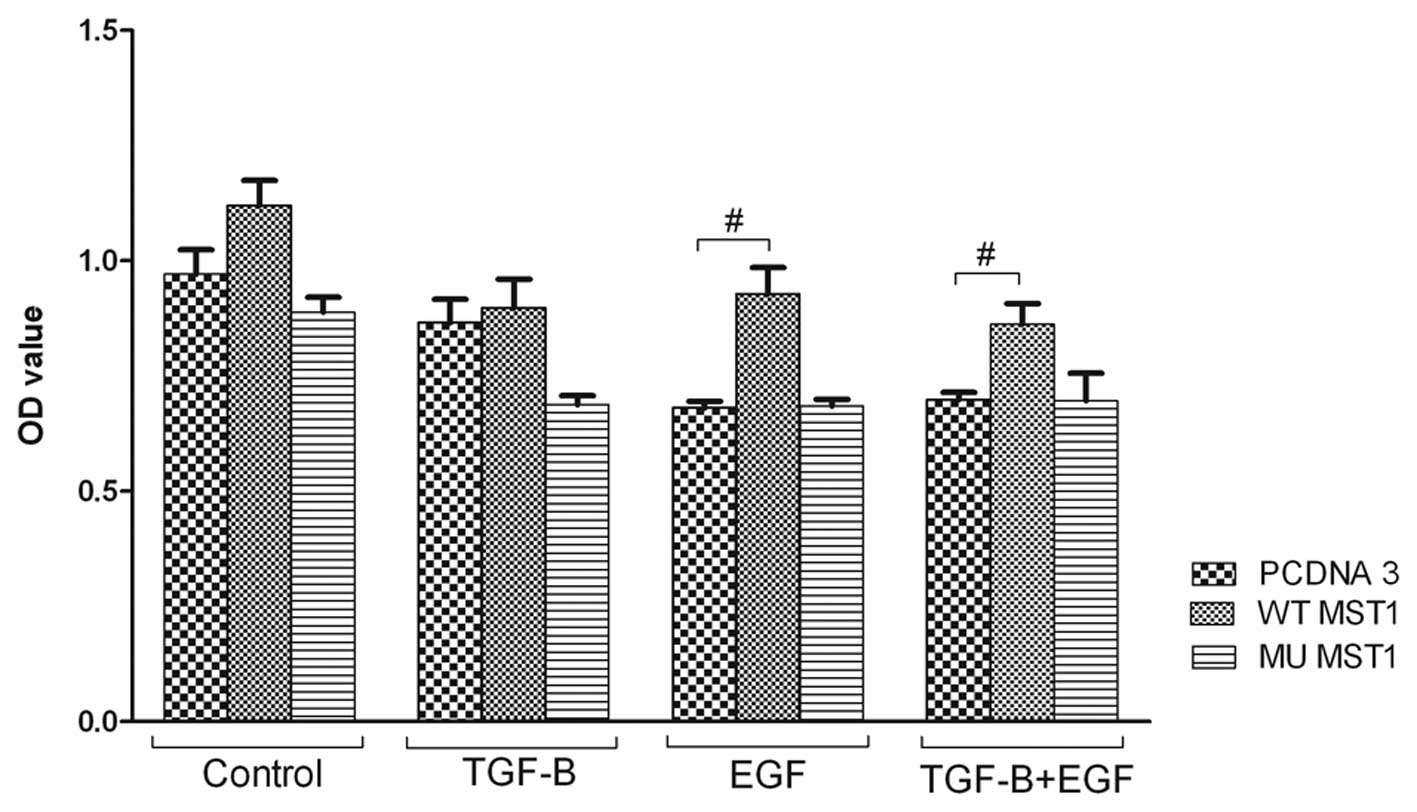
MST1 does not affect apoptosis of HEC-1-A
cells
One of the cellular mechanisms in cancer is cell
death. Therefore, we studied weather MST1 kinase affects cell
apoptosis. Expression of MST1 promoted apoptosis activity of the
cells upon treatment with EGF and TGFβ1, and showed the stimulatory
tendency for non-treated cells (Fig.
3). However, these effects were not strong, and did not affect
cellular growth. Therefore, we concluded that the effect of MST1 on
the cells death was not pronounced (Fig. 3).
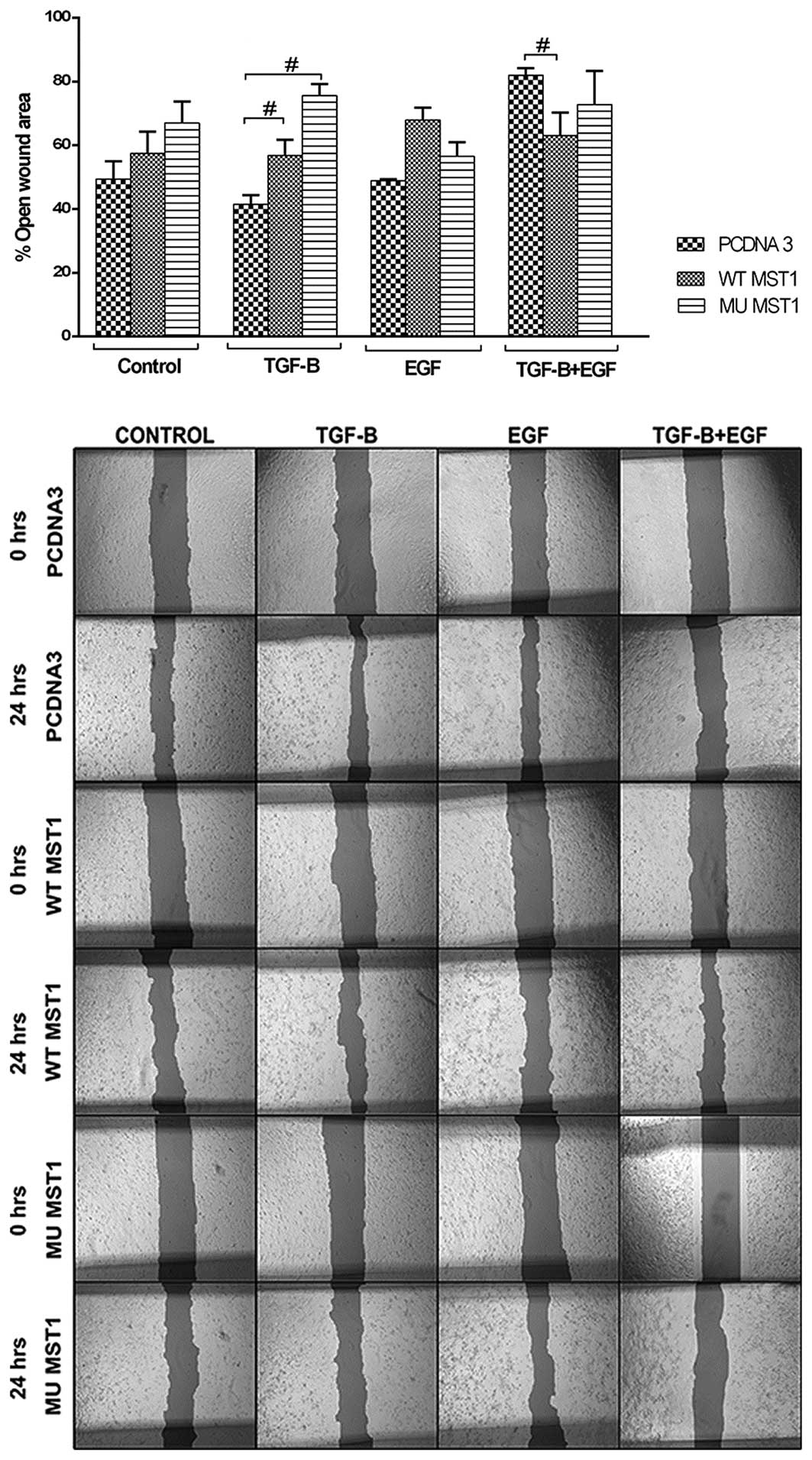
MST1 affects cell migration regulated by
TGFβ1 and EGF
To explore the effects of MST1 on cell migration, we
performed wound healing and membrane migration assays. These assays
explore cells in different conditions, e.g., in a confluent
monolayer and in a sparse culture. However, in both assays the
cells are prompted to migrate and this migration is then
measured.
Wound healing assay showed that expression of MST1
constructs inhibited cell migration in non-treated and TGFβ1 or EGF
treated cells (Fig. 4). When cells
were treated with both TGFβ1 and EGF, a significant inhibition was
observed (Fig. 4). This inhibition
was slightly counteracted upon expression of the wild-type MST1.
The most pronounced effect of TGFβ1 and EGF treatment on cell
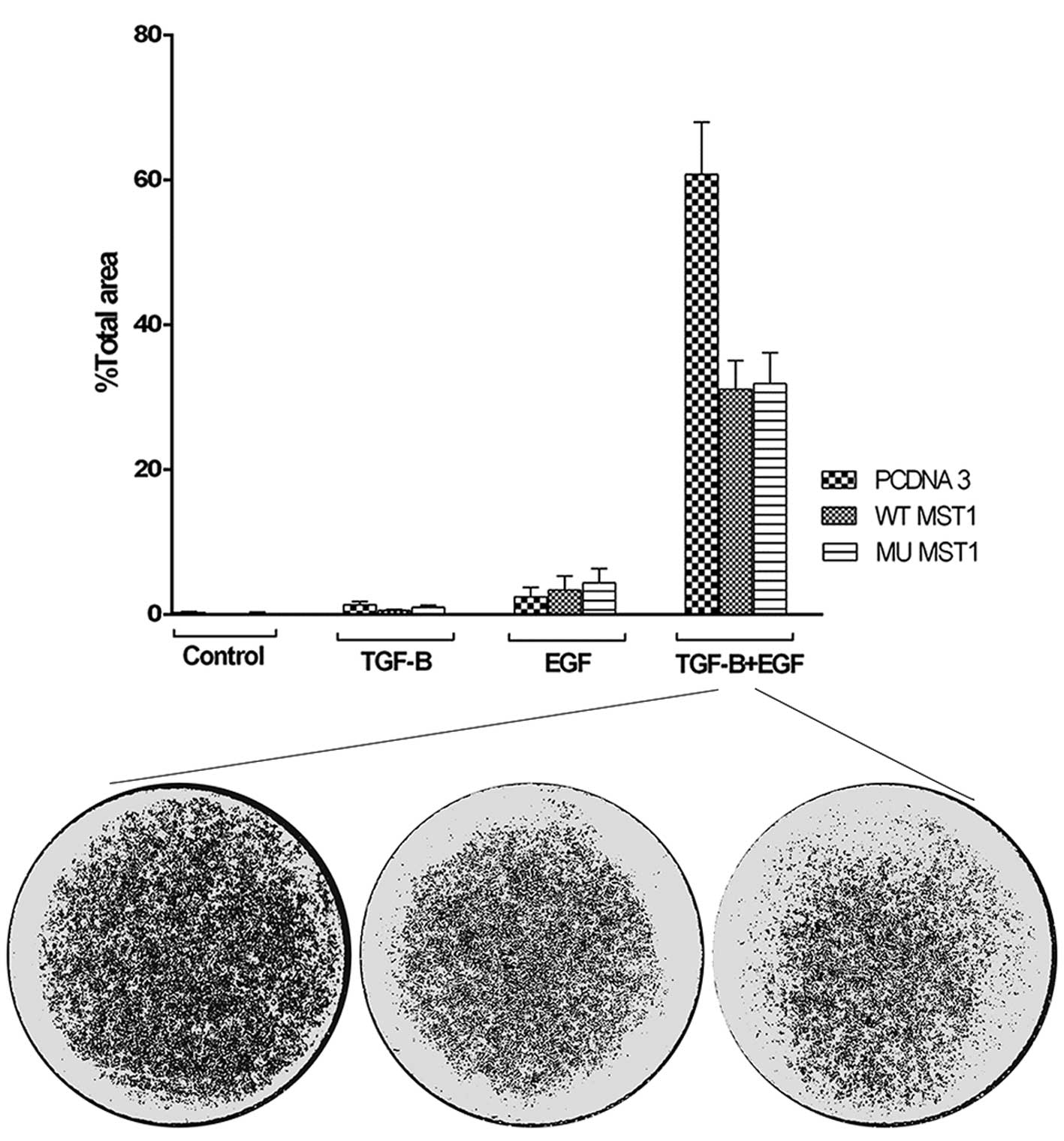
migration was observed in a membrane migration assay. We observed
that combined treatment with TGFβ1 and EGF strongly induced cell
migration, while single treatments had only marginal effect
(Fig. 5). Expression of both MST1
constructs resulted in much lower migration rate of the cells.
Mutant MST1 significantly increases cell
invasiveness
Invasiveness of cells is one of the features with
high importance for tumorigenesis. We explored invasiveness of the
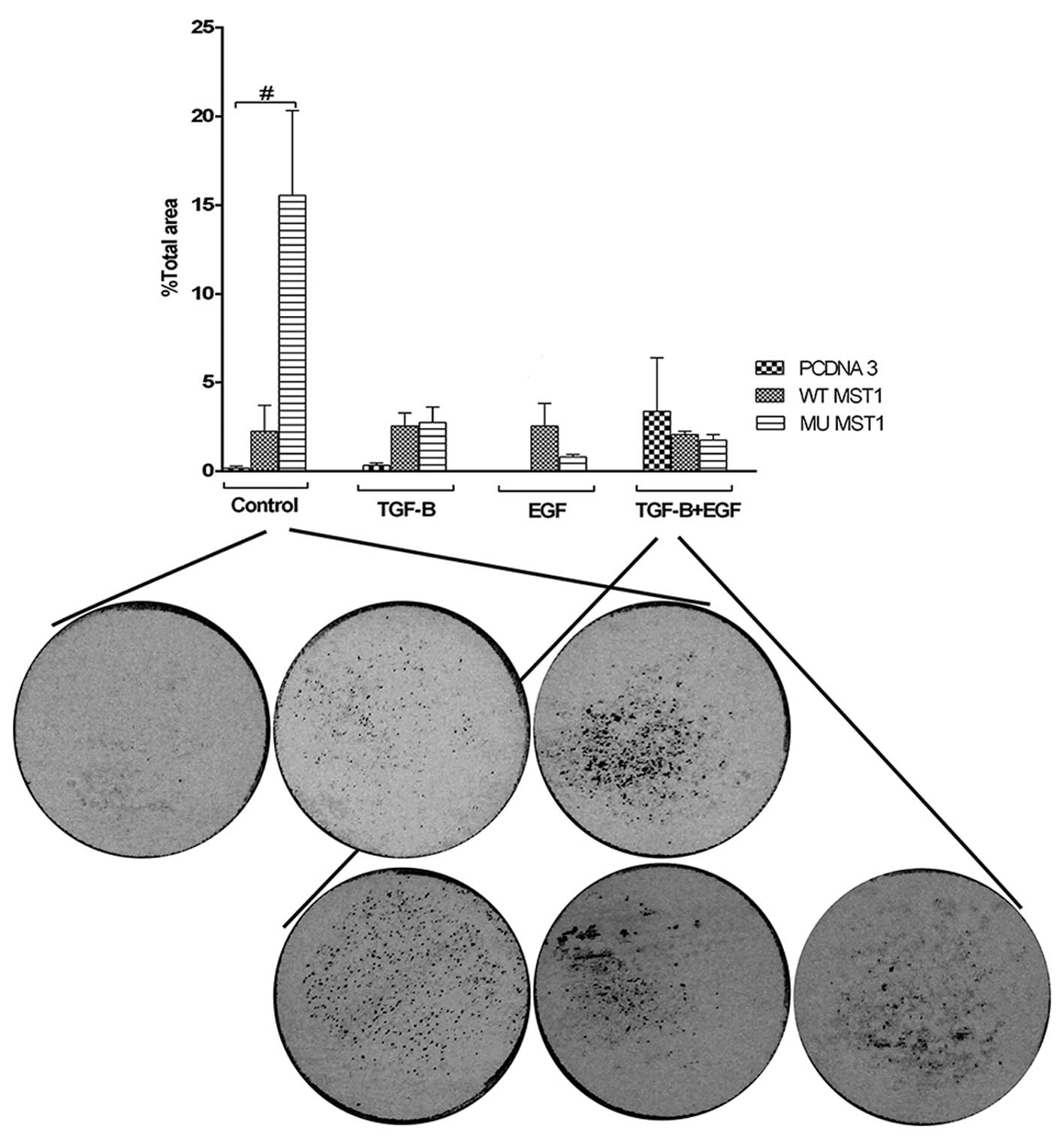
cells through a layer of denaturated collagen, i.e., gelatin. We
observed that transfection of MST1 constructs led to enhanced
invasiveness of cells, although the level of induction was
different under different treatment conditions (Fig. 6). The strongest stimulatory effect
was observed upon expression of the mutant MST1 in non-treated
cells. This stimulatory effect was abolished under treatment of
cells with TGFβ1 and/or EGF. We observed that combined treatment of
cells with TGFβ and EGF was decreased upon expression of the MST1
constructs. This observation indicates that the abrogation of
dimerization and subsequent inhibition of the kinase activity of
MST1 was not essential when cells were treated, but was of
importance for response of non-treated cells (Fig. 6). Thus, MST1 constructs
counteracted stimulatory effect on cell invasiveness of the
combined treatment with TGFβ1 and EGF.
MST1 modulation of invasiveness
correlates with phosphorylation of FAK, but not with
phosphorylation of Smad2, Erk1/2, pRb or expression of vimentin and
E-cadherin
TGFβ and EGF employ many different signal
transducers, with convergence on many common targets. To explore at
which level of the signal transduction MST1 may interfere with TGFβ
and EGF signaling, we measured expression and/or activation of
Smad2, pRb, FAK, Erk1/2 and expression of vimentin and
E-cadherin.
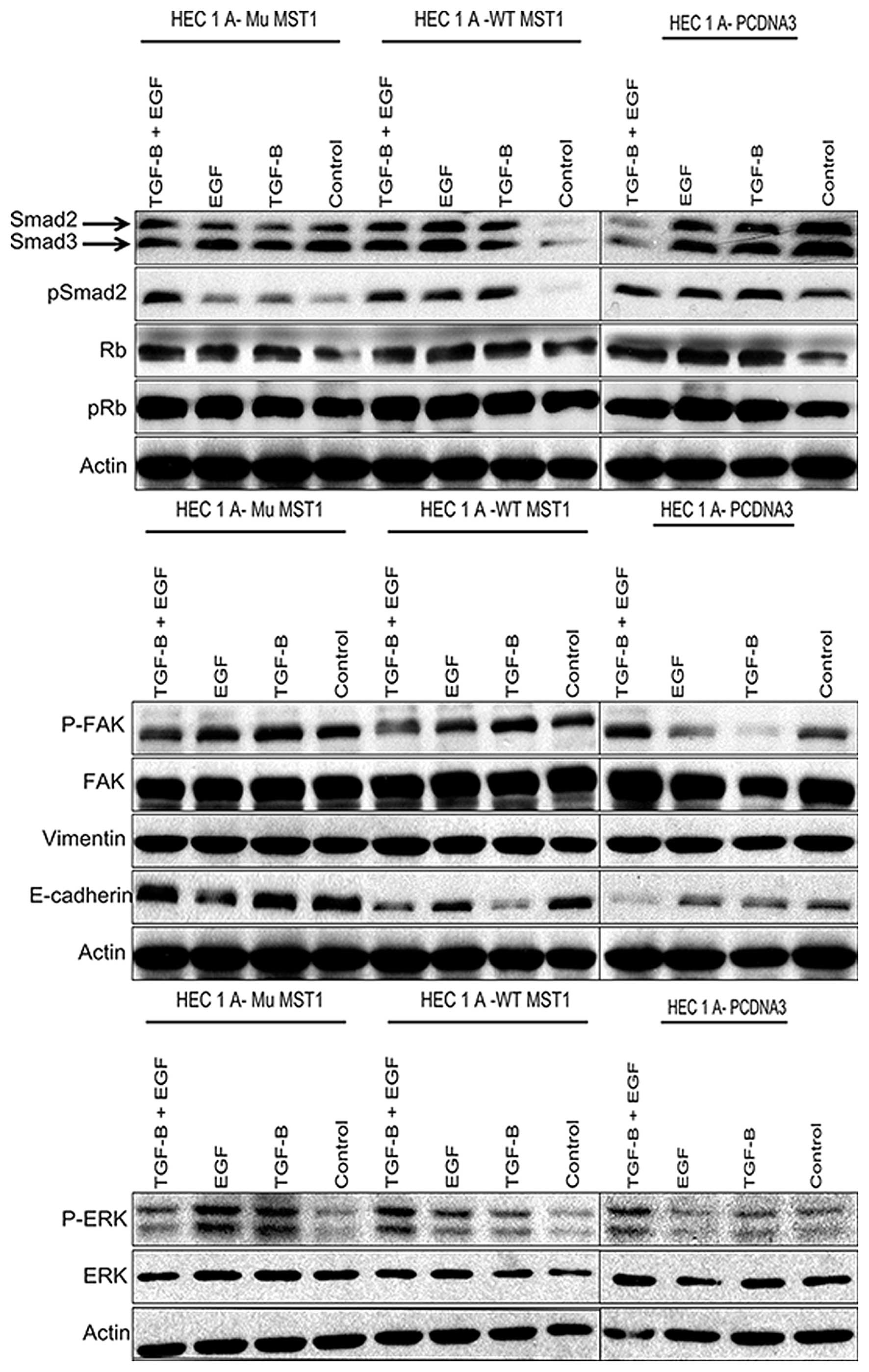
Phosphorylation of Smad2 at its C-terminal serine
residues reflects activation of signaling downstream of TGFβ
receptors (16). We observed that
only expression of the wild-type MST1 had a significant inhibitory
effect on expression of Smad2 and Smad3, and phosphorylation of
Smad2 (Fig. 7). In all other
conditions, effects on Smads were weak or not significant. This
indicates that MST1 does not affect TGFβ signaling events via Smad2
protein.
We observed no significant effect on pRb expression
and its phosphorylation on Serine780 residue in cells treated as
shown in Fig. 7. This observation
is not in line with the results of the MTT proliferation assay
(Fig. 2), and suggests that pRb
expression and Ser780 phosphorylation did not correlate with the
MST1 impact. Phosphorylation of Erk1/2 kinase correlates often with
the proliferation rate of cells. As in the case with pRb, we did
not observe a correlation of Erk1/2 phosphorylation (Fig. 7) with the results of the
proliferation assay (Fig. 2).
However, the expression of the MST1 constructs did modulate Erk1/2
phosphorylation upon treatments with TGFβ1 and/or EGF. This
suggests that Erk1/2 phosphorylation is modulated by MST1, but this
impact is not reflected in the proliferation rates of the
cells.
Measuring phosphorylation and expression of focal
adhesion kinase (FAK) allows monitoring cytoskeleton rearrangements
involved in cell migration. We observed that FAK phosphorylation
correlated with enhanced invasiveness of HEC-1-A cells transfected
or not with MST1, and treated with different combinations of TGFβ1
and EGF (Fig. 7). For the results
of migration assay, correlation was observed for all conditions,
except the non-transfected cells which showed high phosphorylation
of FAK, while no migration through the membrane was observed
(Fig. 5).
E-cadherin and vimentin are markers of
epithelial-mesenchymal phenotype of cells. They are also used as
markers to evaluate invasiveness-related epithelial-mesenchymal
transition (EMT). HEC-1-A cells show detectable expression levels
of vimentin and E-cadherin (Fig.
7). Expression of either of MST1 constructs did not modulate
vimentin expression while expression of the wild-type MST1 reduced
E-cadherin levels upon single TGFβ1 and double TGFβ1 and EGF
treatments. TGFβ-dependent inhibition of E-cadherin expression is
known as a part of TGFβ-induced EMT. The results of immunoblot
analysis for E-cadherin indicate that MST1 has modulatory impact on
TGFβ and/or EGF regulated expression of E-cadherin, but this impact
has to be combined with other regulatory processes, which then
would result in an impact on cell proliferation, migration and
invasiveness. Among the markers of intracellular signaling
pathways, studied by us, phosphorylation of FAK showed correlation
with the pattern of cell invasiveness.
Discussion
Cellular functions are controlled by combinations of
different regulators. Here we described the impact of MST1 on
functional interaction between TGFβ and EGF in regulation of cell
invasiveness, migration and proliferation. Recent studies showed
that MST1 regulates cell death, differentiation and proliferation
(17). Aberrations in MST1
expression have been observed in tumorigenesis, with indication
that MST1 may have a tumor suppressive role (6,18).
Mechanisms of MST1 intracellular signaling mechanisms are currently
under investigation in many laboratories, and one of the key
conclusions is that MST1 may play role of a coordinator between
different pathways.
EGF and TGFβ are two well-studied regulators of
tumorigenesis. EGF is predominantly tumor-promoting factor, due to
its strong pro-mitogenic activity (3). TGFβ on the contrary is a strong
inhibitor of proliferation of epithelial cells. However, TGFβ has a
double role in tumorigenesis (19,20).
At the early stages it inhibits tumor growth, while at the later
stages it promotes metastases. Extensive cross-talk between EGF and
TGFβ has been described. Intracellular regulators, which first were
considered as specific for TGFβ or EGF pathways, later were shown
to be shared between these pathways (19,20).
Identification of MST1 as a protein with potential
involvement in cross-talk of TGFβ and EGF (Fig. 1A) prompted us to explore whether
MST1 indeed affects EGF and TGFβ dependent regulation of cell
invasiveness, migration and proliferation. MST1 has been reported
to induce cell death (10,21,22).
We observed only marginal cell death induction upon transfection of
the wild-type MST1 in HEC-1-A cells (Fig. 3). Our results showed that MST1 may
act as a negative regulator of combined action of TGFβ and EGF on
cell invasiveness and migration, while its effect is not pronounced
when cells are challenged with each of the growth factors
separately (Figs. 5 and 6). This observation underlines the
importance of exploring combinatorial treatments. The challenge of
such exploration is in the high number of intracellular regulators
which have to be tested. We monitored some of the proteins which
may reflect activation of TGFβ and EGF signaling, i.e., Smad2 and
Erk1/2, and proteins reflecting migratory and invasive mechanisms,
i.e., FAK, vimentin and E-cadherin (Fig. 7). The reported exploration of the
cellular responses to the combination of TGFβ, EGF and MST1,
together with the evaluation of protein markers of signaling
pathways provide a rationale for further more elaborate mechanistic
studies. Our data include also MST1 in the network of TGFβ and EGF
signaling, which may improve prediction of responses to EGF and
TGFβ targeting drugs already used or in clinical trials for
treatment of cancer.
Acknowledgements
We are grateful to Dr Zengqiang Yuan for the gift of
the MST1 constructs. We are also grateful to the Oves Minnesfond
for support and encouragement. This study is supported in part by
grants from the Radiumhemmet research funds (#121202), the Swedish
Cancer Society, the Swedish Research Council, the Swedish
Institute, INTAS, Erasmus KI-UWM and STINT to S.S., and grants from
the regional agreement on medical training and clinical research
(ALF) between Stockholm County Council and the Karolinska Institute
and Swedish Labour Market Insurance (AFA) to M.M.
References
|
1
|
Jakowlew SB: Transforming growth
factor-beta in cancer and metastasis. Cancer Metastasis Rev.
25:435–457. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dunfield LD and Nachtigal MW: Inhibition
of the antiproliferative effect of TGFbeta by EGF in primary human
ovarian cancer cells. Oncogene. 22:4745–4751. 2003. View Article : Google Scholar
|
|
3
|
Scaltriti M and Baselga J: The epidermal
growth factor receptor pathway: a model for targeted therapy. Clin
Cancer Res. 12:5268–5272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lurje G and Lenz H-J: EGFR signaling and
drug discovery. Oncology. 77:400–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Creasy CL and Chernoff J: Cloning and
characterization of a member of the MST subfamily of Ste20-like
kinases. Gene. 167:303–306. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minoo P, Zlobec I, Baker K, Tornillo L,
Terracciano L, Jass JR and Lugli A: Prognostic significance of
mammalian sterile20-like kinase 1 in colorectal cancer. Mod Pathol.
20:331–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cinar B, Collak FK, Lopez D, et al: MST1
is a multifunctional caspase-independent inhibitor of androgenic
signaling. Cancer Res. 71:4303–4313. 2011. View Article : Google Scholar
|
|
8
|
Steinmann K, Sandner A, Schagdarsurengin U
and Dammann RH: Frequent promoter hypermethylation of tumor-related
genes in head and neck squamous cell carcinoma. Oncol Rep.
22:1519–1526. 2009.PubMed/NCBI
|
|
9
|
Seidel C, Schagdarsurengin U, Blümke K, et
al: Frequent hypermethylation of MST1 and MST2 in soft tissue
sarcoma. Mol Carcinog. 46:865–871. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiao M, Wang Y, Xu X, et al: Mst1 is an
interacting protein that mediates PHLPPs’ induced apoptosis. Mol
Cell. 38:512–523. 2010.PubMed/NCBI
|
|
11
|
Song H, Mak KK, Topol L, et al: Mammalian
Mst1 and Mst2 kinases play essential roles in organ size control
and tumor suppression. Proc Natl Acad Sci USA. 107:1431–1436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gebäck T, Schulz MMP, Koumoutsakos P and
Detmar M: TScratch: a novel and simple software tool for automated
analysis of monolayer wound healing assays. Biotechniques.
46:265–274. 2009.PubMed/NCBI
|
|
13
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012.PubMed/NCBI
|
|
14
|
Attarha S, Andersson S, Mints M and
Souchelnytskyi S: Individualised proteome profiling of human
endometrial tumours improves detection of new prognostic markers.
Br J Cancer. 109:704–713. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bi W, Xiao L, Jia Y, et al: c-Jun
N-terminal kinase enhances MST1-mediated pro-apoptotic signaling
through phosphorylation at serine 82. J Biol Chem. 285:6259–6264.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Souchelnytskyi S, Tamaki K, Engström U,
Wernstedt C, ten Dijke P and Heldin CH: Phosphorylation of Ser465
and Ser467 in the C terminus of Smad2 mediates interaction with
Smad4 and is required for transforming growth factor-beta
signaling. J Biol Chem. 272:28107–28115. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin F, Tian J, Zhou D and Chen L: Mst1 and
Mst2 kinases: regulations and diseases. Cell Biosci. 3:312013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ng Y-K, Lau W-S, Lui VWY, et al:
Full-length Mst1 exhibits growth promoting function in human
hepatocellular carcinoma cells. FEBS Lett. 587:496–503. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Katsuno T, Ochi M, Tominaga K, et al:
Mesenchymal stem cells administered in the early phase of
tumorigenesis inhibit colorectal tumor development in rats. J Clin
Biochem Nutr. 53:170–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Massagué J: TGF-β signaling in development
and disease. FEBS Lett. 586:18332012.
|
|
21
|
Graves JD, Gotoh Y, Draves KE, et al:
Caspase-mediated activation and induction of apoptosis by the
mammalian Ste20-like kinase Mst1. EMBO J. 17:2224–2234. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin Y, Khokhlatchev A, Figeys D and Avruch
J: Death-associated protein 4 binds MST1 and augments MST1-induced
apoptosis. J Biol Chem. 277:47991–8001. 2002. View Article : Google Scholar : PubMed/NCBI
|