Introduction
Multiple myeloma (MM) is a clonal plasma cell
disorder affecting both the immune system and bone metabolism, and
it remains incurable even with high dose chemotherapy (1,2).
Chemoresistance of MM could be, in part, attributed to the
overexpression of anti-apoptotic protein, BCL-2 (3–5). In
fact, >80% of the patients with MM overexpress BCL-2 protein,
and increased level of BCL-2 is associated with poor overall
survival (6).
Human BCL-2 gene is located on chromosome 18,
at the breakpoint of t(14;18), a chromosomal translocation that was
first discovered in follicular lymphoma (7,8).
This translocation juxtaposes BCL-2 gene to the enhancer of
immunoglobulin heavy chain gene, resulting in the overexpression of
BCL-2 protein. Notably, overexpression of BCL-2 is not only
observed in tumors with t(14;18), but frequently seen in a variety
of hematological malignancies without t(14;18) (9,10).
Increased BCL-2 protein in tumor cells stabilizes mitochondrial
membrane and prevents the release of cytochrome c from
mitochondria, consequently interrupting the intrinsic apoptotic
signaling cascade (11–13). Therefore, downregulation of BCL-2
protein can restore intrinsic apoptotic pathways and resensitize
tumor cells to apoptosis (10). In
fact, current therapeutic approaches to boost apoptosis in
malignant cells often target BCL-2 family members, and such
approach is particularly effective for tumors overexpressing Bcl-2
including MM (14).
Gossypol (Gos) is a promising anticancer agent
presently under clinical trial (15). Gos is a natural polyphenol compound
extracted from cottonseeds (16),
which was initially investigated in China as a male contraceptive
agent (17). Gos is a natural BH3
mimetics, acting as a small molecule inhibitor for the interaction
between anti-apoptotic Bcl-2/Bcl-XL/Mcl-1 and
pro-apoptotic BH3-only proteins such as BIM, BID, BAD or BIK,
thereby induces apoptosis in cancer cells (18). Recent observations indicated
antiproliferative or antimetastatic activity of Gos in several
tumor types, including human breast carcinoma, colon carcinoma,
leukemia, adrenocortical carcinoma, glioma, and prostate cancer
(19–26). Treatment of tumor cells with Gos
resulted in cell cycle arrest at G0/G1 phase, and one report
suggested that Gos induced apoptotic cell death in leukemia cells,
possibly through protein kinase C (PKC) pathway (27). However, studies on Gos-induced
apoptosis are still limited and precise molecular circuitry of
Gos-induced apoptosis, particularly in MM cells, remains
elusive.
In the present study, we tried to elucidate the
signaling pathways regulating Gos-induced apoptosis in MM cells.
Besides a role as BH3 mimetics, we found that Gos inhibits
interleukin (IL)-6 signaling, thereby dephosphorylates Bcl-2 and
downregulates Mcl-1. This suggests that inhibition of IL-6
signaling may be an alternative mechanism for Gos-induced apoptosis
in MM cells besides the interference of BH3-dependent
interaction.
Materials and methods
Cells and cultures
Human myeloma cell line (OPM2) was obtained from the
Japan Cancer Research Resources Bank (Tokyo, Japan). Cells were
maintained in RPMI-1640 medium (Sigma, St. Louis, MO, USA)
supplemented with 10% fetal bovine serum (FBS; Sigma), 100 U/ml
penicillin, and 100 μg/ml streptomycin in a humidified atmosphere
with 5% CO2. Cell morphology was examined by staining
cytospin preparation of the cells with Giemsa solution. Viability
of the cells was evaluated by trypan blue dye exclusion method.
Reagents
Gossypol was purchased from Sigma and dissolved in
DMSO at a stock concentration of 100 mM, which was stored at −30°C.
The pan-caspase inhibitor Z-VAD-FMK, and JAK2 inhibitor AG490 were
from Calbiochem (La Jolla, CA, USA).
Antibodies
The antibodies for caspases-3, caspase-8, STAT3,
pTyr705-STAT3, pSer727-STAT3, pSer70-bcl2, Bid, Bad, Akt, p38MAPK,
pThr180/Tyr182-p38MAPK, cytochrome c, were from Cell
Signaling Technology (Beverly, MA, USA). Antibodies for Bax and
p21CIP1 were from MBL (Nagoya, Japan). Antibodies for
β-actin, Rb, Mcl-1, bcl-2, bcl-xL, p-gp130, JAK2, Cyclin D2, cyclin
E, CDK2, CDK4, PKCα, PP2A/Aα, PP2A/B56α, PP2A/C were from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for p-JAK2 or
p27kip1 were from Upstate (Waltham, MA, USA) or BD
Transduction Laboratories, respectively. Anti-ERK1/2 or p-ERK1/2
antibody was from Sigma. Secondary antibodies conjugated with
horseradish peroxidase were obtained from GE Healthcare (Tokyo,
Japan).
Apoptosis assay
Apoptosis was examined by cellular morphology or
staining cells with Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) by Annexin V staining kit (BD Bioscience
Pharmingen, San Diego, CA, USA) according to the manufacturer’s
protocol. Stained cells were analyzed by FACSCalibur
(Becton-Dickinson, San Jose, CA, USA) with CellQuest software
(Becton-Dickinson).
For DNA fragmentation assay, cells were harvested
and incubated in a lysis buffer [10 nM Tris-HCl (pH 7.4), 10 mM
EDTA, 0.5% Triton 100-X] at 4°C, which were then centrifuged at
15,000 rpm for 15 min at 4°C. Supernatants were collected and
incubated with RNase A (Sigma) at 50 μg/ml and proteinase K (Sigma)
for 1 h at 37°C. DNA samples were subjected to 2% agarose gel and
were visualized by ethidium bromide staining.
For pharmacological inhibition of apoptosis, cells
were pre-incubated with 20 μM of pan-caspase inhibitor, Z-VAD-FMK
for 2 h prior to addition of Gossypol (5 μM). The final
concentration of DMSO in the experiment did not exceed 0.1%. Effect
of Z-VAD-FMK was assessed by DNA fragmentation assay.
Assay for mitochondrial transmembrane
potential (MMP)
Cells were washed with PBS and subjected to staining
with 40 nM of DioC6 (Sigma-Aldrich Japan, Tokyo, Japan) for 30 min
at 37°C. The stained cells were washed with PBS and analyzed by
flow cytometry.
Cell cycle analysis
Cells were suspended in hypotonic solution [0.1%
Triton X-100, 1 mM Tris-HCl (pH 8.0), 3.4 mM sodium citrate, 0.1 mM
EDTA] and stained with 50 μg/ml of PI. Cells were analyzed by flow
cytometry and the population of cells in each cell cycle phase was
determined using ModiFIT software (Becton-Dickinson).
Immunoblotting
Cells were lysed in a lysis buffer [1% NP40, 1 mM
phenylmethylsulfonyl fluoride (PMSF), 40 mM Tris-HCl (pH 8.0) and
150 mM NaCl] at 4°C for 15 min. Mitochondria and cytosol were
fractionated using the Mitochondria/Cytosol Fractionation kit
(BioVision Inc., Mountain View, CA, USA). Cell lysates (15 μg of
protein/lane) were fractionated in SDS-polyacrylamide gels and were
transferred onto the nylon membranes (Immobilon-P; Millipore,
Bedford, MA, USA). Membranes were probed with primary antibodies
and horseradish peroxidase labeled secondary antibodies as
described previously. Bound antibodies were detected by enhanced
chemiluminescence (ECL) kit (Amersham, Buckinghamshire, UK).
Statistical analysis
Data are expressed as mean ± standard deviation (SD)
Statistical analyses were performed by unpaired Student’s t-test.
P-values <0.05 were considered statistically significant.
Results
Gossypol induces apoptosis in multiple
myeloma cells
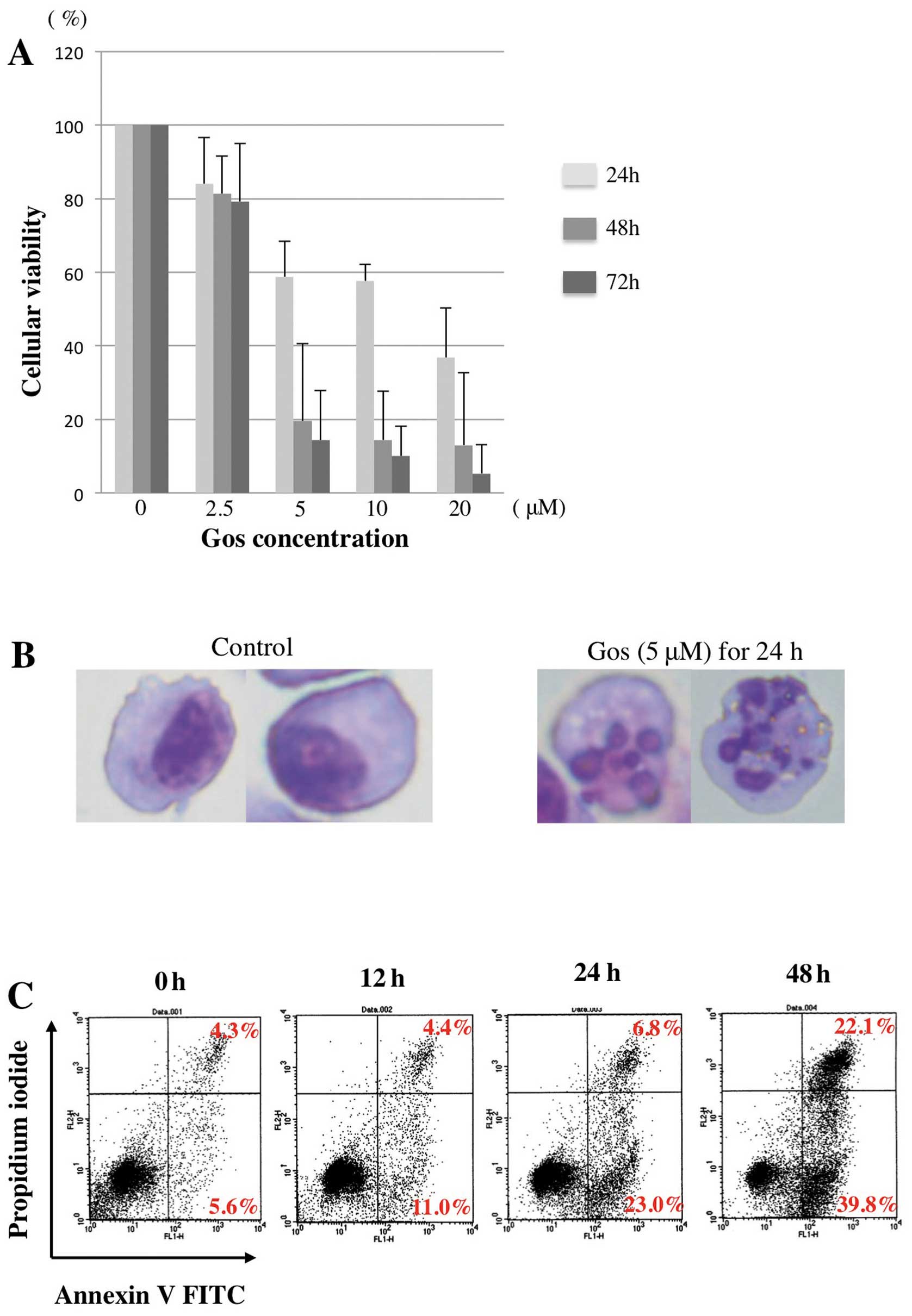
We first examined the effect of Gos on the
proliferation of MM cell line, OPM2 cells. As shown in Fig. 1A, Gos inhibited the proliferation
of OPM2 cells in a dose- and time-dependent manner. Morphological
examination revealed that treatment of OPM2 cells with 5 μM of Gos
for 24 h clearly induced cell death with nuclear fragmentation,
typical appearance of apoptosis (Fig.
1B). Furthermore, proportions of Annexin
V+/PI− as well as Annexin
V+/PI+ cells were strikingly increased by
48-h treatment of Gos (Fig. 1C).
Taken together, these results indicated that Gos induces apoptosis
in OPM2 cells.
Effect of Gos on cell cycle progression
in myeloma cells
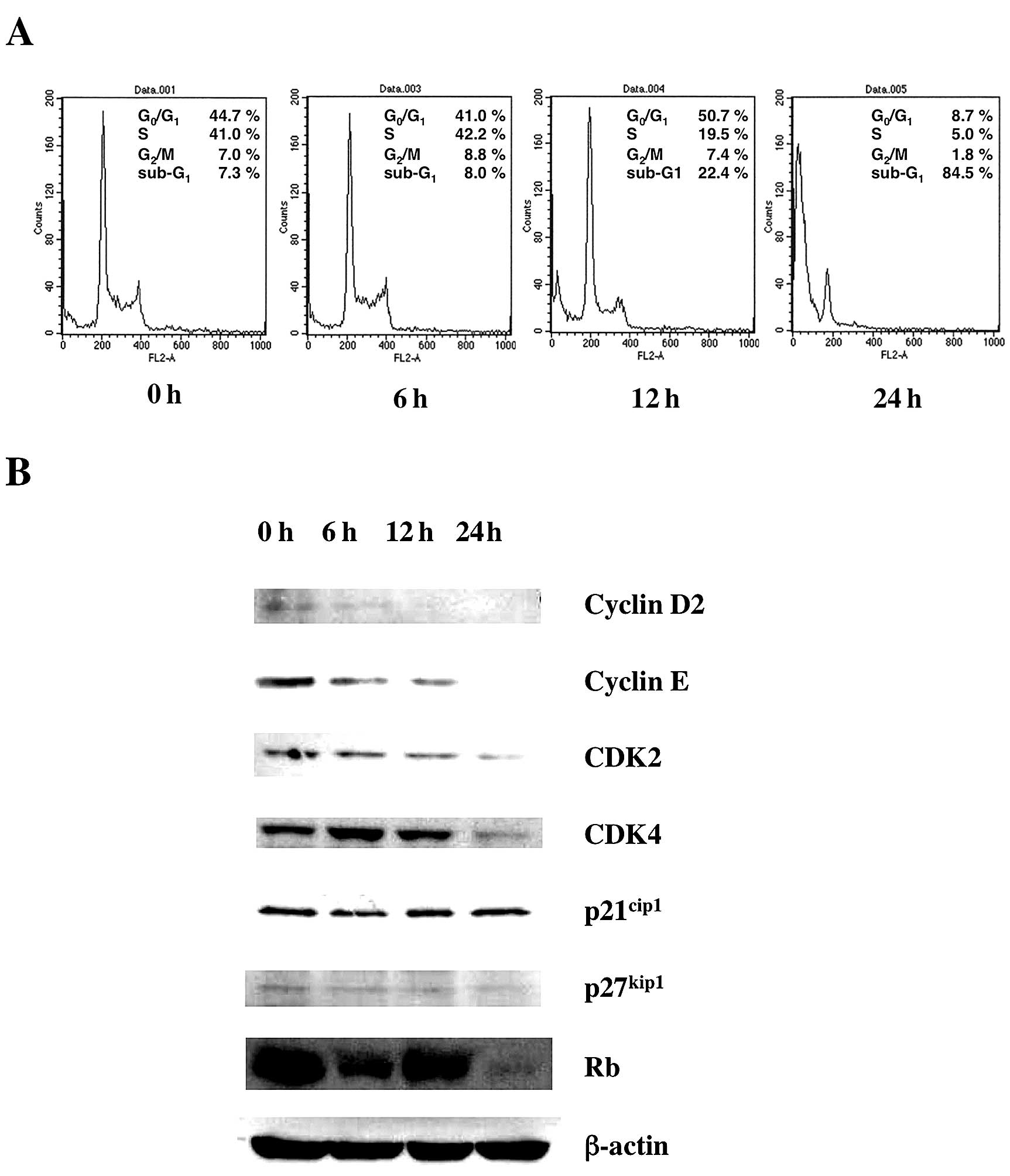
Next we examined the effect of Gos on the cell cycle
status of OPM2 cells. Cells were incubated with Gos and the cell
cycle status was examined by flow cytometry at various time-points
(Fig. 2A). The results
demonstrated that Gos induced depletion of cells in S/G2/M phase
and dramatic increase of cells in sub-G1 after 24 h of treatment.
These results suggest that Gos induced cell cycle arrest at G1
followed by apoptosis in OPM2 cells.
To investigate the molecular mechanism of
Gos-induced cell cycle arrest in OPM2 cells, the expression of cell
cycle-associated genes was examined by western blotting. As shown
in Fig. 2B, the expressions of
Cyclin D2, Cyclin E, CDK2, CDK4 and Rb were decreased by
Gos-treatment, whereas p21 and p27 were unchanged. These results
indicate that Gos affected molecules driving cell cycle progression
rather than inhibitors of cell cycle.
Gossypol-induced apoptosis involves
activation of caspase-3 and -8
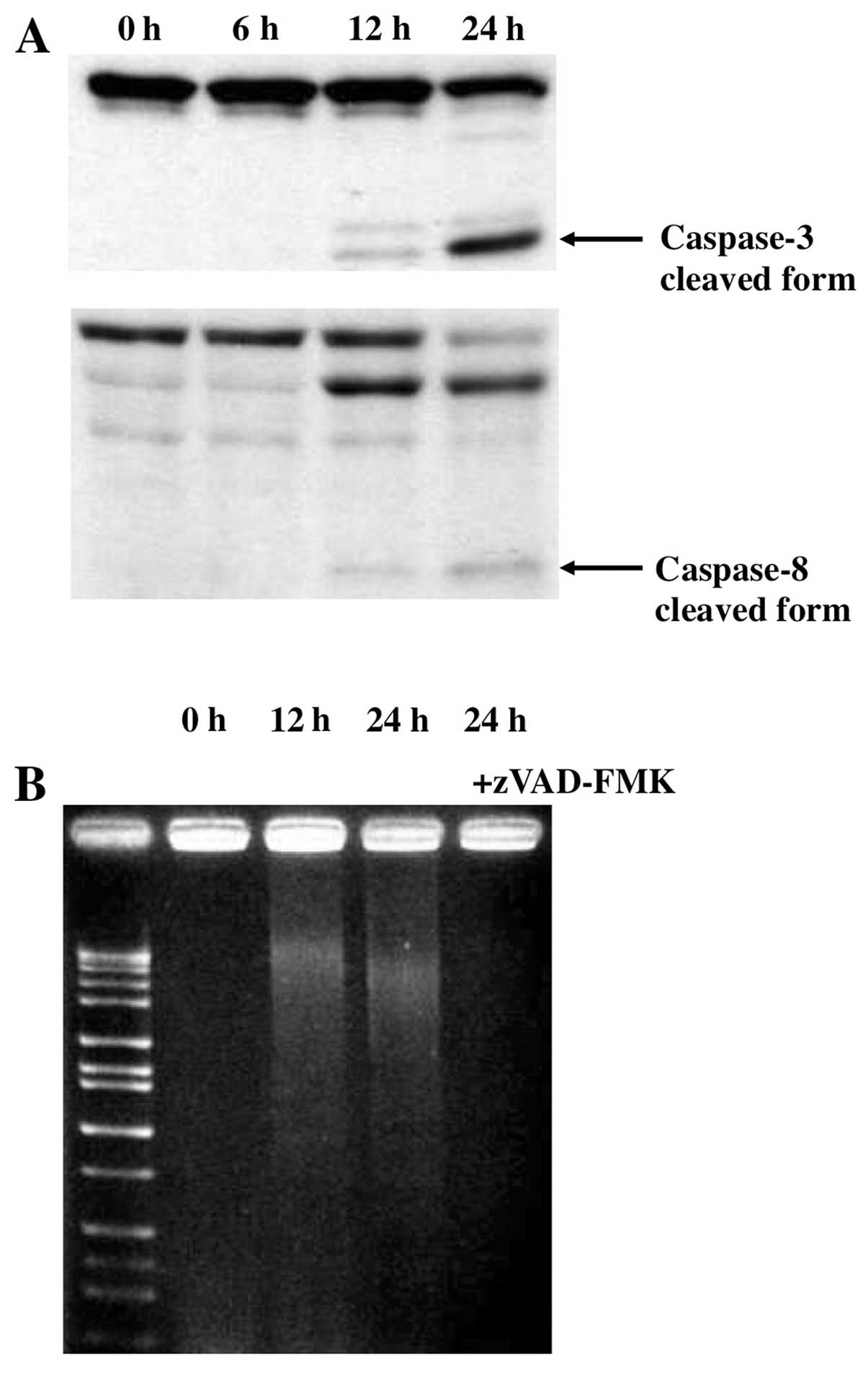
It is well known that the activation of caspases
plays a pivotal role in the apoptosis-signaling pathway (28). We therefore examined the activation
of caspases-3, a common effector caspase that integrates various
death signals, during Gos-induced apoptosis in OPM2 cells. Active
cleaved form of caspase-3 appeared from 12 h of Gos-treatment and
became evident at 24 h (Fig. 3A).
Furthermore, we found that Gos activated caspase-8, a mediator of
death receptor-mediated apoptosis pathways, as shown by cleavage of
the pro-form into the active form (Fig. 3A). The caspase activation was
associated with fragmentation of DNA as shown by Fig. 3B. To further examine the role of
caspases in Gos-induced apoptosis, we asked if caspase inhibitor
could suppress the apoptotic processes in OPM2 cells. As expected,
Gos-induced apoptosis in OPM2 cells as assessed by DNA
fragmentation was completely inhibited by a pan-caspase inhibitor,
Z-VAD-FMK (Fig. 3B). Of note,
Z-VAD-FMK, when used alone, did not exert any effect on the
proliferation of OPM2 cells (data not shown).
Mitochondrial changes and decrease of Bid
protein during Gos-induced apoptosis
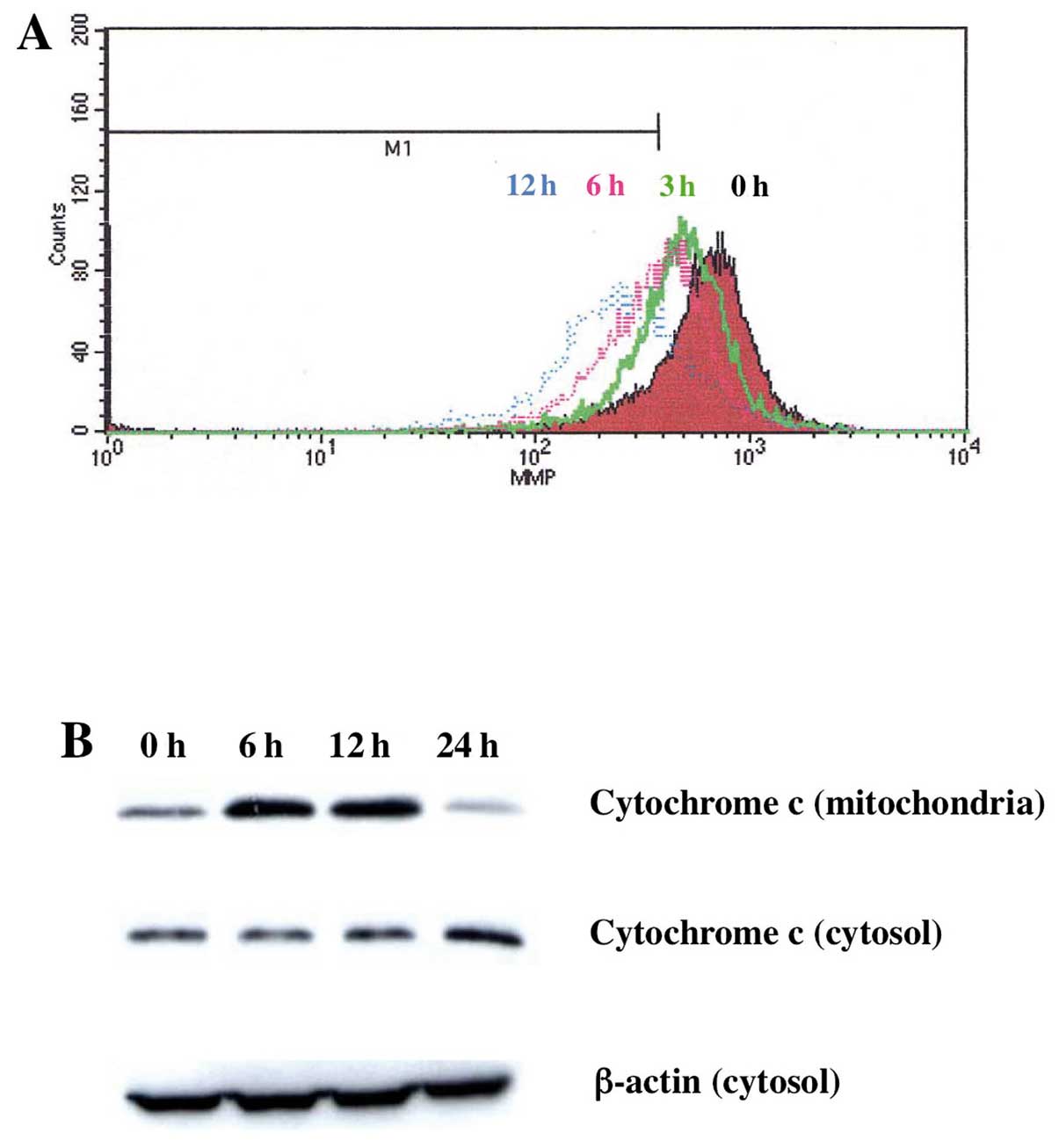
To examine the activation of mitochondrial apoptosis
pathway by Gos, we next examined the mitochondrial changes evoked
by Gos treatment. Mitochondrial changes during apoptosis include
permeability transition pore opening and the collapse of the
mitochondrial transmembrane potential (ΔΨm), which
results in the release of cytochrome c into the cytosol and
subsequent activation of caspases. In order to examine these
processes, we measured changes of mitochondrial ΔΨm in
OPM2 cells treated with Gos by flow cytometry using DioC6. By
treatment with 5 μM of Gos, ΔΨm of mitochondria
decreased in a time-dependent manner in OPM2 cells (Fig. 4A). Furthermore, the decrease of
ΔΨm was accompanied by the release of cytochrome
c from mitochondria to cytosol (Fig. 4B). These results suggested that
Gos-induced apoptosis is, at least in part, mediated through a
mitochondria-dependent pathway.
We have also reconfirmed the activation of death
receptor mediated pathways as shown by the activation of caspase-8
in Gos-induced apoptosis by checking the expression of Bid protein,
since Bid is a substrate of activated caspase-8. As shown in
Fig. 5A, Bid was clearly
downregulated during the Gos-treatment. This result, together with
the activation of caspase 8, indicated that the death
receptor-mediated signaling pathway was also operational in the
Gos-induced apoptosis in MM cells. Taken together, these results
showed that Gos-induced apoptosis involved both mitochondria- and
death receptor-dependent pathways.
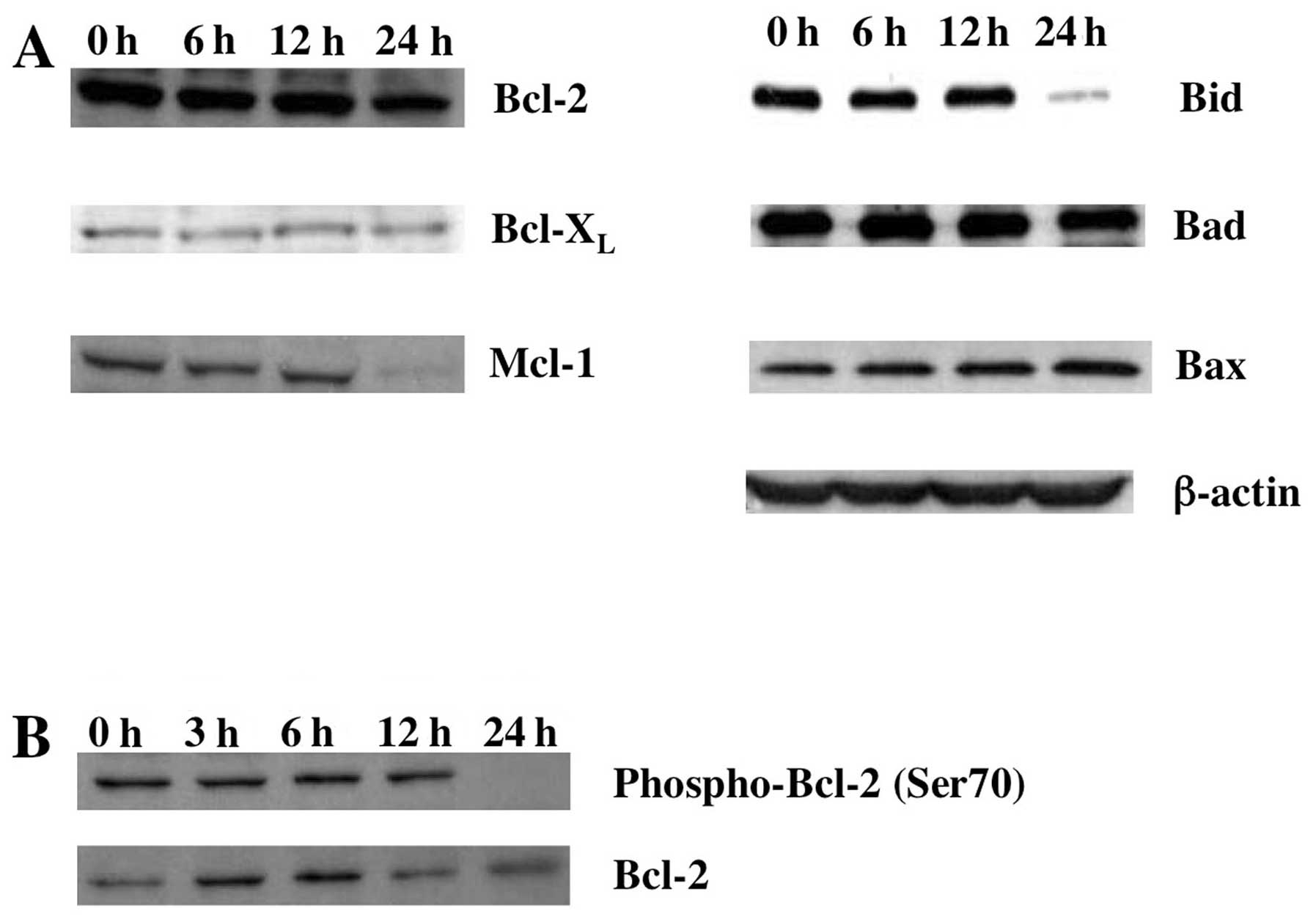
Expression of apoptosis-associated
proteins in Gos-treated OPM2 cells
We next examined the expression of
apoptosis-associated proteins during Gos-induced apoptosis in OPM2
cells. In addition to the decrease of Bid protein as shown above,
MCL-1, one of the major anti-apoptotic proteins in hematopoietic
cells, clearly decreased by 24 h of Gos-treatment. In contrast,
however, Gos did not affect the levels of pro-apoptotic Bax and Bad
protein as well as anti-apoptotic Bcl-2 and Bcl-XL
proteins (Fig. 5A).
Although protein levels did not change by
Gos-treatment, we suspected that BCL-2 might be functionally
impaired, since anti-apoptotic function of BCL-2 can be regulated
by phosphorylation at serine 70 (Ser70) (29,30).
In fact, Ser70 phosphorylation of BCL-2 was downregulated by 24 h
of Gos-treatment (Fig. 5B),
indicating that anti-apoptotic function of BCL-2 was perturbed by
Gos-treatment. Taken together, these results suggest that
downregulation of MCL1 and dephosphorylation of BCL-2 are
responsible for Gos-induced apoptosis.
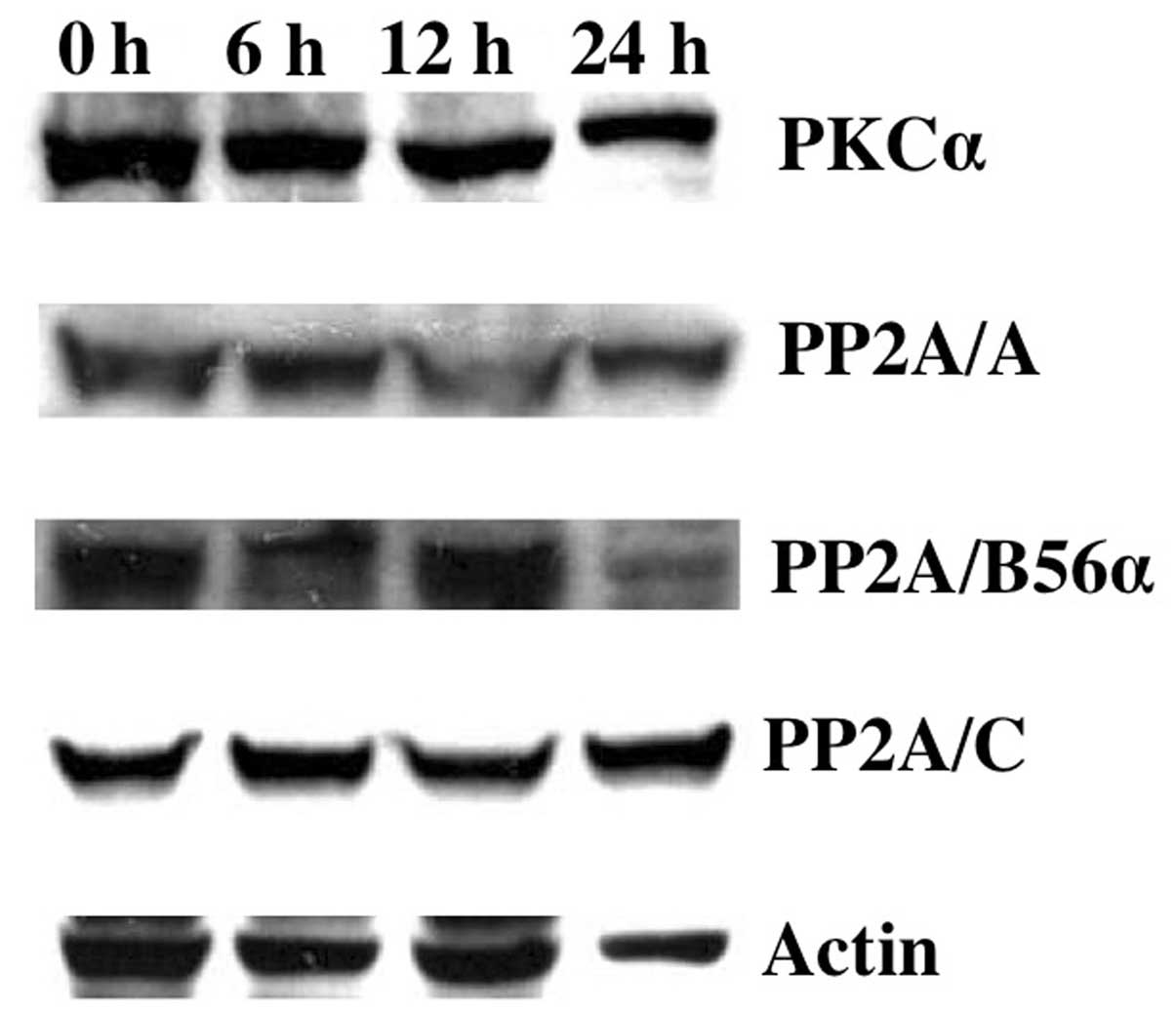
Downregulation of ERK1/2 signaling by Gos
may be responsible for BCL-2 dephosphorylation
Recent studies have shown that ERK1/2, p38MAPK,
protein kinase C (PKC) or protein phosphatase (PP) 2A were involved
in the phosphorylation or dephosphorylation of BCL-2, respectively
(31,32). To investigate the possible
molecules regulating BCL-2 dephosphorylation during Gos-treatment,
we first analyzed the expression levels of PKCα and PP2A in OPM2
cells. The results showed that protein levels of PKCα, PP2A/A,
PP2A/B56α or PP2A/C did not change during 24 h of Gos-treatment
(Fig. 6), suggesting that these
enzymes did not affect the phosphorylation status of BCL-2 protein.
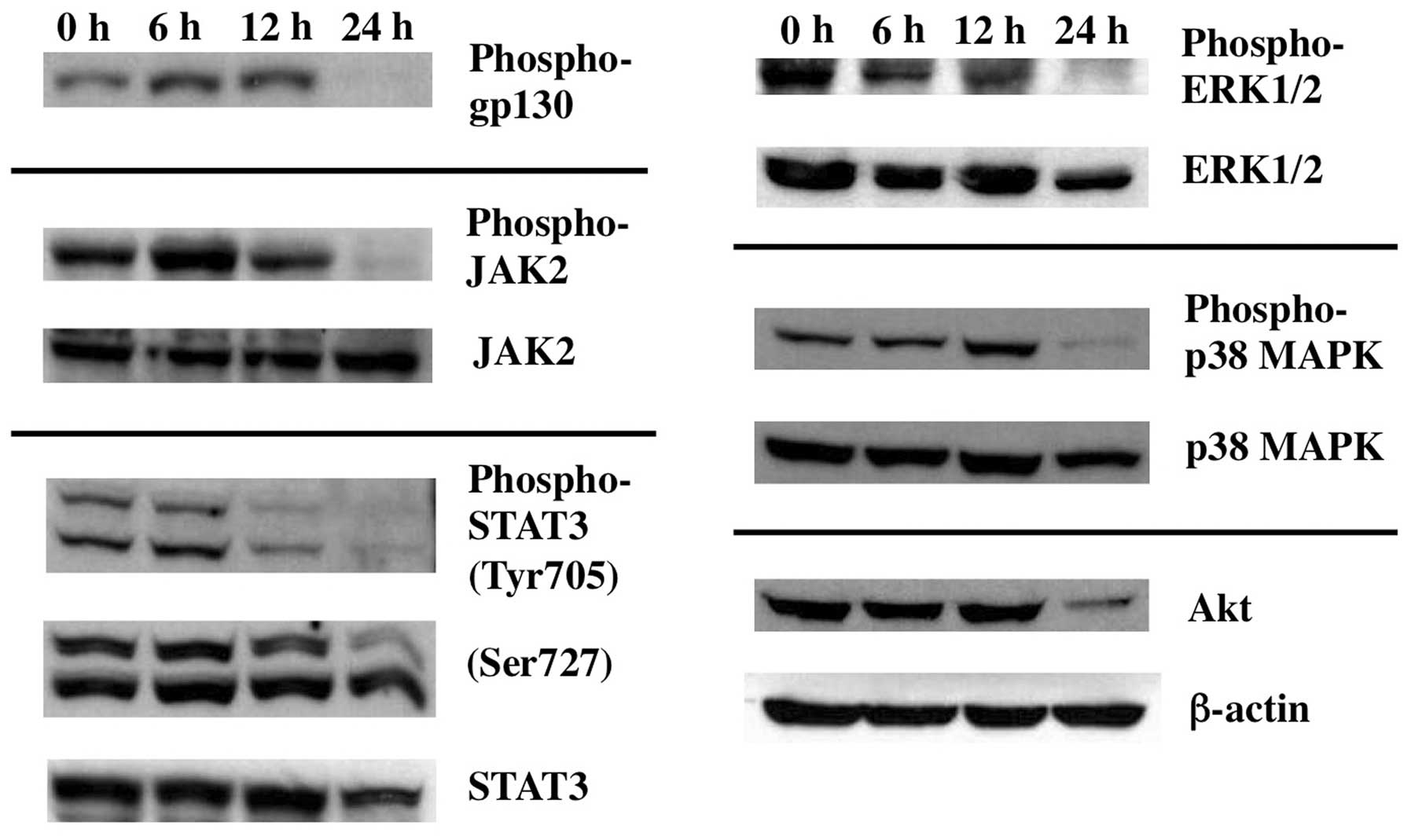
In contrast, however, activation of ERK1/2 as examined by its
phosphorylated form, was clearly decreased at 24 h of Gos-treatment
(Fig. 7). Taken together, these
results suggest that Gos induces dephosphorylation of BCL-2 through
inhibition of ERK1/2.
Inhibition of interleukin-6 signaling by
Gos
It is well known that interleukin (IL)-6 plays a
critical role in the proliferation of myeloma cells including OPM2
(33). To further gain insight
into the upstream signaling pathways regulating Gos-induced
apoptosis, we investigated the effect of Gos on the IL-6 signaling
in OPM2 cells. IL-6 regulates cellular survival through three major
signaling pathways: JAK2/STAT3, Ras/Raf/MEK/MAPK, and
phosphoinositide-3 kinase (PI-3K)/Akt (34). Interestingly, treatment of OPM2
cells with Gos for 24 h inhibited phosphorylation of JAK2 and IL-6
signaling receptor, gp130, as well as the downstream signaling
effectors such as STAT3, p38MAPK and ERK1/2 as mentioned earlier
(Fig. 7). Downregulation of
PI-3K/Akt pathway was also evident, since protein level of Akt
itself decreased by Gos-treatment. These results suggest that Gos
inhibits IL-6 signaling at a level between the ligand-receptor
interaction and the activation of JAK2, which then leads to
apoptosis in OPM2 cells.
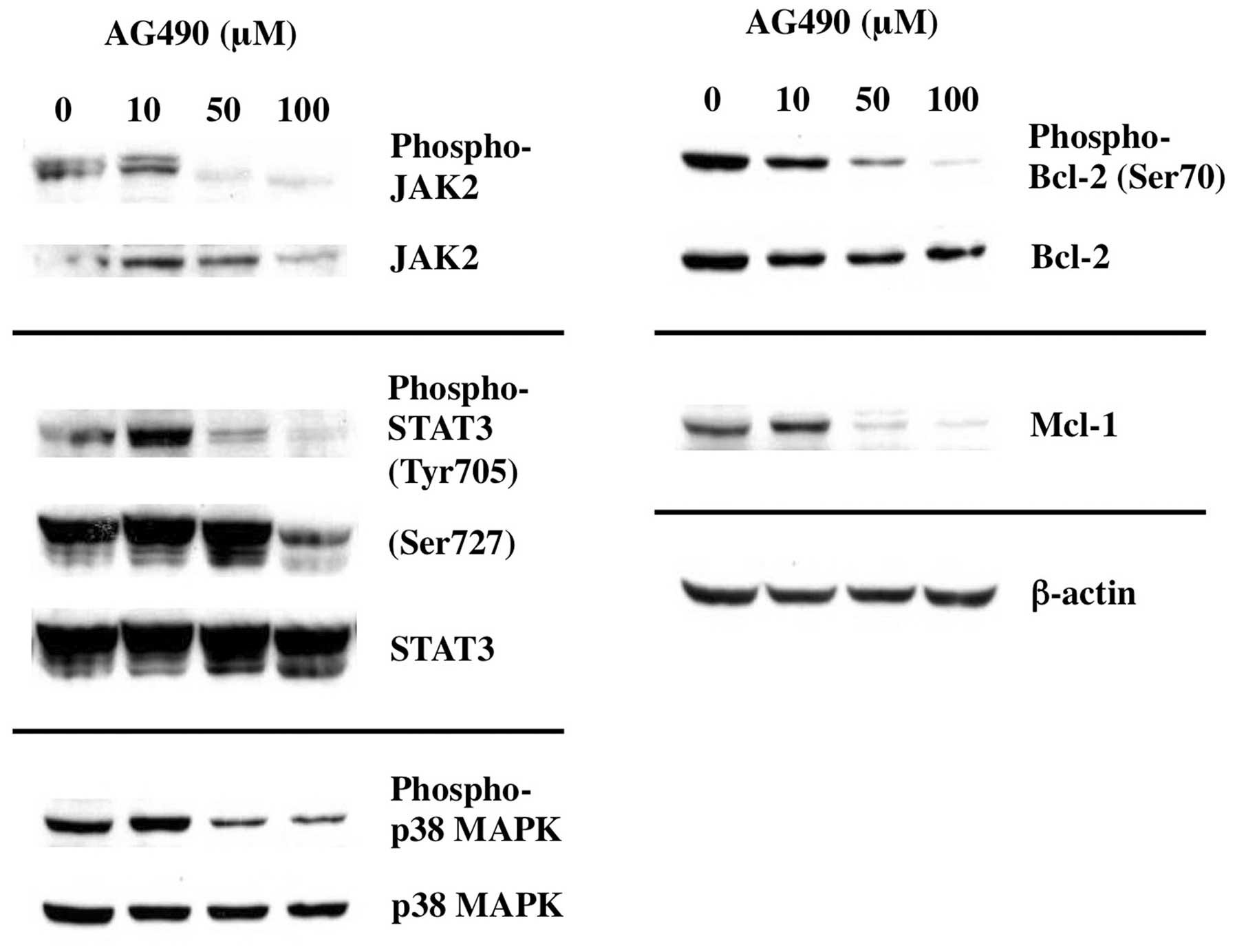
Inhibition of JAK2 results in
dephosphorylation of BCL-2 and downregulation of MCL-1
To elucidate the acting point of Gos-mediated
inhibition of IL-6 signaling, we examined whether inhibition of
JAK2 was sufficient to cause deregulation of apoptosis-associated
proteins evoked by Gos stimulation. As shown in Fig. 8, treatment of OPM2 cells with JAK2
inhibitor AG490 inhibited the phosphorylation of JAK2 as well as
the downstream effectors such as STAT3 and p38MAPK. As expected,
dephosphorylation of BCL-2 and decrease of MCL-1 were induced by
AG490 treatment in parallel to JAK2 inhibition.
These data suggest that Gos-induced deregulation of
Bcl-2 and Mcl-1 was mediated through inhibition of IL-6 signaling
at the level leading to JAK2 activation.
Discussion
Gos is a natural small molecule inhibitor for Bcl-2
or Bcl-XL that has been shown to act as a potent inducer
of apoptosis in multiple tumor cell types. Previous studies have
shown that Gos downregulated the expression of
Bcl-2/Bcl-XL/Mcl-1 proteins in multiple tumor cell
lines. In contrast, however, our data indicated that Gos treatment
did not affect the protein levels of Bcl-2 or Bcl-XL.
Instead, Gos attenuated Ser-70 phosphorylation of Bcl-2, which is
critical for executing its full and potent anti-apoptotic effects
(29,30). It has been reported that Ser70 of
Bcl-2 was phosphorylated by ERK1/2 kinases (29,35),
and we demonstrated that activated form of ERK1/2 (phospho-ERK1/2)
was in fact downregulated by Gos treatment. We went on to show that
Gos also downregulated phosphorylated forms of JAK2, gp130, and
STAT3, critical components of active IL-6 signaling. Taken
together, these results indicate that Gos induces dephosphorylation
of Bcl-2 at Ser-70 through global inhibition of IL-6 signaling.
We also demonstrated that another anti-apoptotic
protein, Mcl-1, was downregulated by Gos treatment. This should be
due to the inhibition of IL-6 signaling by Gos as well, since Mcl-1
is a downstream target of IL-6 (36,37).
Mcl-1 is frequently overexpressed in MM cells (38), and has been shown to be critical
for their survival (39). Some
reports even demonstrated that Mcl-1, rather than Bcl-2 or
Bcl-XL, plays a primary role in the survival of MM cells
(40). Therefore, downregulation
of Mcl-1 is definitely one of the major pathways of Gos-induced
apoptosis.
Gos shares a structural profile of BH3 mimetics with
other inhibitors for Bcl-2/Bcl-XL such as Genasense,
TW-37, Obatoclax or ABT-263 that are presently under clinical
trials (10,28). It has been therefore recognized
that displacement of BH3-only proteins from Bcl-2/Bcl-XL
or direct activation of BAX or BAD through BH3-mediated interaction
is a major molecular mechanism for Gos-induced apoptosis in cancer
cells (10). However, as described
above, we demonstrated that Gos inhibits IL-6 signaling in MM cell
line and induces apoptosis through downregulation of Mcl-1 and
Bcl-2 dephosphorylation. These results propose inhibition of IL-6
signaling as a novel mechanism for Gos-induced apoptosis in MM
cells.
Molecular mechanism of Gos-mediated inhibition of
IL-6 signaling is not clear at present. In cytokine signaling,
ligand-induced dimerization of receptor chains provokes activation
of Jak kinases, which then leads to phosphorylation of receptor
chains and initiates downstream signaling cascades (41,42).
In this study, we demonstrated that Gos inhibited phosphorylation
of JAK2 as well as major downstream molecules of IL-6 signaling
such as STAT3 and ERK1/2. These results suggest that inhibition of
JAK2 is the primary effect of Gos on IL-6 signaling in MM cells. In
support of this notion, pharmacological inhibition of JAK2 in OPM2
cells showed the biochemical effects highly similar to
Gos-treatment, such as dephosphorylation of Bcl-2 and decrease of
Mcl-1. Inhibition of JAK2 activation by Gos could be occurring at
the level of ligand binding, receptor dimerization, or JAK2 itself.
Elucidating precise molecular mechanism how Gos inhibits JAK2
activation requires further study.
During the preparation of this report, we noticed
that another group reported apoptosis inducing effect of Gos in MM
cells (43). In contrast to our
findings, they showed that Gos decreased protein levels of Bcl-2
and Bcl-XL by flow cytometry, although they did not
examine the phosphorylation status of Bcl-2. One possible reason
for this discrepancy could be the different concentrations of Gos
used in either study. They used Gos at 25 μM, while we took 5 μM in
most experiments. In fact, we observed moderate decrease of Bcl-2
protein when cells were treated with Gos at 10 μM or higher for 24
h (data not shown). However, our data clearly indicate that
Gos-induced apoptosis occurs at 5 μM, a concentration that readily
induces Bcl-2 dephosphorylation (Figs.
1–4). This strongly suggests
that decrease of Bcl-2/Bcl-XL protein levels is not the
primary cause for apoptosis induction by Gos in MM cells.
In conclusion, we demonstrated that Gos, a natural
BH3 mimetics, induces apoptosis in MM cells not only through
displacement of BH3-only proteins from Bcl-2/Bcl-XL, but
also via the inhibition of IL-6 signaling. Future study will focus
on the mechanism how Gos inhibits IL-6 signaling in MM cells. It is
expected that this allows us to obtain vital information on novel
strategies for IL-6 inhibition, which leads to a development of new
drugs for MM treatment.
Acknowledgements
The authors would like to thank Chika Saito for
excellent technical assistance. This study was supported by grants
from the Ministry of Education, Culture, Sports and Technology of
Japan; the Ministry of Health, Labor, and Welfare of Japan (KAKENHI
24591409). This study was also supported in part by the National
Cancer Research and Development Fund (26-A-4).
References
|
1
|
Hideshima T and Anderson KC: Molecular
mechanisms of novel therapeutic approaches for multiple myeloma.
Nat Rev Cancer. 2:927–937. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hideshima T, Bergsagel PL, Kuehl WM and
Anderson KC: Advances in biology of multiple myeloma: clinical
applications. Blood. 104:607–618. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pettersson M, Jernberg-Wiklund H, Larsson
LG, et al: Expression of the bcl-2 gene in human multiple myeloma
cell lines and normal plasma cells. Blood. 79:495–502.
1992.PubMed/NCBI
|
|
4
|
Oancea M, Mani A, Hussein MA and Almasan
A: Apoptosis of multiple myeloma. Int J Hematol. 80:224–231. 2004.
View Article : Google Scholar
|
|
5
|
van de Donk NW, Bloem AC, van der Spek E
and Lokhorst HM: New treatment strategies for multiple myeloma by
targeting BCL-2 and the mevalonate pathway. Curr Pharm Des.
12:327–340. 2006.PubMed/NCBI
|
|
6
|
Guo S, Zhi Y, Yang H, et al: Bcl-2
expression is associated with poor prognosis of solitary
plasmacytoma of bone. Ann Hematol. 93:471–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kridel R, Sehn LH and Gascoyne RD:
Pathogenesis of follicular lymphoma. J Clin Invest. 122:3424–3431.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bende RJ, Smit LA and van Noesel CJ:
Molecular pathways in follicular lymphoma. Leukemia. 21:18–29.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weyhenmeyer B, Murphy AC, Prehn JH and
Murphy BM: Targeting the anti-apoptotic Bcl-2 family members for
the treatment of cancer. Exp Oncol. 34:192–199. 2012.PubMed/NCBI
|
|
10
|
Juin P, Geneste O, Gautier F, Depil S and
Campone M: Decoding and unlocking the BCL-2 dependency of cancer
cells. Nat Rev Cancer. 13:455–465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shamas-Din A, Kale J, Leber B and Andrews
DW: Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb
Perspect Biol. 5:a0087142013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Keshmiri-Neghab H and Goliaei B:
Therapeutic potential of gossypol: an overview. Pharm Biol.
52:124–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X, Howell CP, Chen F, Yin J and Jiang
Y: Gossypol - a polyphenolic compound from cotton plant. Adv Food
Nutr Res. 58:215–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lopez LM, Grimes DA and Schulz KF:
Nonhormonal drugs for contraception in men: a systematic review.
Obstet Gynecol Surv. 60:746–752. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oliver CL, Bauer JA, Wolter KG, et al: In
vitro effects of the BH3 mimetic, (-)-gossypol, on head and neck
squamous cell carcinoma cells. Clin Cancer Res. 10:7757–7763. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stein RC, Joseph AE, Matlin SA, Cunningham
DC, Ford HT and Coombes RC: A preliminary clinical study of
gossypol in advanced human cancer. Cancer Chemother Pharmacol.
30:480–482. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le Blanc M, Russo J, Kudelka AP and Smith
JA: An in vitro study of inhibitory activity of gossypol, a
cottonseed extract, in human carcinoma cell lines. Pharmacol Res.
46:551–555. 2002.PubMed/NCBI
|
|
21
|
Kapoor S: Attenuating effect of gossypol
on tumor growth in systemic malignancies. Cell Biochem Biophys.
67:1551–1552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu P, Ye W, Jen R, Lin SH, Kuo CT and Lin
YC: Mitogenic activity of zeranol in human breast cancer cells is
enhanced by leptin and suppressed by gossypol. Anticancer Res.
29:4621–4628. 2009.PubMed/NCBI
|
|
23
|
Anderson MA, Huang DC and Roberts AW: BH3
mimetic therapy: an emerging and promising approach to treating
chronic lymphocytic leukemia. Leuk Lymphoma. 54:909–911. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Voss V, Senft C, Lang V, et al: The
pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in
malignant glioma. Mol Cancer Res. 8:1002–1016. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng Y, Tang W, Dai Y, et al: Natural BH3
mimetic (-)-gossypol chemosensitizes human prostate cancer via
Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol
Cancer Ther. 7:2192–2202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Volate SR, Kawasaki BT, Hurt EM, et al:
Gossypol induces apoptosis by activating p53 in prostate cancer
cells and prostate tumor-initiating cells. Mol Cancer Ther.
9:461–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang LH, Hu JQ, Tao WQ, et al: Gossypol
inhibits phosphorylation of Bcl-2 in human leukemia HL-60 cells.
Eur J Pharmacol. 645:9–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruvolo PP, Deng X and May WS:
Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia.
15:515–522. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deng X, Kornblau SM, Ruvolo PP and May WS
Jr: Regulation of Bcl2 phosphorylation and potential significance
for leukemic cell chemoresistance. J Natl Cancer Inst Monogr.
30–37. 2001.PubMed/NCBI
|
|
31
|
Tamura Y, Simizu S and Osada H: The
phosphorylation status and anti-apoptotic activity of Bcl-2 are
regulated by ERK and protein phosphatase 2A on the mitochondria.
FEBS Lett. 569:249–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sahin F, Avci CB, Gunduz C, Sezgin C,
Simsir IY and Saydam G: Gossypol exerts its cytotoxic effect on
HL-60 leukemic cell line via decreasing activity of protein
phosphatase 2A and interacting with human telomerase reverse
transcriptase activity. Hematology. 15:144–150. 2010. View Article : Google Scholar
|
|
33
|
Heinrich PC, Behrmann I, Haan S, Hermanns
HM, Muller-Newen G and Schaper F: Principles of interleukin
(IL)-6-type cytokine signalling and its regulation. Biochem J.
374:1–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Catlett-Falcone R, Landowski TH, Oshiro
MM, et al: Constitutive activation of Stat3 signaling confers
resistance to apoptosis in human U266 myeloma cells. Immunity.
10:105–115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Subramanian M and Shaha C: Up-regulation
of Bcl-2 through ERK phosphorylation is associated with human
macrophage survival in an estrogen microenvironment. J Immunol.
179:2330–2338. 2007. View Article : Google Scholar
|
|
36
|
Puthier D, Bataille R and Amiot M: IL-6
up-regulates Mcl-1 in human myeloma cells through JAK/STAT rather
than ras/MAP kinase pathway. Eur J Immunol. 29:3945–3950. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Puthier D, Derenne S, Barille S, et al:
Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells.
Br J Haematol. 107:392–395. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wuilleme-Toumi S, Robillard N, Gomez P, et
al: Mcl-1 is over-expressed in multiple myeloma and associated with
relapse and shorter survival. Leukemia. 19:1248–1252. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang B, Gojo I and Fenton RG: Myeloid
cell factor-1 is a critical survival factor for multiple myeloma.
Blood. 99:1885–1893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Derenne S, Monia B, Dean NM, et al:
Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L)
is an essential survival protein of human myeloma cells. Blood.
100:194–199. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ihle JN, Thierfelder W, Teglund S, et al:
Signaling by the cytokine receptor superfamily. Ann NY Acad Sci.
865:1–9. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nakashima K and Taga T: gp130 and the IL-6
family of cytokines: signaling mechanisms and thrombopoietic
activities. Semin Hematol. 35:210–221. 1998.PubMed/NCBI
|
|
43
|
Lin J, Wu Y, Yang D and Zhao Y: Induction
of apoptosis and antitumor effects of a small molecule inhibitor of
Bcl-2 and Bcl-xl, gossypol acetate, in multiple myeloma in
vitro and in vivo. Oncol Rep. 30:731–738.
2013.PubMed/NCBI
|