Introduction
Human melanoma, an aggressive skin cancer, accounts
for 10% of all skin cancers, but was estimated to be involved in
>80% of deaths from skin cancers (1). In Western countries, skin cancer
melanoma is becoming more common and resulting in increased
mortality (2). In the USA, the
incidence of melanoma has increased by 15-fold in the last 40 years
(1,3). In individuals of European origin, the
incidence of melanoma is still rising (4). Survival ratio for metastatic melanoma
is low and the 10-year survival rate for patients with metastatic
melanoma is <10% (5,6). It was reported that human melanoma is
highly resistant to conventional chemotherapy (7). Currently, the effective treatment of
human melanoma such as surgery, radiation, chemotherapy or a
combination of radiotherapy with chemotherapy is not satisfactory.
Thus, numerous studies had focused on finding novel potent drugs
from natural products to combat this disease.
In nature, insects produce different defensive
molecules against predators, and these molecules may be clinically
used as medicinal drugs for therapeutic purposes (8). The dried body of mylabris
(Mylabris phalerata Pallas) has been used in Chinese
traditional medicine for the treatment of cancer (9). Cantharidin (CTD), a terpenoid, was
isolated from mylabris (blister beetles) and other insects and was
shown to induce cancer cell apoptosis in leukemia (10), myeloma (11), bladder (12), breast (13), colon (14), liver (15), pancreatic (16) and lung (17). It was reported that CTD inhibits
migration and invasion of A549 human lung cancer cells via the
inhibition of matrix metalloproteinase 2 (18). Recently, we also found that CTD
induces cell apoptosis through mitochondria-dependent pathways
(18) and induced DNA damage and
inhibits DNA repair-associated protein levels in NCI-H460 human
lung cancer cells (19).
Numerous studies have shown that CTD induced
cytotoxic effects in many human cancer cell lines through the
induction of apoptosis, however, there is no available information
to show CTD-induced apoptosis in human skin cancer cells.
Therefore, in the present study, A375.S2 human melanoma cells were
selected for use as a cell model to investigate the anti-melanoma
potential of CTD in vitro. The results indicated that CTD
induced G2/M phase arrest and cell apoptosis in A375.S2 cells via
the caspase- and mitochondrial-dependent signaling pathways.
Materials and methods
Chemicals and reagents
CTD, propidium iodide (PI), Trypsin-EDTA, dimethyl
sulfoxide (DMSO) and DAPI were purchased from Sigma Chemical Co.
(St. Louis, MO, USA). CTD was dissolved in DMSO to make a stock
solution. Minimum essential medium (MEM), fetal bovine serum (FBS),
L-glutamine and penicillin-streptomycin were purchased from
Gibco®/Invitrogen Life Technologies (Carlsbad, CA, USA).
Primary antibody such as WEE1, Cdc25c, Cyclin A, CDK1, p21, Fas,
Fas-L, AIF, Endo G, cytochrome c, caspase-3, -8 and -9, Bax,
Bid, Bcl-2, Bcl-x, XBP-1, GADD153, GRP78, caspase-12, IRE1β, ATF6α
and Calpain 1 and peroxidase conjugated secondary antibodies were
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
The enhanced chemiluminescence (ECL) detection system was obtained
from Amersham Life Science, Inc. (Arlington Heights, IL, USA).
Cell culture
The A375.S2 human malignant melanoma cancer cells
were obtained from the Food Industry Research and Development
Institute (Hsinchu, Taiwan). The cells were cultured in MEM
supplemented with 10% FBS, 1% antibiotics (100 U/ml penicillin and
100 μg/ml streptomycin) and 2 mM L-glutamine
(Gibco®/Invitrogen Life Technologies, Grand Island, NY,
USA) and maintained at 37°C with 5% CO2 in a humidified
atmosphere. The medium was changed every 2 days (20–22).
Observation of morphological changes and
measurement of viable cells
A375.S2 cells (2×105 cells/well) were
seeded into 12-well plates for 24 h. CTD diluted in DMSO was then
individually added to a final concentration of 0, 1, 2, 3, 4 and 5
μM, and an equal amount of DMSO was added to the well as the
control group for 48 h. The cellular morphology was observed and
photographed by using a phase contrast microscope at magnification
of ×200. Cells from each well were harvested for the measurement of
percentage of viability using a flow cytometric method (BD
Bioscience FACSCalibur flow cytometer; Becton-Dickinson, San Jose,
CA, USA) as described previously (20).
Measurement of cell cycle distribution by
flow cytometry
A375.S2 cells (2×105 cells/well) were
seeded into 12-well culture plates for 24 h and then were incubated
with 0, 1, 2, 3, 4 and 5 μM of CTD, or only with vehicle (DMSO, 1%
in culture media) for 24 and 48 h. Cells were harvested by
centrifugation and washed with phosphate-buffered saline (PBS).
Then cells were fixed with 70% ethanol overnight at least for 24 h
at 4°C and were washed twice with PBS and stained with 1 ml PI
working solution (100 μg/ml RNase A, 40 μg/ml PI and 0.1% Triton
X-100) for cellular staining at room temperature for 30 min in the
dark. Analysis of cell cycle distribution was performed by a flow
cytometer and analyzed by Cell Quest software package (BD
Bioscience FACSCalibur flow cytometer; Becton-Dickinson) as
described previously (20). Each
experiment was repeated three times.
Reactive oxygen species (ROS),
intracellular Ca2+ and mitochondrial membrane potential
(ΔΨm) assays
Flow cytometry was used for measuring the levels of
ROS, Ca2+ and Δψm in A375.S2 cells. Briefly, A375.S2
cells (2×105 cells/well) placed in 12-well plates for 24
h were then treated with 4 μM of CTD for various time periods. The
cells were collected from each timer point and then re-suspended in
500 μl of DCFH-DA (10 μM) for 30 min for ROS
(H2O2) measurement, re-suspended in 500 μl of
DiOC6 (4 μM) for 30 min for the levels of Δψm
measurement and re-suspended in 500 μl of Fluo-3/AM (2.5 μg/ml) for
30 min for intracellular Ca2+ measurement and all
samples were analyzed by flow cytometry as described
previously.
Caspase-3, -8 and -9 activity assay
A375.S2 cells (2×105 cells/well) were
seeded onto 12-well plates for 24 h and then were pre-treated with
Z-VAD-FMK, Z-IETD-FMK, Z-LEHD-FMK and Z-DEVD-FMK (inhibitors of
caspase-pan, -8, -9 and -3, respectively) and then treated with 4
μM of CTD for 0, 6, 24 and 48 h. Then cells were harvested and
washed with PBS, and were re-suspended in 50 μl of 10 μM substrate
solution of caspase-8, -9 and -3 substrates (CaspaLux8-L1D2,
CaspaLux9-M1D2 and PhiPhiLux-G1D2, respectively) (OncoImmunin,
Inc., Gaithersburg, MD, USA) for 30 min in the dark. Cells were
measured for the activities of caspase-8, -9 and -3 by using flow
cytometric assay as described previously (20).
Western blotting
A375.S2 cells (1×106 cells/dish) were
placed in 10 cm dish for 24 h and then were incubated with or
without 4 μM CTD for 0, 6, 12, 24 and 48 h then lysed in an
ice-cold lysis buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM
EGTA, 0.3 mM PMSF, 0.2 mM sodium orthovanadate, 0.1% SDS, 1 mM
EDTA, 1% NP-40, 10 mg/ml leupeptin, and 10 mg/ml aprotinin],
followed by denaturation. Then centrifuged at 13,000 rpm for 20 min
at 4°C, before getting the supernatant to measure protein
concentration by using a Bio-Rad protein assay kit (Bio-Rad,
Hercules, CA, USA). Protein (30 μg) was electrophoresed in 12%
SDS-PAGE gel at 4°C, steady flow (10 mA in composition gel, 15 mA
in separation gel) followed by transfer onto nitrocellulose
membranes. The membranes were blocked with 5% skim milk in TBST (20
mmol/l Tris-HCl at pH 8.0, 150 mmol/l NaCl, and 0.05% Tween-20) for
1 h at room temperature, and then probed with relevant primary
antibodies (anti-WEE1, Cdc25c, Cyclin A, CDK1, p21, Fas, Fas-L,
AIF, Endo G, cytochrome c, caspase-3, -8 and -9, Bax, Bid,
Bcl-2, Bcl-x, XBP-1, GADD153, GRP78, caspase-12, IRE1β, ATF6α and
Calpain 1) overnight at 4°C followed by peroxidase-conjugated
secondary antibody for 1 h at 25°C. Proteins on the membrane were
visualized by ECL detection (Amersham Biosciences ECL™) and exposed
to X-ray film and bands obtained were quantified using NIH Image
analyzer (NIH, Bethesda, MD, USA). β-actin staining served as the
internal standard for the membranes. All of the western blots were
performed at least three times (21,22).
Confocal laser scanning microscopy
assay
A375.S2 cells (5×104 cells/well) were
placed on 4-well chamber slides and incubated with or without 4 μM
CTD for 48 h and then fixed in 4% formaldehyde in PBS for 15 min,
and they were permeablized using 0.3% Triton X-100 in PBS for 1 h,
followed using 2% BSA for blocking non-specific binding sites.
Cells were stained by primary antibodies such as anti-Endo G,
anti-cytochrome c and anti-AIF (all in green fluorescence)
overnight and then washed with PBS. Cells were incubated with
fluorescein isothiocyanate-conjugated second antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) followed by mitotracker (red
fluorescence) staining for nuclein examination. The stained cells
were analyzed with Leica TCS SP2 Confocal Spectral Microscope as
described previously (23,24).
Statistical analysis
All data were expressed as mean ± SD from triplicate
experiments. Statistically significant of differences between the
CTD-treated and -untreated (control) groups were assessed by
Student’s t-test with SPSS 11.0 statistic software. P<0.05 was
considered statistically significant.
Results
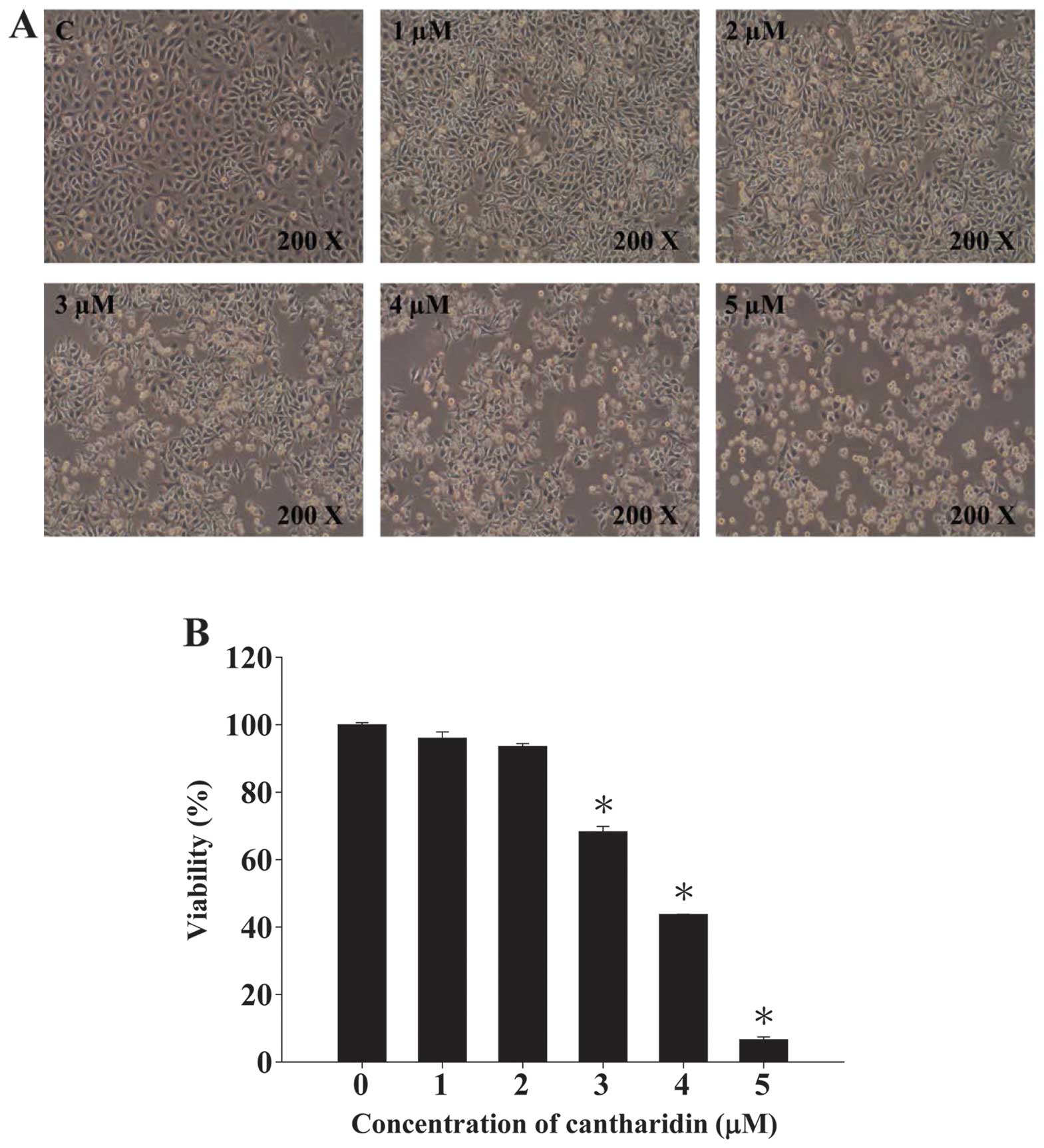
CTD induced cell morphological changes
and decreased the cell viability of A375.S2 cells
A375.S2 cells were pre-incubated with 0, 1, 2, 3, 4
and 5 μM of CTD for 48 h then cells were photographed by phase
contrast microscopy and were harvested for the total percentage of
viable cells and the results are shown in Fig. 1A and B. Fig. 1A shows that CTD induced cell
morphological changes. Fig. 1B
shows a significant dose-dependent reduction of living cells with
CTD treatment when compared to the control groups in A375.S2 cells
and these effects are dose-dependent.
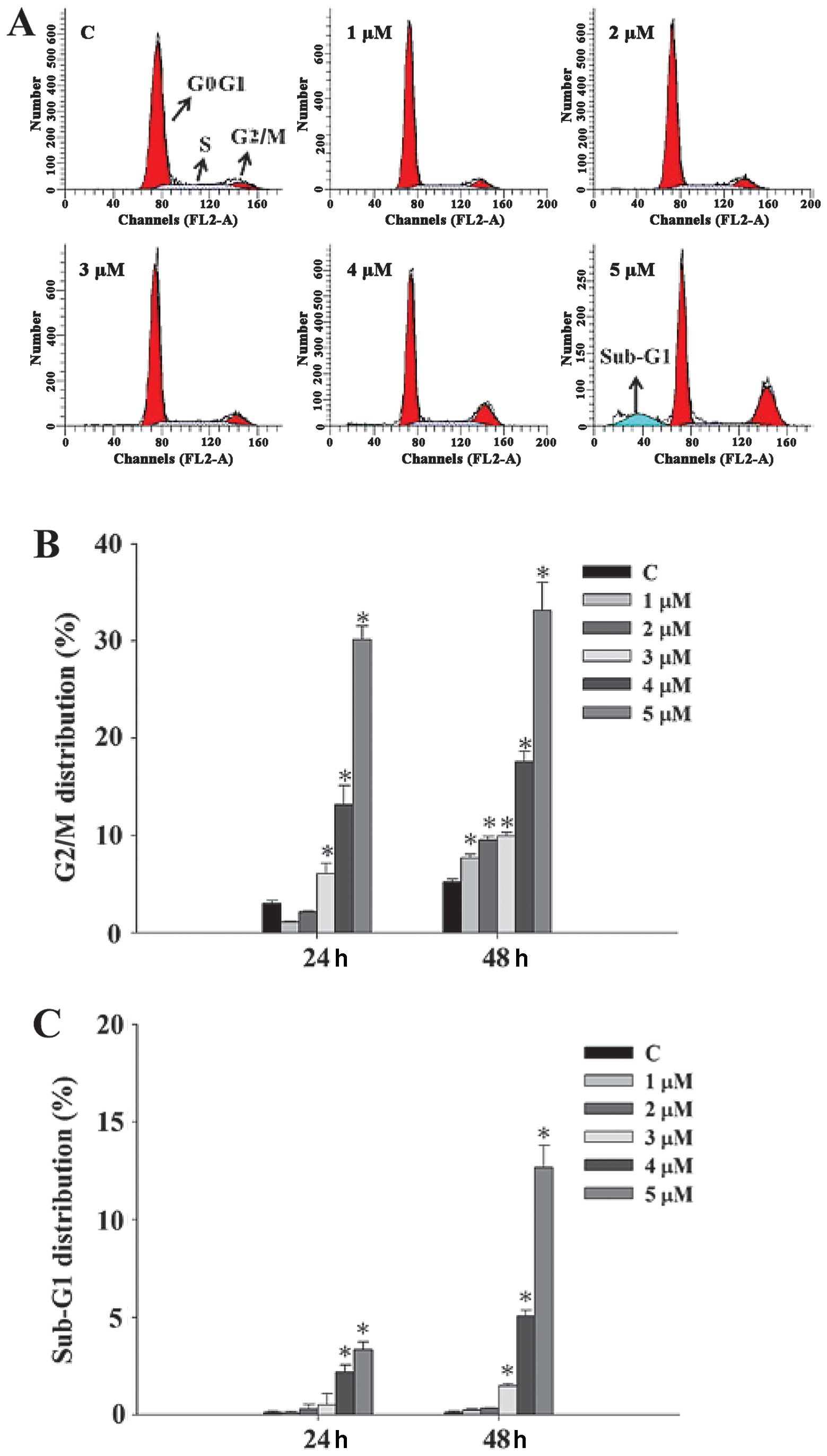
CTD induces G2/M phase arrest and sub-G1
phase (apoptosis) of A375.S2 cells
A375.S2 cells were treated with various doses of CTD
for 24 and 48 h before the cells were examined for sub-G1 phase in
cell cycle assay by flow cytometry and the results are shown in
Fig. 2A–C. Fig. 2A shows representative profiles from
flow cytometry assay indicating that CTD induced sub-G1 phase and
G2/M phase arrest in A375.S2 cells. Data in Fig. 2B and C indicate that CTD induced
G2/M phase arrest and induced sub-G1 phase development,
respectively, and these effects are dose-dependent. At the 48-h
treatment of CTD, a higher percentage of G2/M phase arrest and
sub-G1 phase (apoptosis) was recorded than that of control
groups.
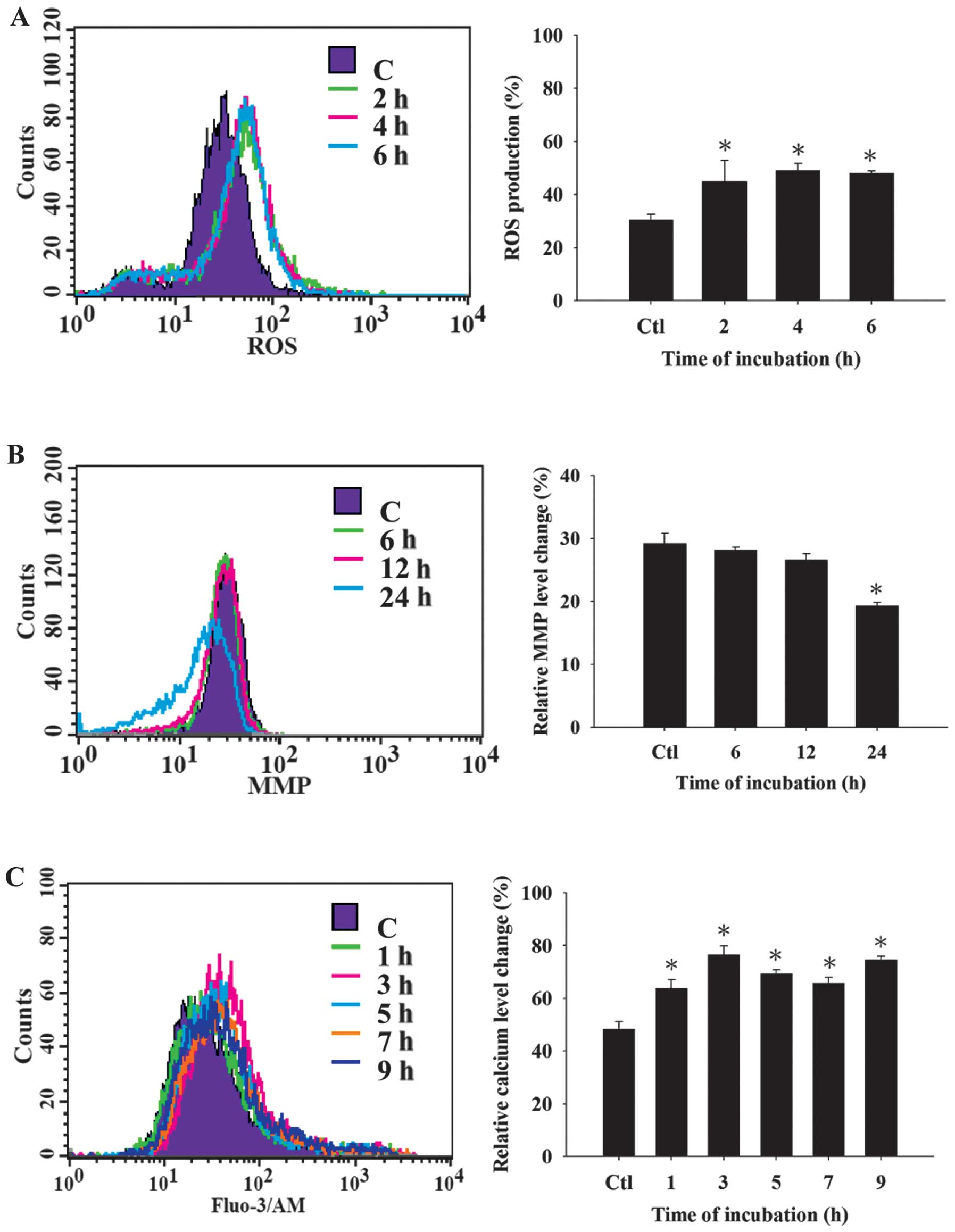
CTD induces ROS and Ca2+
production and decreases the levels of Δψm in A375.S2 cells
In order to confirm whether CTD induced apoptotic
cell death in A375.S2 cells via the production of ROS and
Ca2+ or dysfunction of mitochondria, cells were treated
with CTD then analyzed by flow cytometry and the results are shown
in Fig. 3A–C. Fig. 3A shows that CTD increased ROS
production at 2–6 h of treatment. Furthermore, CTD induced
Ca2+ production (Fig.
3C) from 1–9 h of treatment in A375.S2 cells and these effects
are time-dependent. However, Fig.
3B indicates that CTD decreased the levels of Δψm at 24-h of
treatment and shows that CTD-induced apoptosis of A375.S2 cells is
associated with dysfunction of mitochondria.
CTD affects the activities of caspase-8,
-9 and -3 in A375. S2 cells
To confirm whether CTD induced apoptosis through the
activation of caspase-8, -9 and -3 in A375. S2 cells, cells were
pre-treated with or without the inhibitors (Z-VAD-FMK, Z-IETD-FMK,
Z-LEHD-FMK and Z-DEVD-FMK: caspase-pan, -8, -9 and -3,
respectively) and then were treated with 4 μM of CTD and were
harvested and assessed by flow cytometric assay and the results are
shown in Fig. 4A–D. Results from
Fig. 4A–C indicate that CTD
increased the activities of caspase-8, -9 and -3 and these effects
are time-dependent. Cells were pre-treated with the inhibitors of
caspase-pan, -8, -9 and -3 and then were treated with CTD and the
total percentage of viable cells were measured and the results
(Fig. 4D) show increased
percentage of viable cells when compared to the treatment without
the inhibitor. These results showed that CTD induced apoptosis via
the caspase-dependent pathway.
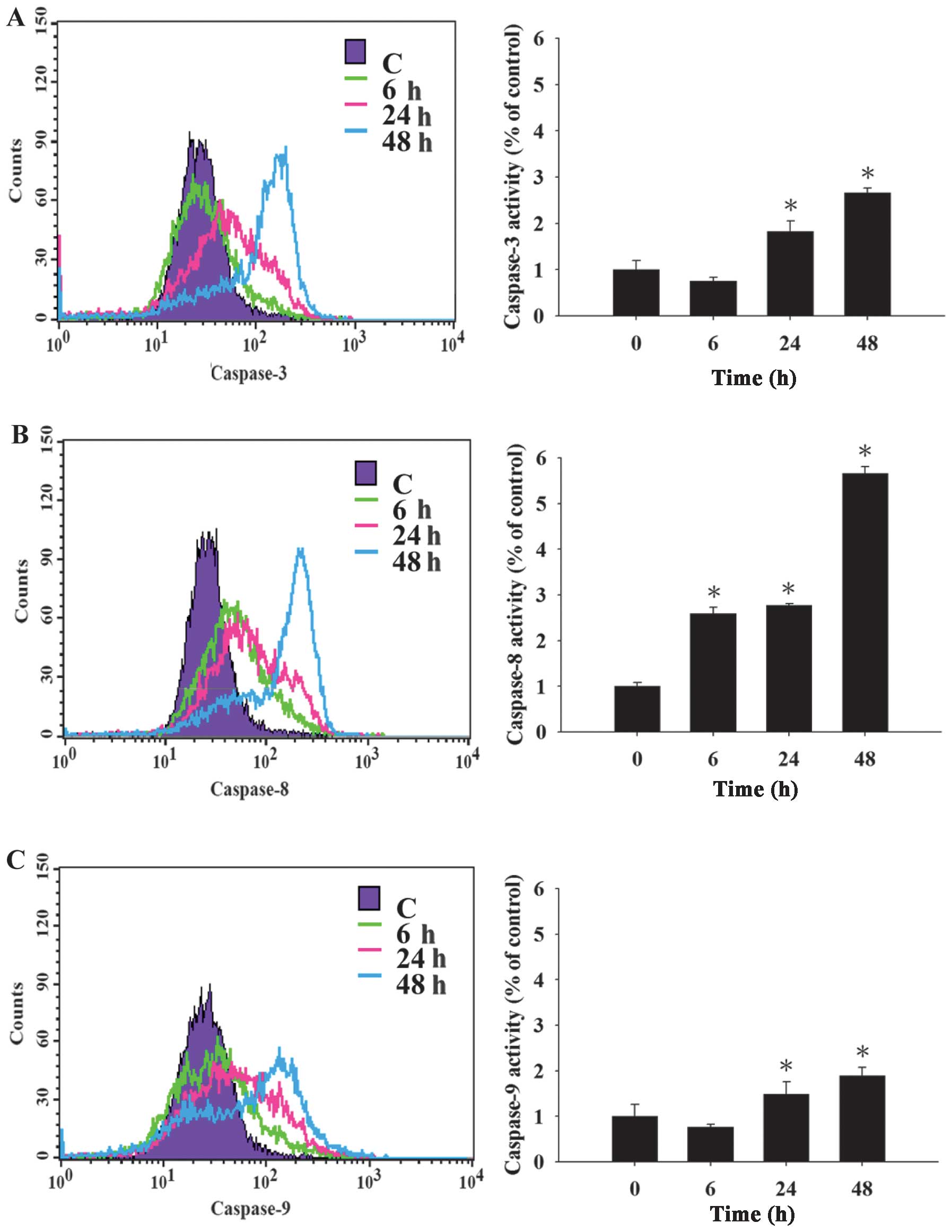 | Figure 4Cantharidin (CTD) affects caspase-3,
-8 and -9 activities in A375.S2 cells. A375.S2 cells
(2×105 cells/well) were pre-treated with or without
Z-VAD-FMK, Z-IETD-FMK, Z-LEHD-FMK and Z-DEVD-FMK (inhibitors of
caspase-pan, -8, -9 and -3, respectively) then were incubated 4 μM
CTD for different time periods. After harvesting and washing, the
cells were re-suspended in 50 μl of 10 μM substrate solution of
caspase-8, -9 and -3 substrates (CaspaLux8-L1D2, CaspaLux9-M1D2 and
PhiPhiLux-G1D2), respectively, then the activities of (A)
caspase-3, (B) caspase-8, (C) caspase-9 and (D) percentage of
viable cells were measured by using flow cytometry as described in
Materials and methods. The results are shown as a mean ± SD (n=3);
*P<0.05, significant difference between CTD-treated
groups and the control as analyzed by Student’s t-test. |
CTD affects G2/M phase arrest and
apoptosis-associated protein expression in A375.S2 cells
To further investigate whether CTD induced G2/M
phase arrest and apoptosis in A375.S2 cells through the presented
alterations of G2/M phase and apoptosis-associated protein, cells
were treated with 4 μM of CTD for 0, 6, 12, 24 and 48 h and then
the associated protein alterations were examined by western
blotting and the results are shown in Fig. 5A–D. Fig. 5A indicates that CTD inhibited
Cdc25c, Cyclin A and CDK1 but increased p21 and WEE1 proteins in
A375.S2 cells. These cellular proteins were known to respond to
G2/M phase in cell cycle progression. We inferred that CTD could
bring about the proteolytic activations of various G2/M phase
proteins to obstruct the cell cycle progression of A375.S2 cells.
Results in Fig. 5C show that CTD
significantly increased the expression of Fas, Fas-L, and caspase-8
that is associated with the extrinsic pathway. Furthermore, results
show that CTD increased cytochrome c, AIF, Endo G, caspase-9
and -3 (Fig. 5C), increased Bid,
Bax, but decreased the levels of Bcl-2, Bcl-x and XBP-1 (Fig. 5B) that are all associated with the
intrinsic apoptotic pathway. Additionally, Fig. 5D indicates that CTD increased ER
stress-associated protein expression such as GADD153, GRP78, IRE1β,
Calpain 1, ATF6α and caspase-12. These results indicate that CTD
induced G2/M phase arrest via inhibited cell cycle
progression-associated protein and induced apoptosis through the
extrinsic, intrinsic and ER stress pathways in A375.S2 cells.
 | Figure 5Cantharidin (CTD) affects G2/M phase
and apoptosis-associated protein expression in A375.S2 cells.
A375.S2 cells were treated with 4 μM of CTD for 0, 6, 12, 24 and 48
h and then total proteins were quantitated and apoptosis-associated
proteins were examined by western blotting as described in
Materials and methods. (A) WEE1, Cdc25c, Cyclin A, CDK1 and p21;
(B) Bcl-2, Bcl-x, Bid, Bax and XBP-1; (C) Fas, Fas-L, caspase-8,
AIF, Endo G, cytochrome c, caspase-3 and -9; (D) GADD153,
GRP78, caspase-12, calpain 1, IRE1β and ATF6α. β-actin,
control. |
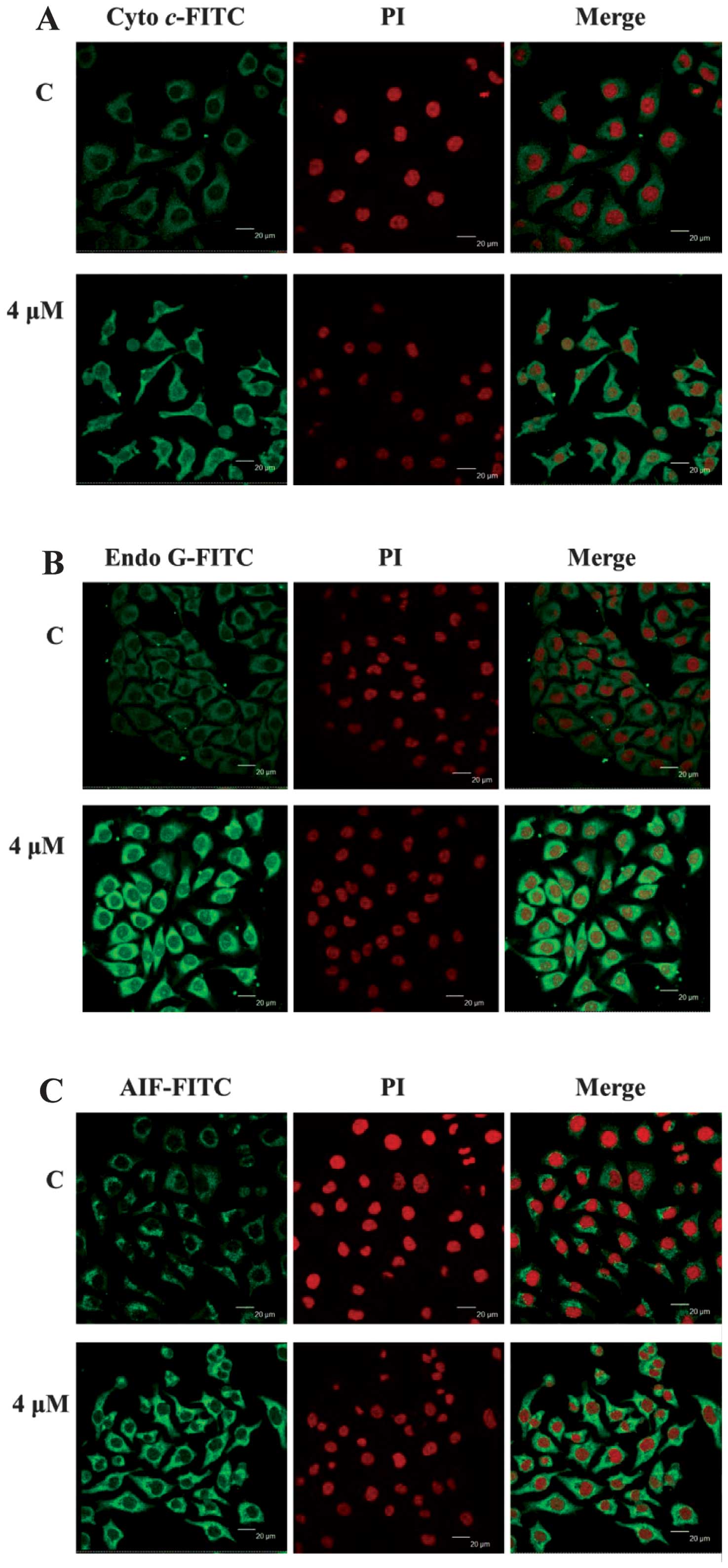
CTD affected the translocation of
apoptotic associated proteins in A375.S2 cells
To further confirm that CTD affects the
translocation of cytochrome c, AIF, and Endo G involved in
apoptosis in A375.S2 cells, cells were treated with 4 μM of CTD for
48 h and then stained by anti-cytochrome c, AIF and Endo G
to examine and photograph by confocal laser microscopic systems.
Results show that CTD promoted Endo G (Fig. 6B), cytochrome c (Fig. 6A), AIF (Fig. 6C) releases from mitochondria in
A375.S2 cells when compared to untreated (control) groups that
indicated CTD induced apoptosis via the mitochondria-dependent
pathway.
Discussion
It was reported that >50% of anti-cancer drugs
used in patients are directly or indirectly derived from natural
plants (24). CTD, a natural
active compound isolated from various insects, was found to have
in vitro antitumor activity against many human cancer cell
lines (10–17). In this study, for the first time,
we demonstrated that CTD induced G2/M phase arrest via the
inhibition of Cdc25c and cyclin A and induced apoptosis was through
death receptor (extrinsic), intrinsic (mitochondria) and ER stress
pathways in A375. S2 cells. Furthermore, results indicated that CTD
induced cell morphological changes (Fig. 1A) and decreased the percentage of
viable cells (Fig. 1B) via the
induction of G2/M phase arrest, sub-G1 phase (apoptosis) (Fig. 2).
It is well known that cells undergo cell cycle from
G0/G1, S, and G2/M phase that are controlled by
checkpoint-associated proteins (23,25)
and agents including anticancer drugs can affect checkpoint
proteins distributing the progression of cell cycle then leading
the cells to undergo apoptosis (26,27).
Herein, we found CTD induced G2/M phase arrest in A375.S2 cells, it
also inhibited the protein expression of Cdc25c, Cyclin A and CDK1
(Fig. 6A) that are associated with
G2/M arrest.
It is well documented that the induction of
apoptosis triggered by anticancer drugs has been recognized as the
best strategy for anticancer therapy (28,29).
It was reported that intracellular ROS generation plays an
important role in physiological and pathological processes.
Furthermore, higher ROS is involved in apoptotic cell death
(30). Mitochondria plays a
critical role in cell apoptosis (31,32)
and has been suggested to act as the central executioner in
apoptotic signaling pathways (33). We found that CTD increased the
production of ROS (Fig. 3A)
time-dependently and decreased the levels of Δψm (Fig. 3B) in A375.S2 cells. It was reported
that the mitochondria-derived ROS is caused by the dysfunction of
mitochondrial electron transport chain (34). These observations indicated the
mitochondrial dysfunction occurred during CTD-induced A375.S2 cell
apoptosis. At 48-h of CTD treatment, it led to mitochondria
dysfunction and results from western blotting also showed that CTD
increased the release of cytochrome c, AIF and Endo G
(Fig. 5C) release. Furthermore, it
increased Bid and Bax but decreased Bcl-2 and Bcl-x (Fig. 5B) in A375. S2 cells. Bcl-2 gene
family is divided mainly into the Bax, Bcl-2, and Bid proteins. Bax
is an apoptosis-promoting protein, while Bcl-2 is an anti-apoptotic
protein that plays a critical role in regulating cell apoptosis
(35,36). By western blotting we found that
the expression of Bax was increased and that of Bcl-2 was reduced
in A375.S2 cells when treated with CTD, therefore increasing the
Bax/Bcl-2 ratio significantly.
Other studies have shown that oxidative stress
stimulates translocation of Bax from cytosol to mitochondria
causing cytochrome c release inside the cytoplasm during
liver apoptosis (37). CTD-induced
ROS generation, and we suggest that CTD-induced apoptosis might be
modulated by the ROS-mediated pathways in A375.S2 cells.
It was well known that cysteine-containing
aspartate-specific proteases (caspases) are involved in cell
apoptosis (38,39). Caspase-8 is related to extrinsic
pathway and caspase-9 is involved in the intrinsic pathway,
however, caspase-3 is related to the common pathway of cell
apoptosis and it is a key executor of cell apoptosis. In our study,
the results in Fig. 5 show that
increased activation of caspase-8, -9 and -3 (Fig. 4), and expression of protein
(Fig. 6B) are associated with cell
apoptosis. Furthermore, cells were pre-treated with the inhibitors
of caspase-8, -9 and -3 and then treated with CTD leading to
increase in the percentage of viable cells when compared to CTD
only treated cells.
In conclusion, caspase-pathway activation,
mitochondria dysfunction and oxidative stress (ROS generation)
induced by CTD contribute to the activation of the apoptotic
pathway in CTD-treated A375.S2 cells. Furthermore, the modulating
expression and translocation of apoptotic proteins induced the
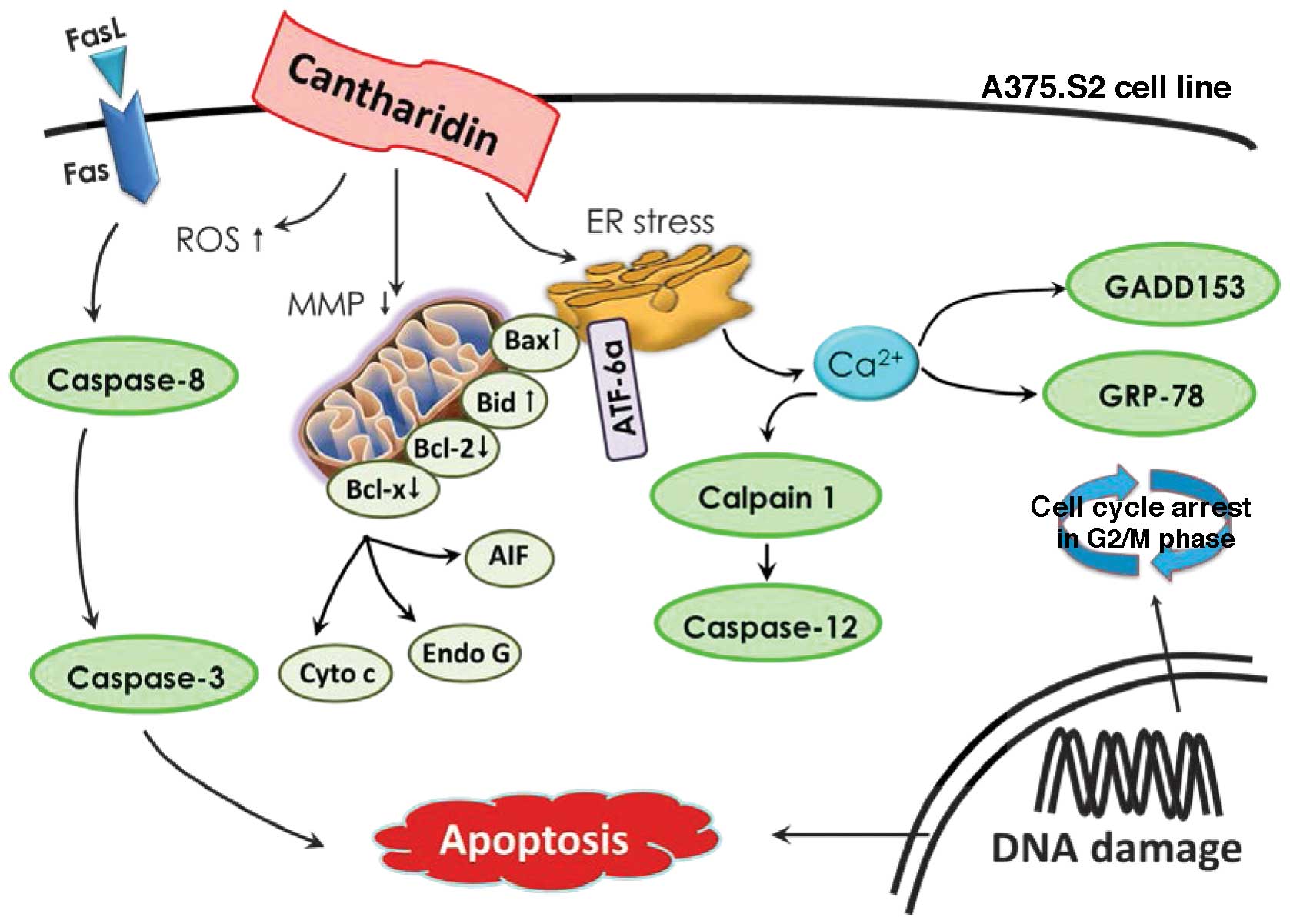
mitochondrial pathways in A375.S2 cells as shown in Fig. 7. Based on these observations, CTD
inhibits human skin cancer A375.S2 cellular growth and our studies
provide a better understanding of the molecular mechanism of CTD
function.
Acknowledgements
This study was supported in part by a research grant
from China Medical University [CMU102-ASIA-20]. Experiments and
data analysis were performed in part through the use of the Medical
Research Core Facilities Center, Office of Research and Development
at China medical University, Taichung, Taiwan, R.O.C.
References
|
1
|
Postovit LM, Margaryan NV, Seftor EA and
Hendrix MJ: Role of nodal signaling and the microenvironment
underlying melanoma plasticity. Pigment Cell Melanoma Res.
21:348–357. 2008. View Article : Google Scholar
|
|
2
|
Bergomi M, Pellacani G, Vinceti M, et al:
Trace elements and melanoma. J Trace Elem Med Biol. 19:69–73. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chudnovsky Y, Khavari PA and Adams AE:
Melanoma genetics and the development of rational therapeutics. J
Clin Invest. 115:813–824. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thompson JF, Scolyer RA and Kefford RF:
Cutaneous melanoma in the era of molecular profiling. Lancet.
374:362–365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miller AJ and Mihm MC Jr: Melanoma. N Engl
J Med. 355:51–65. 2006. View Article : Google Scholar
|
|
6
|
Bhatia S, Tykodi SS and Thompson JA:
Treatment of metastatic melanoma: an overview. Oncology (Williston
Park). 23:488–496. 2009.
|
|
7
|
Gava B, Zorzet S, Spessotto P, Cocchietto
M and Sava G: Inhibition of B16 melanoma metastases with the
ruthenium complex imidazolium
trans-imidazoledimethylsulfoxide-tetra-chlororuthenate and
down-regulation of tumor cell invasion. J Pharmacol Exp Ther.
317:284–291. 2006. View Article : Google Scholar
|
|
8
|
Newman DJ, Cragg GM and Snader KM: The
influence of natural products upon drug discovery. Nat Prod Rep.
17:215–234. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang GS: Medical uses of mylabris in
ancient China and recent studies. J Ethnopharmacol. 26:147–162.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu B: The inf luence of several anticancer
agents on cell proliferation, differentiation and the cell cycle of
murine erythroleukemia cells. Am J Chin Med. 9:268–276. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sagawa M, Nakazato T, Uchida H, Ikeda Y
and Kizaki M: Cantharidin induces apoptosis of human multiple
myeloma cells via inhibition of the JAK/STAT pathway. Cancer Sci.
99:1820–1826. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuo JH, Chu YL, Yang JS, et al:
Cantharidin induces apoptosis in human bladder cancer TSGH 8301
cells through mitochondria-dependent signal pathways. Int J Oncol.
37:1243–1250. 2010.PubMed/NCBI
|
|
13
|
Williams LA, Möller W, Merisor E, Kraus W
and Rösner H: In vitro anti-proliferation/cytotoxic activity of
cantharidin (Spanish Fly) and related derivatives. West Indian Med
J. 52:10–13. 2003.PubMed/NCBI
|
|
14
|
Huang WW, Ko SW, Tsai HY, et al:
Cantharidin induces G2/M phase arrest and apoptosis in human
colorectal cancer colo 205 cells through inhibition of CDK1
activity and caspase-dependent signaling pathways. Int J Oncol.
38:1067–1073. 2011.
|
|
15
|
Wang CC, Wu CH, Hsieh KJ, Yen KY and Yang
LL: Cytotoxic effects of cantharidin on the growth of normal and
carcinoma cells. Toxicology. 147:77–87. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li W, Chen Z, Zong Y, et al: PP2A
inhibitors induce apoptosis in pancreatic cancer cell line PANC-1
through persistent phosphorylation of IKKα and sustained activation
of the NF-κB pathway. Cancer Lett. 304:117–127. 2011.
|
|
17
|
Hsia TC, Yu CC, Hsu SC, et al: Cantharidin
induces apoptosis of H460 human lung cancer cells through
mitochondria-dependent pathways. Int J Oncol. 45:245–254. 2014.
|
|
18
|
Kim YM, Ku MJ, Son YJ, Yun JM, Kim SH and
Lee SY: Anti-metastatic effect of cantharidin in A549 human lung
cancer cells. Arch Pharm Res. 36:479–484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hsia TC, Lin JH, Hsu SC, et al:
Cantharidin induces DNA damage and inhibits DNA repair-associated
protein levels in NCI-H460 human lung cancer cells. Environ
Toxicol. Mar 17–2014.(Epub ahead of print).
|
|
20
|
Huang SH, Hsu MH, Hsu SC, et al: Phenethyl
isothiocyanate triggers apoptosis in human malignant melanoma A375.
S2 cells through reactive oxygen species and the
mitochondria-dependent pathways. Hum Exp Toxicol. 33:270–283. 2014.
View Article : Google Scholar
|
|
21
|
Chang Y-M, Velmurugan BK, Kuo W-W, et al:
Inhibitory effect of alpinate Oxyphyllae fructus extracts on Ang
II-induced cardiac pathological remodeling-related pathways in H9c2
cardiomyoblast cells. BioMedicine. 3:148–152. 2013. View Article : Google Scholar
|
|
22
|
Lin M-C, Tsai S-Y, Wang F-Y, Liu F-H, Syu
J-N and Tang F-Y: Leptin induces cell invasion and the upregulation
of matrilysin in human colon cancer cells. BioMedicine. 3:174–180.
2013. View Article : Google Scholar
|
|
23
|
Das KC and Ravi D: Altered expression of
cyclins and cdks in premature infant baboon model of
bronchopulmonary dysplasia. Antioxid Redox Signal. 6:117–127. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee KH: Anticancer drug design based on
plant-derived natural products. J Biomed Sci. 6:236–250.
1999.PubMed/NCBI
|
|
25
|
Das KC and Wasnick JD: Biphasic response
of checkpoint control proteins in hyperoxia: exposure to lower
levels of oxygen induces genome maintenance genes in experimental
baboon BPD. Mol Cell Biochem. 395:187–198. 2014. View Article : Google Scholar
|
|
26
|
Salameh A, Galvagni F, Anselmi F, De
Clemente C, Orlandini M and Oliviero S: Growth factor stimulation
induces cell survival by c-Jun. ATF2-dependent activation of
Bcl-XL. J Biol Chem. 285:23096–23104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shirali S, Aghaei M, Shabani M, Fathi M,
Sohrabi M and Moeinifard M: Adenosine induces cell cycle arrest and
apoptosis via cyclinD1/Cdk4 and Bcl-2/Bax pathways in human ovarian
cancer cell line OVCAR-3. Tumour Biol. 34:1085–1095. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Strasser A, Cory S and Adams JM:
Deciphering the rules of programmed cell death to improve therapy
of cancer and other diseases. EMBO J. 30:3667–3683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laurent A, Nicco C, Chéreau C, et al:
Controlling tumor growth by modulating endogenous production of
reactive oxygen species. Cancer Res. 65:948–956. 2005.PubMed/NCBI
|
|
31
|
Gillies LA and Kuwana T: Apoptosis
regulation at the mitochondrial outer membrane. J Cell Biochem.
115:632–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z, Cai F, Chen X, Luo M, Hu L and Lu
Y: The role of mitochondria-derived reactive oxygen species in
hyperthermia-induced platelet apoptosis. PLoS One. 8:e750442013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Crompton M: The mitochondrial permeability
transition pore and its role in cell death. Biochem J. 341:233–249.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: Bcl-2/Bax: a rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
36
|
Lindsay J, Esposti MD and Gilmore AP:
Bcl-2 proteins and mitochondria - specificity in membrane targeting
for death. Biochim Biophys Acta. 1813:532–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guha M, Kumar S, Choubey V, Maity P and
Bandyopadhyay U: Apoptosis in liver during malaria: role of
oxidative stress and implication of mitochondrial pathway. FASEB J.
20:1224–1226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Galluzzi L, Vitale I, Abrams JM, et al:
Molecular definitions of cell death subroutines: recommendations of
the Nomenclature Committee on Cell Death 2012. Cell Death Differ.
19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|