Introduction
Many biological processes show a circadian rhythm,
which is controlled by an endogenous clock that synchronizes with
day and night phases of the solar day. The periodical rhythm is
generated by a molecular oscillator located in the suprachiasmatic
nuclei of the hypothalamus, which controls peripheral oscillators
in virtually any other cell (1).
The circadian clock is organized through a complex network of
transcription-translational feedback loops that drive rhythmic
expression patterns of core clock components in mammals (2).
The Timeless (TIM) protein interacts with clock
proteins and is essential for generation of a circadian rhythm in
flies (3). In addition,
phylogenetic sequence analysis revealed the presence of a paralogue
in D. melanogaster (4),
which is not involved in the core clock machinery, but is important
for the maintenance of chromosome integrity, growth control, and
development. In contrast, a single TIM gene has been identified in
mammals, which acquired all of these functions as indicated by its
role in DNA damage response, replication, and circadian rhythm
(5–9). Consistently, TIM knockout resulted in
embryonic lethality in mice (10).
In addition, TIM and other clock genes have been linked to human
carcinogenesis (11–14). Using integrative molecular
profiling we have recently identified that inactivation of Period
homolog 3, a component of the clock machinery, occurs in human HCC
indicating that dysregulation of this regulatory network may
contribute to hepatocarcinogenesis (15).
The eukaryotic elongation factor 1-α (EEF1A), a
member of the G protein family, represents one of the four subunits
that constitute the eukaryotic elongation factor 1 (16). In humans, two EEF1A isoforms are
known. While EEF1A1 is expressed in almost all tissues, EEF1A2
expression was only reported in heart, brain and skeletal muscle
(17,18). However, EEF1A2 exerts an
anti-apoptotic function, which may explain its role in
tumorigenesis (19). We have
previously identified EEF1A2 as a candidate oncogene in human
hepatocarcinogenesis and demonstrated that EEF1A2 overexpression
was strongly associated with the survival probability of HCC
patients (20,21).
Here we analyzed the potential protumorigenic
function of TIM in human hepatocarcinogenesis. Our data show that
TIM overexpression promotes tumor cell proliferation via inhibition
of CHEK2 phosphorylation. In addition, TIM directly interacts with
the eukaryotic elongation factor 1A2 (EEF1A2), which promotes tumor
cell migration and affects ribosomal protein biosynthesis.
Materials and methods
Tumor material and patient
characteristics
Expression profiles were generated from 40 human
HCCs as described previously (15). The specimen included 22 liver
resections and 15 explant livers; median age at surgery was 56
years (range 16–78) and the male/female ratio was 3:1. Human tissue
samples were provided by the tissue bank of the National Center for
Tumor Diseases Heidelberg. All diagnoses were confirmed by
histological re-evaluation and use of the samples was approved by
the local ethics committee. According to the vote an informed
consent was not required because only long-term archived (>5
years), pseudonymized samples were used for this study. From three
patients two HCC nodules were included that previously showed
different aCGH data indicating independent tumor development.
Etiology was determined as previously described (20). The underlying etiologies were HBV
(n=8), HCV (n=8), alcohol (n=7), cryptogenic (n=10), genetic
hemochromatosis (n=3), and α1-antitrypsin deficiency (n=1). The
patient characteristics are shown in Table I.
 | Table IThe patient characteristics of the
HCC cohort used for expression profiling. |
Table I
The patient characteristics of the
HCC cohort used for expression profiling.
| Gender |
| Male | 27 (73%) |
| Female | 10 (27%) |
| Median age
(range) | 56 (16–78) |
| Etiology |
| HBV | 8 (21.6%) |
| HCV | 8 (21.6%) |
| Alcohol | 7 (18.9%) |
| Cryptogenic | 10 (27.0%) |
| Genetic
hemochromatosis | 3 (8.1%) |
| Others | 1 (2.7%) |
| Grading |
| Well
differentiated HCC | 4 (10.0%) |
| Moderately
differentiated HCC | 31 (77.5%) |
| Poorly
differentiated HCC | 5 (12.5%) |
| Tumor size |
| ≤5.0 cm | 18 (48.6%) |
| >5.0 cm | 19 (51.4%) |
| UICC stage |
| I | 24 (64.9%) |
| II | 11 (29.7%) |
| III | 0 (0%) |
| IV | 2 (5.4%) |
| Vascular
invasion |
| Present | 10 (27.0%) |
| None | 27 (73.0%) |
| Liver
cirrhosis |
| Present | 22 (59.4%) |
Reverse transcription and polymerase
chain reaction
RNA was isolated from 100 mg of snap-frozen tissue
after histological validation using the RNeasy Midi-Kit (Qiagen,
Hilden, Germany) according to the manufacturer’s instructions. One
microgram of total RNA was reversely transcribed with the
RevertAid™ H minus Reverse Transcriptase (Fermentas, St. Leon-Rot,
Germany) and analyzed using the ABI PRISM 7300 Real-Time PCR System
(Sequence Detection software v1.2.2, Applied Biosystems, Foster
City, CA, USA) with Absolute SYBR Green ROX Mix (ABgene, Epsom,
UK). Calculations of efficacy, normalization, and relative
quantification versus 18s rRNA were done according to published
algorithms (22). The following
primer sequences were used (Eurofins MWG Operon, Ebersberg,
Germany): TIM-fw 5′-GCC CTC AAT GTG AGG CTC TT-3′, TIM-rev 5′-CCC
GAA GCA GGT GAT CCT TT-3′, MDM4-fw 5′-CAG CAG GTG CGC AAG GTG
AA-3′, MDM4-rev 5′-CTG TGC GAG AGC GAG AGT CTG-3′, MDM2-fw 5′-TCT
GTG AGT GAG AAC AGG TGT CAC-3′, MDM2-rev 5′-ACA CAC AGA GCC AG GCT
TTC-3′, p21-fw 5′-CAC CGA GAC ACC ACT GGA GG-3′, p21-rev 5′-GAG AAG
ATC AGC CGG CGT TT-3′, puma-fw 5′-AAA CGG CTA CCA CAT CCA AG-3′,
puma-rev 5′-CCT CCA ATG GAT CCT CGT TA-3′, bax-fw 5′-TGG AGC TGC
AGA GGA TGA TTG-3′, bax-rev 5′-AAA CAT GTC AGC TGC CAC TCG-3′,
EEF1A2-fw 5′-AGA TGT CGA TGG TGA TGC-3′, EEF1A2-rev 5′-AGA TGT CGA
TGG TGA TGC-3′, 18s-fw 5′-AAA CGG CTA CCA CAT CCA AG-3′, 18s-rev
5′-CCT CCA ATG GAT CCT CGT TA-3′. Reaction temperatures and periods
were used according to the manufacturer’s instructions
(ABgene).
DNA microarray hybridization and
analysis
Quality and integrity of the total RNA was
controlled using an Agilent Technologies 2100 Bioanalyzer (Agilent
Technologies, Waldbronn, Germany). Total RNA (200 ng) was applied
for Cy3-labelling reaction using the one color Quick Amp Labeling
protocol (Agilent Technologies). Labeled cRNA was hybridized to
Agilent human 8×60k microarrays at 68°C for 16 h and scanned using
the Agilent DNA Microarray Scanner (Agilent Technologies).
Expression values were calculated by the software package Feature
Extraction 10.5.1.1. Complete data are available online (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50579).
Western blot analysis and
immunoprecipitation
Cell samples were homogenized in lysis buffer (no.
9803, Cell Signaling Technology, Danvers, MA, USA) containing the
complete protease inhibitor cocktail (Roche, Mannheim, Germany) and
sonicated. Protein concentrations were determined using Nanodrop
ND1000 (Thermo Scientific, Waltham, MA, USA). For Western blotting,
aliquots of 100 μg were denatured by boiling in Tris-glycine SDS
sample buffer (Invitrogen, Karlsruhe, Germany), separated by
SDS-PAGE, and blotted onto nitrocellulose membranes (Invitrogen).
Membranes were blocked in 5% non-fat dry milk in Tris-buffered
saline containing 0.1% Tween-20 for 1 h and probed with the
following specific antibodies: mouse monoclonal anti-β-actin
(1:10,000; MP Biomedicals, Santa Ana, CA, USA), rabbit polyclonal
anti-AKT (1:1,000; Cell Signaling Technology), mouse monoclonal
anti-CHK1 (1:1,000; Cell Signaling Technology), rabbit monoclonal
anti-CHK2 (1:1,000; Cell Signaling Technology), rabbit monoclonal
anti-EEF1A2 (1:1,500, Abcam, Cambridge, UK), mouse monoclonal
anti-MDM2 (1:200; Santa Cruz Biotechnology, Dallas, TX, USA),
rabbit polyclonal anti-MDM4 (1:3,000; Abiocode, Agoura Hills, CA,
USA), rabbit monoclonal anti-pAKT (Serine 473, 1:500; Cell
Signaling Technology), rabbit polyclonal anti-PARP (1:500; Cell
Signaling Technology), rabbit monoclonal anti-pCHK1 (Serine 345,
1:1,000; Cell Signaling Technology), rabbit polyclonal anti-pCHK2
(Threonine 68, 1:1,000; Cell Signaling Technology), rabbit
polyclonal anti-TIM (1:2,000; Bethyl Laboratories, Montgomery, TX,
USA), mouse monoclonal anti-α-tubulin (1:2,000; Sigma-Aldrich, St.
Louis, MO, USA). Each incubation with a primary antibody was
followed by incubation with a horseradish peroxidase-conjugated
secondary antibody for 1 h (1:2,000; Cell Signaling Technology) and
visualized using the Super Signal West Pico (Pierce Chemical, New
York, NY, USA). For quantification the protein densities were
calculated by ImageQuaNT 5.1 software (GE Healthcare, Piscataway,
NJ, USA) and normalized to α-tubulin to determine the relative
expression levels. For immunoprecipitation (IP) experiments NP40
buffer (Tris-HCl 50 mM, NACl 150 mM, NP40 1%) was used for protein
isolation. Three milligrams of HCC cell protein lysate were
immunoprecipitated with 6 μg of protein A agarose-bound EEF1A2
rabbit polyclonal antibody (Santa Cruz Biotechnology) at 4°C
overnight. After immunoblotting, the membranes were incubated with
a rabbit monoclonal anti-EEF1A2 antibody (1:1,500; Abcam), rabbit
polyclonal anti-TIM (Bethyl Laboratories) or mouse monoclonal
anti-actin antibody (MP Biomedicals). To exclude unspecific binding
IP was carried out without protein input as negative control.
Respectively, mouse monoclonal anti-rabbit and goat anti-mouse
light chain IgG (1:7,000; Jackson ImmunoResearch Laboratories, West
Grove, PA, USA) were used as secondary antibodies to detect EEF1A2
bound proteins.
Tissue microarrays and
immunohistochemistry
A tissue microarray (TMA) containing tissue from
normal livers (n=27), non-tumor liver tissue of HCC patients
(n=86), and HCCs (n=102; Table
II) was constructed as previously described (23), and immunohistochemistry was
performed on 5-μm sections. From eleven patients two independent
HCC nodules were included. The following primary antibodies were
used for incubation: rabbit monoclonal EEF1A2 (1:1,500; Abcam),
rabbit polyclonal Timeless (1:2,000; Bethyl Laboratories). Antigens
were retrieved using citrate buffer (pH 6.1; Dako, Glostrup,
Denmark). For detection the EnVision method (Dako) was used.
Counterstaining was performed using hemalum. Staining was assessed
using the immunoreactive score as described previously (20): 0, absent; 1–4, weak; 5–8, moderate;
9–12, strong expression.
| Table IIThe patient characteristics of TMA
cohort. |
Table II
The patient characteristics of TMA
cohort.
| Gender |
| Male | 72 (79%) |
| Female | 19 (21%) |
| Median age
(range) | 59 (17–78) |
| Etiology |
| HBV | 17 (18.7%) |
| HCV | 27 (29.7%) |
| Co-infection | 5 (5.5%) |
| Alcohol | 21 (23.1%) |
| Cryptogenic | 18 (19.8%) |
| Genetic
hemochromatosis | 3 (3.3%) |
| Grading |
| Well
differentiated HCC | 19 (18.6%) |
| Moderately
differentiated HCC | 65 (63.7%) |
| Poorly
differentiated HCC | 18 (17.6%) |
| Tumor size |
| <2.0 cm | 8 (8.8%) |
| 2.0–5.0 cm | 51 (56.0%) |
| >5.0 cm | 32 (35.2%) |
| UICC stage |
| I | 39 (42.9%) |
| II | 32 (35.2%) |
| III | 15 (16.5%) |
| IV | 5 (5.5%) |
| Vascular
invasion |
| Present | 32 (35.2%) |
| None | 59 (64.8%) |
| Liver
cirrhosis |
| Present | 58 (63.7%) |
Cell lines, transfection and functional
analyses
HepG2, Hep3B, HLE, HLF, HuH6, HuH7, PLC/PRF/5,
SNU182 and SNU387 cells were cultured either in DMEM, MEM or RPMI
medium, supplemented with 10% fetal bovine serum (PAA, Pasching,
Austria) and 1% penicillin-streptomycin (10 mg/ml, PAA) at 37°C (5%
CO2) and passaged every 3–4 days. All siRNA
transfections were performed using oligofectamine (Invitrogen)
according to the manufacturer’s protocol. The following small
interfering RNAs were used (siRNA, Eurofins MWG Operon): siTIM1
5′-AGA AGA GAA GGA AGA AGA A-dTdT3′, siTIM3: 5′-GCC UAC AUG UGC UAG
AGA U-dTdT3′, neutral control siRNA (siNC) 5′-UUC UCC GAA CGU GUC
ACG U-dTdT3′. Transient transfection experiments of Hep3B cells
with EEF1A2 cDNA in pCMV6-XL5 vector (OriGene Technologies,
Rockville, MD, USA) was performed following the manufacturer’s
protocol using FuGene HD transfection reagent (Promega, Mannheim,
Germany). For functional assays HCC cell lines were seeded at a
density of 6×103 cells in 96-well plates and were
transfected with siRNA after 24 h. Cell viability (MTT-assay;
M2128, Sigma, Deisenhofen, Germany), apoptosis (FACS-assay; FACS
Calibur™, BD Bioscience, Heidelberg, Germany), and migration
(2D-scratch-assay) were determined as described previously
(15,20). For the treatment with chemical
inhibitors, HCC cells were plated at a density of
1.5×105 cells/well in 6-cm plates and were incubated
with the following drugs after 24 h, as indicated: cycloheximide
(protein synthesis inhibitor; Merck, Darmstadt, Germany), MG132
(proteosomal inhibitor, 450 μmol/l; Enzo Life Sciences, Lörrach,
Germany). For all cell based assays, results were confirmed in
three independent experiments.
Immunofluorescence microscopy
After 24 h of culturing on glass coverslips HCC
cells were transfected using siRNAs as described above. Seventy-two
hours after transfection cells were washed repeatedly in PBS
containing 2 mM MgCl2 at 37°C, and fixed with
methanol/acetone. Fixed cells were again washed with PBS for 30
min, permeabilized in PBS with 0.05% Tween-20 for 10 min, and
processed as described previously (24) using a mouse monoclonal β-actin
antibody (1:5,000; MP Biomedicals). Fluorescence was detected using
an Olympus BX40 microscope (Olympus, Hicksville, NY, USA) equipped
with a Leica DFC365FX camera (Leica, Wetzlar, Germany).
Statistical analyses
The correlation between gene expression and
clinicopathological parameters was tested by Wilcoxon-signed rank
tests and measured by Spearman’s rank correlations. P<0.05 was
considered statistically significant. Statistical analyses were
conducted using SPSS 20.0 (SPSS, Chicago, IL, USA).
Results
TIM is upregulated in a subset of human
HCCs
According to our previous expression profiling
(15) upregulation of TIM mRNA
(>2-fold) was observed in 55% of HCCs (n=22), while only 1 tumor
(2.5%) showed lower expression (<50%) compared to normal liver
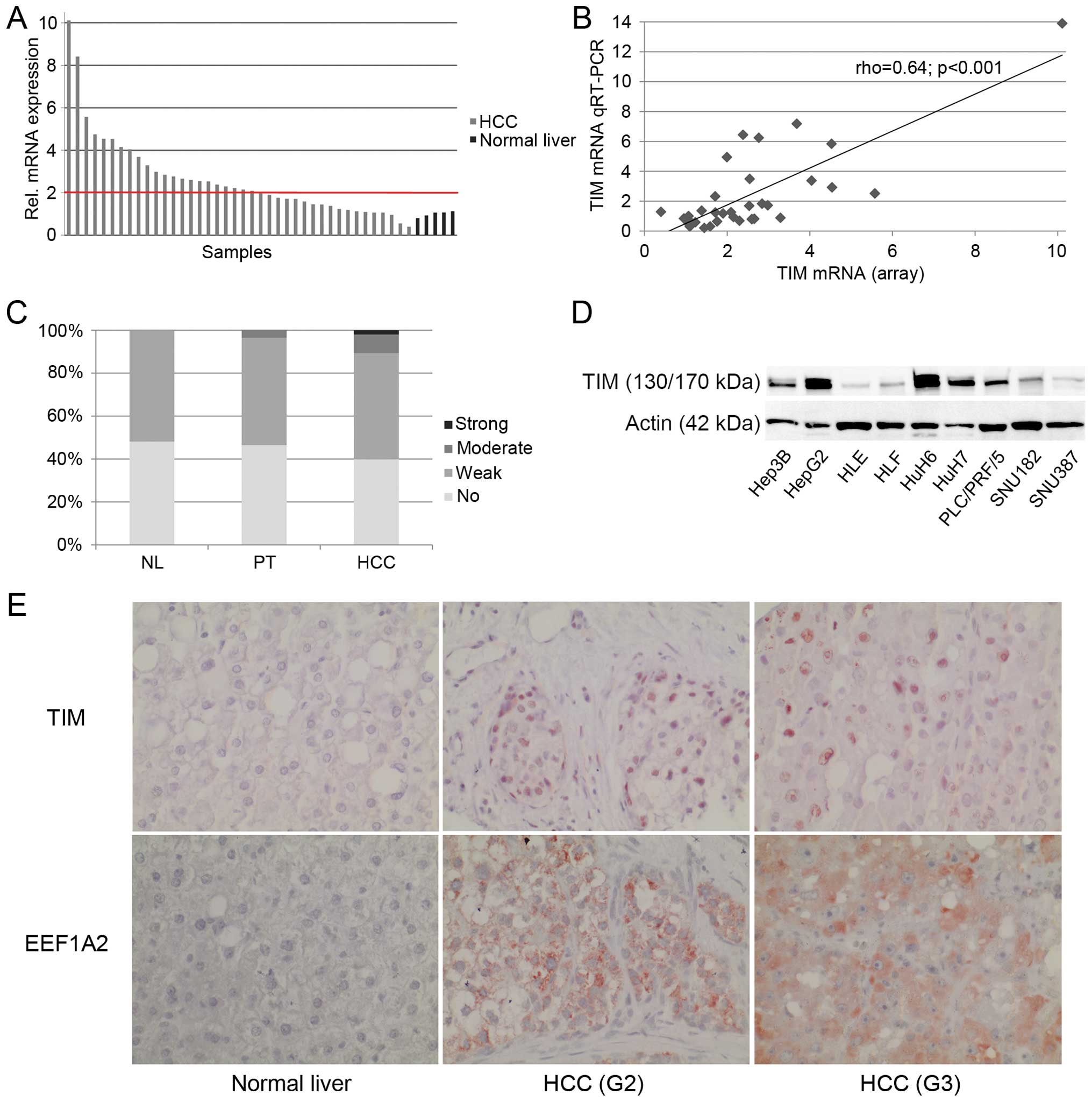
(Fig. 1A). These findings were
validated using quantitative real-time RT-PCR (Fig. 1B). Thus, there was a significant
upregulation of TIM mRNA in human HCCs compared to normal liver
(P=0.004). Tumors measuring >5 cm in diameter showed
significantly higher TIM mRNA levels compared to smaller lesions
(P=0.01). Although there was a trend for increased TIM expression
with tumor dedifferentiation, the results did not reach statistical
significance (rho=0.3, P=0.07). No significant association was
found with gender, etiology, vascular invasion and UICC stage
(P>0.05).
Next we determined the TIM protein expression in
human HCCs using TMA (Fig. 1C and
E). Weak TIM expression was seen in 52% (n=14) of normal liver
tissues and the remaining were negative. In non-tumorous liver
tissues of HCC patients (n=86), 47% displayed no detectable TIM
signal at all, whereas half of the samples showed weak (n=43) and
3% showed moderate expression of TIM. Regarding HCCs (n=103), 40%
did not show any, 50% showed weak, 9% moderate, and 2% showed
strong TIM staining. There was a trend towards higher TIM protein
expression in primary HCCs that had developed extrahepatic
metastasis compared to non-metastasized tumors (P=0.05). No
significant association was found with gender, etiology, vascular
invasion, and tumor size (P>0.05).
TIM promotes cell viability and tumor
cell migration
TIM protein was expressed in all HCC cell lines
analyzed. High TIM expression levels were observed in Hep3B, HepG2,
HuH6, HuH7, and PLC/PRF/5 cells (Fig.
1D). To test whether TIM exerts protumorigenic functions in
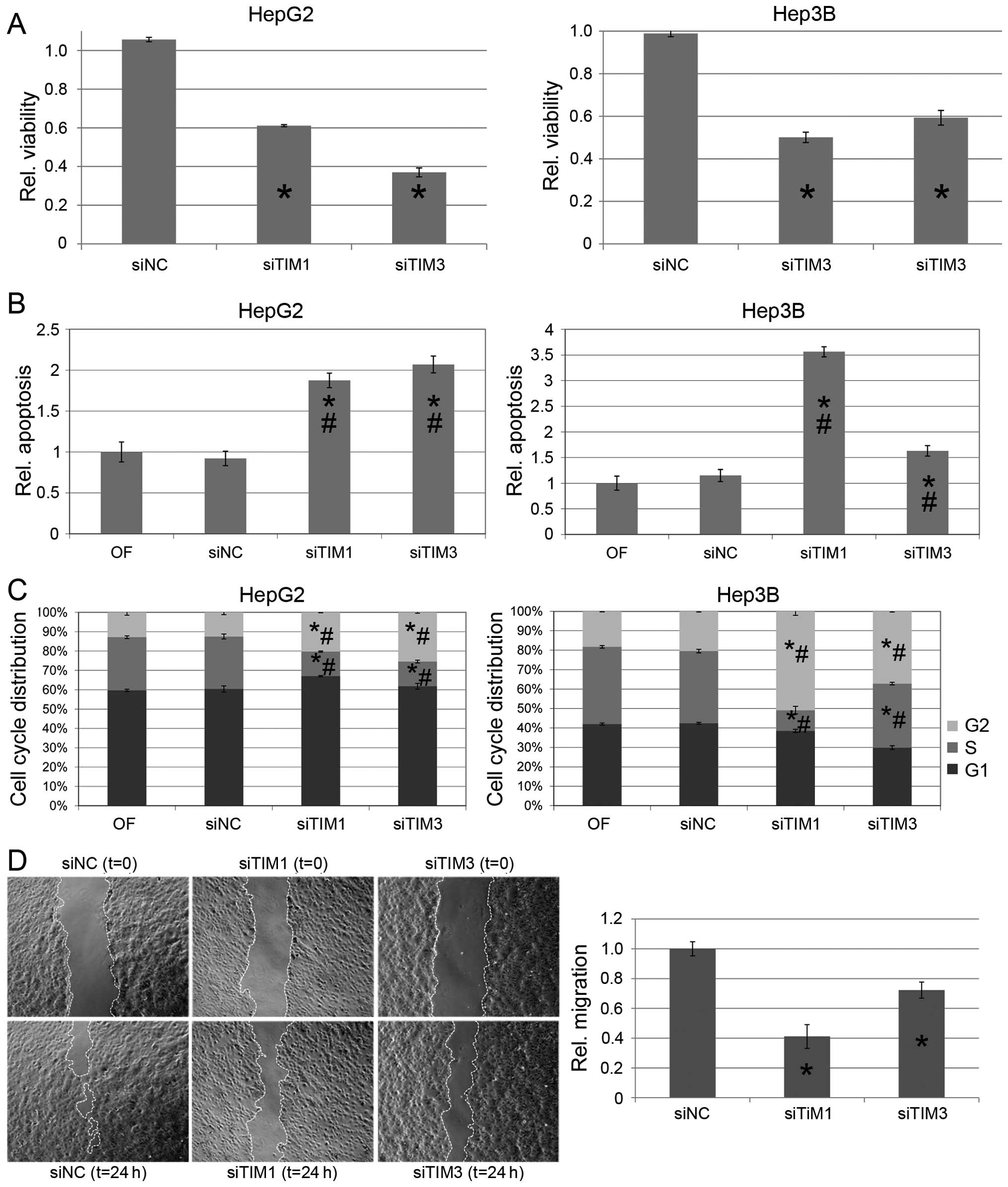
vitro, we used gene-specific siRNAs to knock down TIM
expression in HepG2 and Hep3B cells, which resulted in significant
reduction of cell viability compared to mock transfected cells
[0.61±0.03 (siTIM1) and 0.37±0.11 (siTIM3) in HepG2; 0.50±0.10
(siTIM1) and 0.59±0.17 (siTIM3) in Hep3B cells, P<0.01
respectively; Fig. 2A], and was
associated with a mild induction of apoptosis as shown by FACS
analysis [1.87±0.09-fold (siTiM1) and 2.07±0.10-fold (siTIM3) in
HepG2 and 3.56±0.10-fold (siTIM1) and 1.62±0.10-fold (siTIM3) in
Hep3B compared to Mock transfected cells, each P<0.01; Fig. 2B] as well as cleavage of PARP in
Western immunoblotting (Fig. 3A).
In addition, knockdown of TIM resulted in a significant G2 arrest
in both cells lines analyzed [20.3±0.2% (siTIM1)/25.8±0.5% (siTIM3)
vs. 12.8±1.2% (siNC) in HepG2 and 50.5±2.1% (siTIM1)/37.4±0.4%
(siTIM3) vs. 20.2±0.3% in Hep3B cells, P<0.01 respectively;
Fig. 2C]. Finally, migration was
significantly reduced in TIM-depleted compared to siNC-transfected
Hep3B cells [0.41±0.08 (siTIM1) and 0.72±0.05 (siTIM3), P<0.01
respectively; Fig. 2D]. Due to
their multilayer and colony forming growth characteristics the
migration of HepG2 cells could not be explored in a standardized
manner.
Mechanisms involved in pro-proliferative
and anti-apoptotic function of TIM
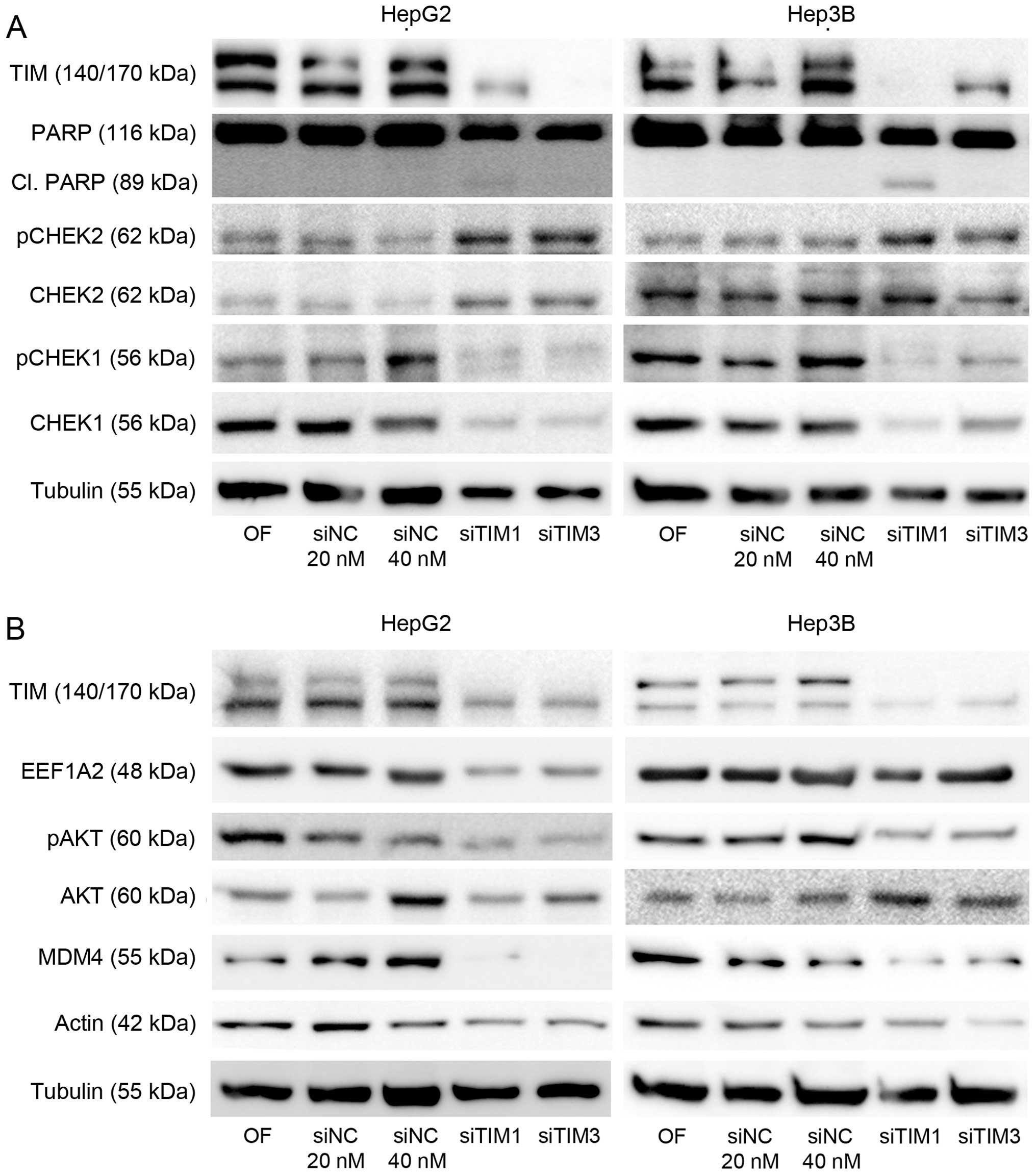
As TIM has been reported to affect cell
proliferation either via CHEK1-or CHEK2-mediated mechanisms
(8,25), we analyzed both expression and
phosphorylation of these proteins. Indeed, both CHEK2 protein
levels as well as CHEK2 phosphorylation were increased in HepG2 and
Hep3B cells after knockdown of TIM expression (Fig. 3A). Interestingly, we observed that
numerous other proteins including CHEK1 (Fig. 3A), EEF1A2, v-akt murine thymoma
viral oncogene homolog (AKT), and β-actin (Fig. 3B) were downregulated following TIM
knockdown suggesting that TIM may activate the protumorigenic
EEF1A2/AKT/MDM4 axis (21). In
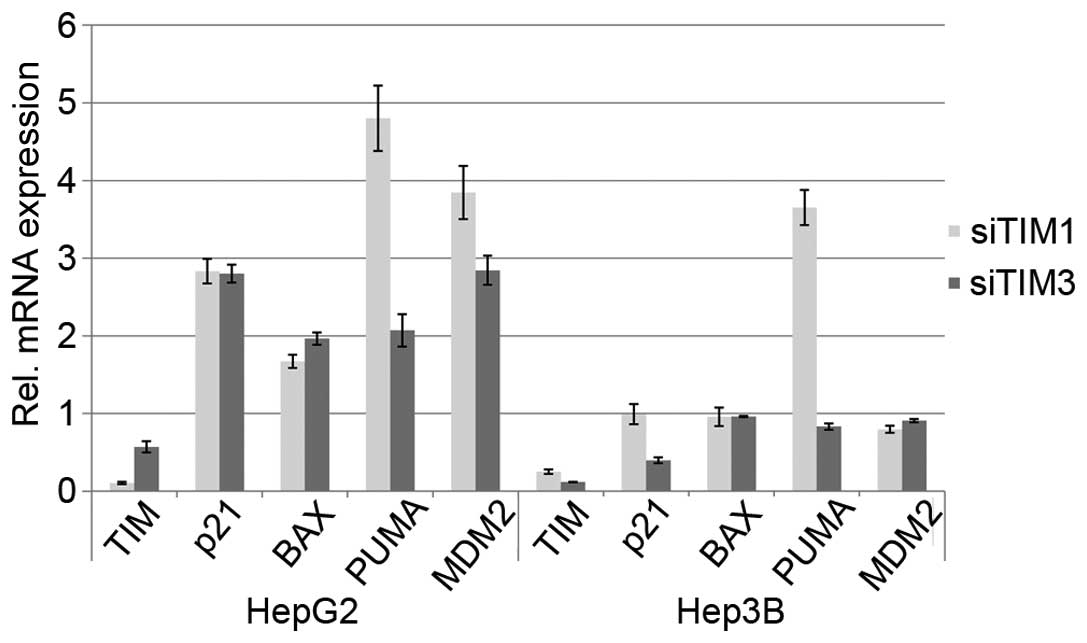
line, transcriptional activation of some p53 target genes was seen
following TIM knockdown in p53-wild-type cells indicating
reactivation of p53 function in these cells (Fig. 4).
EEF1A2 is a crucial mediator of
protumorigenic TIM functions
EEF1A2 has been shown to promote oncogenic functions
in HCC (20) and was downregulated
upon TIM knockdown. In addition, we observed a correlation
(rho=0.37, P>0.001) between EEF1A2 and TIM protein as determined
by immunohistochemistry (Fig. 1E).
As EEF1A2 is involved in ribosomal function as well as actin
dynamics, we hypothesized that EEF1A2 could be a crucial mediator
of the protumorigenic properties of TIM. To explore whether EEF1A2
was able to revert the TIM-induced migratory phenotype, we
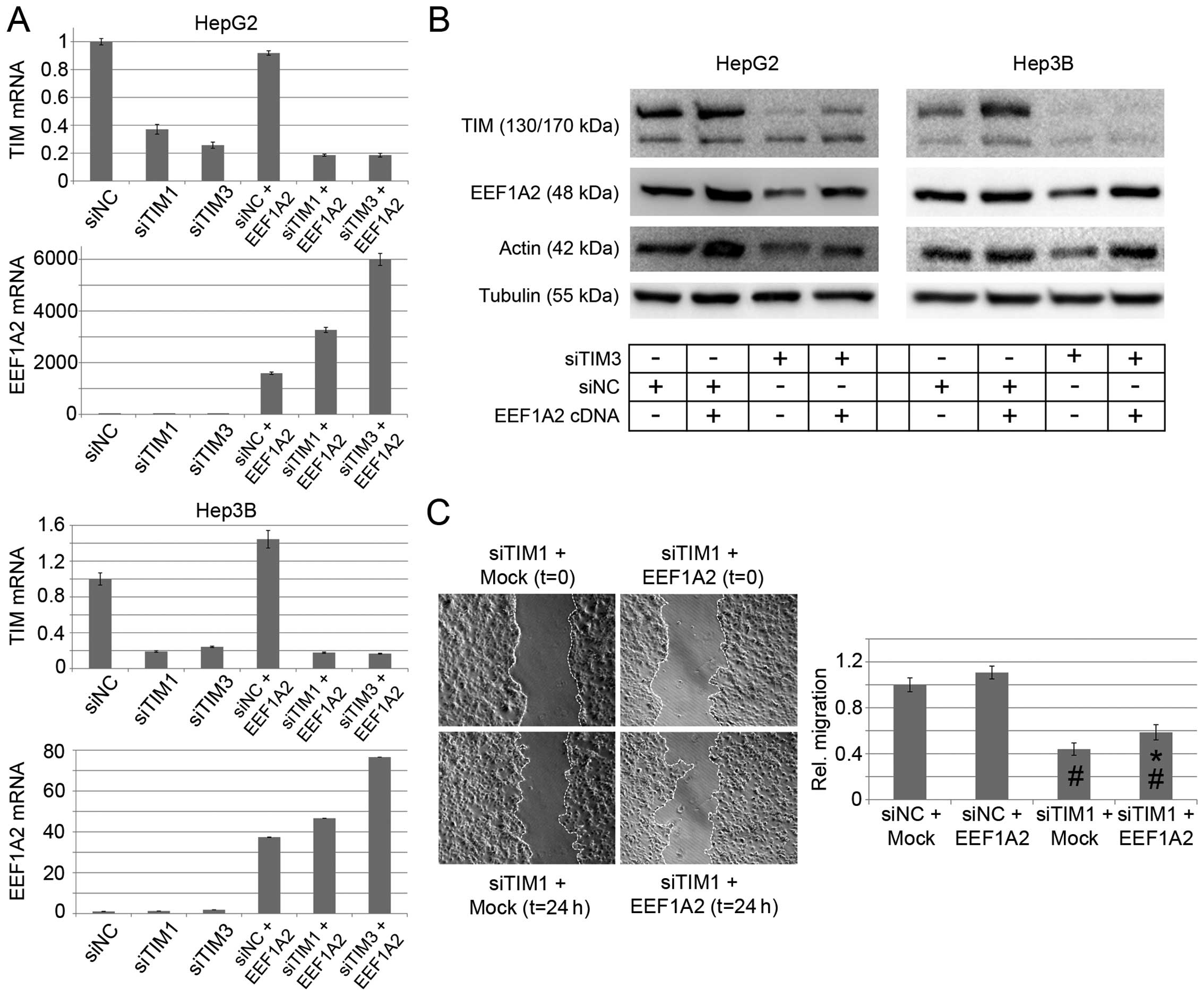
transiently expressed an EEF1A2 cDNA after TIM knockdown. Both,
EEF1A2 mRNA and protein levels were increased (Fig. 5A and B), which was paralleled by an
increase in migratory capacity compared to Mock transfected cells.
However, both the migration of siTIM1-treated Hep3B cells as well
as their EEF1A2 protein level remained lower compared to
siNC-treated Hep3B cells (Fig. 5B and
C).
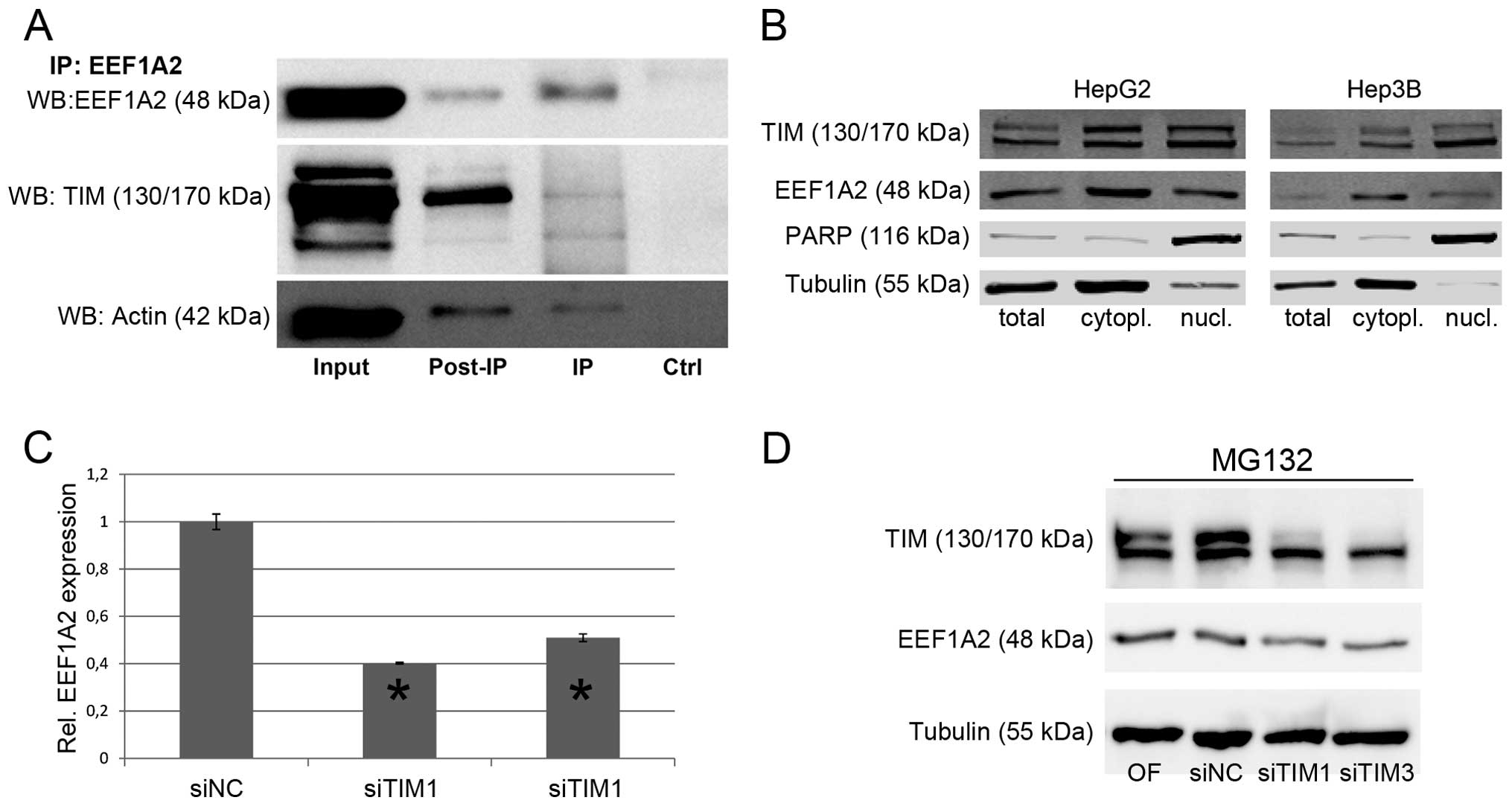
Immunoprecipitation experiments revealed a direct
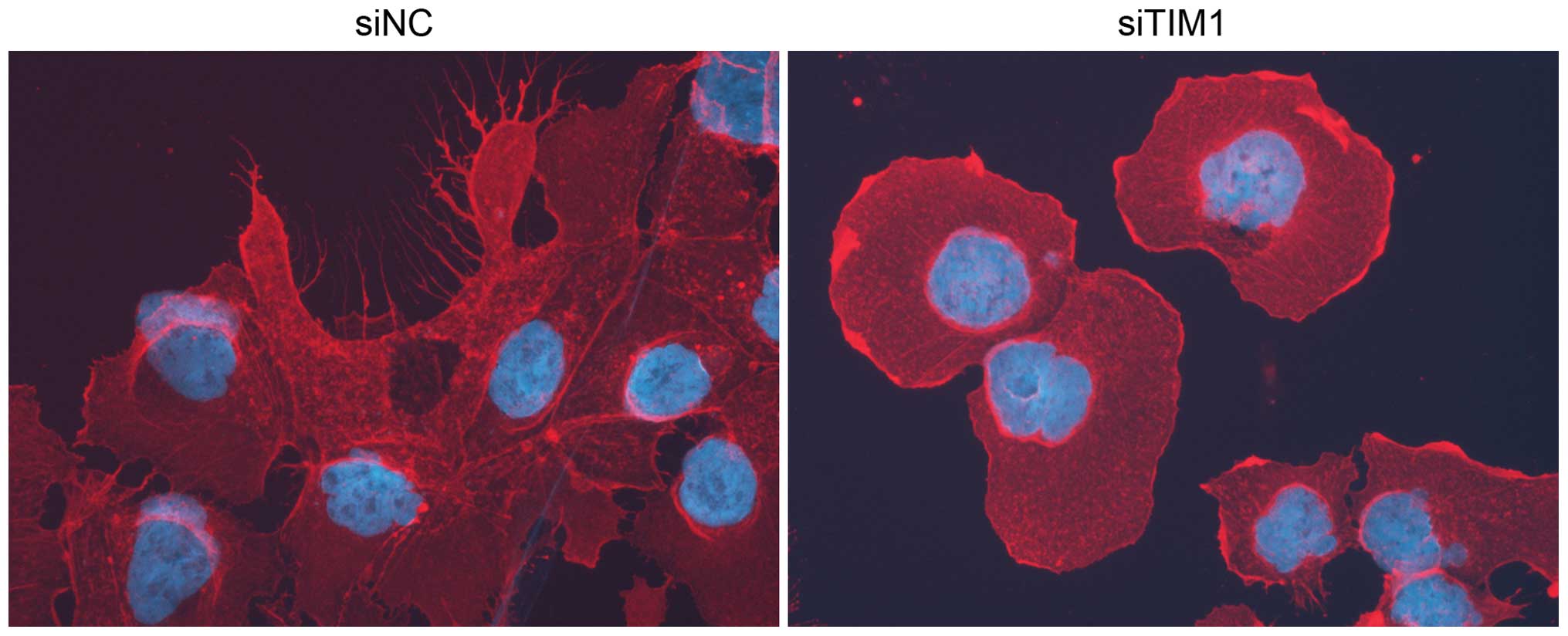
interaction of EEF1A2 with both TIM and β-actin (Fig. 6A). In addition, filopodia formation
was impaired following TIM knockdown (Fig. 7) indicating that the TIM-EEF1A2
interaction may promote actin dynamics and thus tumor cell
migration. As TIM was expressed predominantly in the nucleus, while
EEF1A2 was present in the cytoplasm (Fig. 1E), we further explored in which
cellular compartment TIM and EEF1A2 might interact. Cellular
fractionation confirmed that EEF1A2 was mainly localized in the
cytoplasm, while TIM expression was observed in both compartments
suggesting that the TIM-EEF1A2 interaction occurs in the cytoplasm
(Fig. 6B).
Although TIM seemed to stabilize EEF1A2 via direct
interaction, this finding did not explain the reduction of other
proteins like CHEK1, AKT, or β-actin following TIM knockdown. In
humans, two EEF1A protein isoforms (EEF1A1 and EEF1A2), which have
similar elongation activity, are known (17,18).
Thus, we evaluated the effect of TIM knockdown on protein
biosynthesis. EEF1A2 protein levels were significantly reduced
after cycloheximide treatment in siTIM-compared to siNC-treated
cells (Fig. 6C), while inhibition
of proteasomal degradation using MG132 did not prevent reduction of
EEF1A2 expression following TIM knockdown (Fig. 6D) indicating that impaired protein
biosynthesis, and not increased proteasomal degradation, may be
responsible for this phenotype.
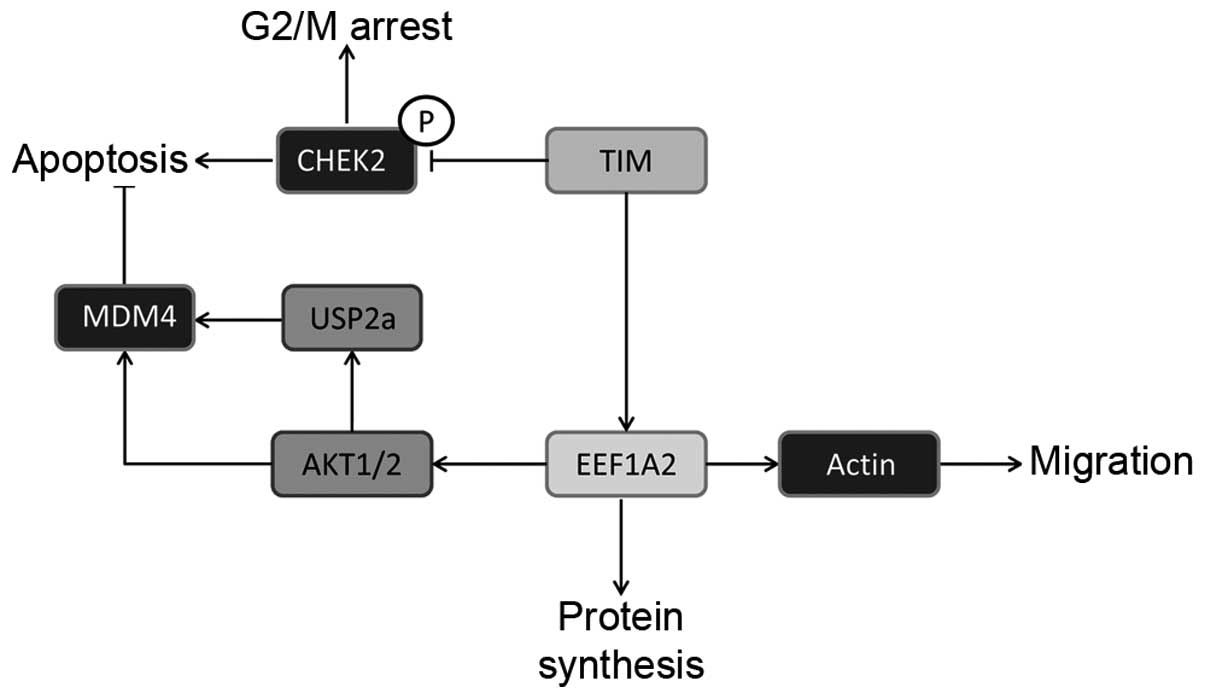
Thus, our data show that TIM exerts protumorigenic
functions in human HCC, which are crucially mediated by physical
interaction with EEF1A2 and downstream activation of the AKT
signaling cascade as well as promotion of tumor cell migration. A
schematic model of the protumorigenic mechanisms activated by TIM
overexpression is shown in Fig.
8.
Discussion
By demonstrating the epigenetic inactivation of
Period homolog 3 we have recently shown that dysregulation of clock
machinery components occurs in human HCC (15). We hypothesized that altered clock
function may be a hallmark of hepatocarcinogenesis and found six
clock genes (CLOCK, CSNK1E, PER1, PER2, RORA and TIM; Gene
Expression Omnibus Series GSE50579) to be differentially expressed
on the mRNA level between HCC and normal liver. We focused on TIM
as it has been shown to be important for the maintenance of
chromosome integrity and growth control (5,6), and
a recent meta-analysis revealed a transcriptional
timeless-dependent network and suggested a role of TIM in
development and progression of various epithelial cancer types
(e.g., bladder, breast, cervical, colorectal, gastric, head and
neck, kidney, lung and prostate) (26). Here, we observed that TIM is
overexpressed in a subset of human HCCs, which is contradictory to
a study reporting downregulation of TIM in HCC (27). Our functional analysis supports an
oncogenic function as TIM knockdown resulted in reduced viability
of HCC cells due to the induction of apoptosis and cell cycle
arrest in vitro. The effect on proliferation may be related
to the fact that TIM-TIPIN complexes have been shown to stabilize
the replication fork during early S phase. Depletion of TIM
resulted in defects of mitotic progression, impaired sister
chromatid cohesion and chromosome fragmentation (28). In line with a previous report
showing that TIM is required for ATM-dependent CHEK2 activation and
G2/M checkpoint control (8), we
observed that siRNA-mediated inhibition of TIM in HCC cells
increased CHEK2 phosphorylation resulting in cell cycle arrest.
Together, these data provide evidence that upregulation of TIM in
HCC cells promotes tumor cell proliferation. Although systemic
targeting of TIM might exert antitumoral activity in overexpressing
tumors, such an approach is probably limited by its physiological
function in the replication fork (28), suggesting that the spectrum of
unwanted side-effects following targeting of TIM might be similar
to conventional chemotherapy due to broad unspecific targeting of
any proliferating cell. Accordingly, specific targeting of the
relevant ‘non-canonical’ mediators of the oncogenic TIM function
might be a more promising therapeutic strategy.
Recent data indicated that EEF1A2 overexpression is
a hallmark of prostate and ovarian cancer and plays an important
role in mammary carcinogenesis (29–31).
We have previously identified EEF1A2 as a candidate oncogene in
human hepatocarcinogenesis (20).
EEF1A2 acts as an upstream inducer of the PI3K/AKT/mTOR axis
leading to functional inactivation of p53 by posttranslational
stabilization of the mouse double minute homolog 4 (MDM4) in human
HCCs (21).
Interaction of EEF1A with the cytoskeleton is
important for efficient translation and EEF1A is involved in the
organization of the actin cytoskeleton in vivo (32,33).
Herein, we demonstrate that EEF1A2 directly
interacts with TIM and β-actin. Since our data indicate that the
TIM-EEF1A2 interaction occurs likely in the cytoplasm, we speculate
that TIM exerts its oncogenic function at least partially in this
compartment. In line, knockdown of TIM reduced EEF1A2 expression
and impaired tumor cell migration. EEF1A2 has been shown to induce
filopodia formation in cell lines and enhance migration in an AKT-
and PI3K-dependent manner (34,35)
indicating that the reduced activation of the EEF1A2/PI3K/AKT/mTOR
axis may contribute to the migratory phenotype observed in this
study.
In addition to its canonical function in
translational elongation EEF1A exerts a plethora of non-canonical
activities including export of proteins from the nucleus (36), a role in viral replication
(37), and interaction with the
proteasome which is increased when translation is inhibited
(38). As EEF1A2 protein levels
could not be rescued by pharmacological inhibition of the
proteasome after TIM knockdown, the latter function is unlikely to
contribute to the decreased expression of various proteins observed
here. The most likely explanation is that the EEF1A2 knockdown
impaired ribosomal function resulting in reduced protein
biosynthesis in HCC cells as indicated by significantly decreased
EEF1A2 protein levels following cycloheximide treatment.
In conclusion, TIM exerts protumorigenic functions
in human HCC, which are crucially mediated by physical interaction
with EEF1A2 and the activation of its downstream signaling cascade
as well as promotion of tumor cell migration via EEF1A2-mediated
activation of actin dynamics. Although targeting of TIM and EEF1A2
was effective in vitro, EEF1A2 seems to be the more
promising target as its physiological expression level is
restricted to non-proliferating cells and de novo expression
is frequently found in human cancers. Future studies are needed to
evaluate both the efficacy as well as potential unwanted effects of
these treatment approaches in vivo.
Acknowledgements
We are grateful to Ariane Eberhardt, Veronika
Geissler, Sara Messard, Verena Kautz, and Eva Eiteneuer for
excellent technical assistance. This study was supported by the
Deutsche Forschungsgemeinschaft DFG SFB/TRR77 (subproject B5 to
R.P, P.S. and T.L.), the Deutsche Krebshilfe (no. 110885 to T.L.
and O.N.), and the Tissue Bank of the National Center for Tumor
Diseases Heidelberg. N.E. received a stipend from the Egyptian
Ministry of Higher Education.
Abbreviations:
|
aCGH
|
array-based comparative genomic
hybridization
|
|
ATM
|
ataxia telangiectasia mutated
|
|
AKT
|
v-akt murine thymoma viral oncogene
homolog
|
|
cDNA
|
complementary DNA
|
|
CHEK1
|
checkpoint kinase 1
|
|
CHEK2
|
checkpoint kinase 2
|
|
CHX
|
cycloheximide
|
|
CSNK1E
|
casein kinase 1ɛ
|
|
Ctrl
|
negative control
|
|
DNA
|
deoxyribonucleic acid
|
|
EEF1A2
|
eukaryotic elongation factor 1A2
|
|
fw
|
forward
|
|
HBV
|
hepatitis B virus
|
|
HCV
|
hepatitis C virus
|
|
HCC
|
hepatocellular carcinoma
|
|
IP
|
immunoprecipitation
|
|
MDM2
|
mouse double minute homolog 2
|
|
MDM4
|
mouse double minute homolog 4
|
|
mTOR
|
mammalian target of rapamycin
|
|
NL
|
normal liver
|
|
OF
|
oligofectamine control
|
|
p21
|
cyclin-dependent kinase inhibitor 1A
(CDKN1A, Cip1)
|
|
p53
|
tumor protein p53
|
|
PI3K
|
phosphoinositide-3-kinase
|
|
PAGE
|
polyacrylamide gel electrophoresis
|
|
PER1
|
period circadian clock 1
|
|
PER2
|
period circadian clock 2
|
|
PT
|
peritumorous, non-neoplastic liver
tissue
|
|
PUMA
|
BCL2 binding component 3
|
|
qRT-PCR
|
quantitative real-time polymerase
chain reaction
|
|
RNA
|
ribonucleic acid
|
|
RORA
|
RAR-related orphan receptor A
|
|
rev
|
reverse
|
|
SDS
|
sodium dodecyl sulfate
|
|
SEM
|
standard error of the mean
|
|
siNC
|
non-target control siRNA
|
|
siRNA
|
short interfering RNA
|
|
siTIM
|
siRNA targeting Timeless
|
|
TIM
|
Timeless
|
|
TIPIN
|
TIMELESS interacting protein
|
|
TMA
|
tissue microarray
|
|
WB
|
western immunoblotting
|
References
|
1
|
Reppert SM and Weaver DR: Coordination of
circadian timing in mammals. Nature. 418:935–941. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ko CH and Takahashi JS: Molecular
components of the mammalian circadian clock. Hum Mol Genet. 15(Spec
No 2): R271–R277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ceriani MF, Darlington TK, Staknis D, et
al: Light-dependent sequestration of TIMELESS by CRYPTOCHROME.
Science. 285:553–556. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benna C, Scannapieco P, Piccin A, et al: A
second timeless gene in Drosophila shares greater sequence
similarity with mammalian tim. Curr Biol. 10:R512–R513. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuo T, Yamaguchi S, Mitsui S, Emi A,
Shimoda F and Okamura H: Control mechanism of the circadian clock
for timing of cell division in vivo. Science. 302:255–259. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Unsal-Kacmaz K, Mullen TE, Kaufmann WK and
Sancar A: Coupling of human circadian and cell cycles by the
timeless protein. Mol Cell Biol. 25:3109–3116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Unsal-Kacmaz K, Chastain PD, Qu PP, et al:
The human Tim/Tipin complex coordinates an Intra-S checkpoint
response to UV that slows replication fork displacement. Mol Cell
Biol. 27:3131–3142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang X, Wood PA and Hrushesky WJ:
Mammalian TIMELESS is required for ATM-dependent CHK2 activation
and G2/M checkpoint control. J Biol Chem. 285:3030–3034. 2010.
View Article : Google Scholar :
|
|
9
|
Barnes JW, Tischkau SA, Barnes JA, et al:
Requirement of mammalian Timeless for circadian rhythmicity.
Science. 302:439–442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gotter AL, Manganaro T, Weaver DR, et al:
A time-less function for mouse timeless. Nat Neurosci. 3:755–756.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wood LD, Parsons DW, Jones S, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fu L, Pelicano H, Liu J, Huang P and Lee
C: The circadian gene Period2 plays an important role in tumor
suppression and DNA damage response in vivo. Cell. 111:41–50. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu A, Leaderer D, Zheng T, Hoffman AE,
Stevens RG and Zhu Y: Genetic and epigenetic associations of
circadian gene TIMELESS and breast cancer risk. Mol Carcinog.
51:923–929. 2012. View
Article : Google Scholar
|
|
14
|
Mazzoccoli G, Panza A, Valvano MR, et al:
Clock gene expression levels and relationship with clinical and
pathological features in colorectal cancer patients. Chronobiol
Int. 28:841–851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Neumann O, Kesselmeier M, Geffers R, et
al: Methylome analysis and integrative profiling of human HCCs
identify novel protumorigenic factors. Hepatology. 56:1817–1827.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee MH and Surh YJ: eEF1A2 as a putative
oncogene. Ann NY Acad Sci. 1171:87–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knudsen SM, Frydenberg J, Clark BF and
Leffers H: Tissue-dependent variation in the expression of
elongation factor-1 alpha isoforms: isolation and characterisation
of a cDNA encoding a novel variant of human elongation-factor 1
alpha. Eur J Biochem. 215:549–554. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee S, Francoeur AM, Liu S and Wang E:
Tissue-specific expression in mammalian brain, heart, and muscle of
S1, a member of the elongation factor-1 alpha gene family. J Biol
Chem. 267:24064–24068. 1992.PubMed/NCBI
|
|
19
|
Ruest LB, Marcotte R and Wang E: Peptide
elongation factor eEF1A-2/S1 expression in cultured differentiated
myotubes and its protective effect against caspase-3-mediated
apoptosis. J Biol Chem. 277:5418–5425. 2002. View Article : Google Scholar
|
|
20
|
Schlaeger C, Longerich T, Schiller C, et
al: Etiology-dependent molecular mechanisms in human
hepatocarcinogenesis. Hepatology. 47:511–520. 2008. View Article : Google Scholar
|
|
21
|
Pellegrino R, Calvisi DF, Neumann O, et
al: EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent
stabilization of MDM4 in hepatocellular carcinoma. Hepatology.
59:1886–1899. 2014. View Article : Google Scholar
|
|
22
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Longerich T, Breuhahn K, Odenthal M,
Petmecky K and Schirmacher P: Factors of transforming growth factor
beta signalling are co-regulated in human hepatocellular carcinoma.
Virchows Arch. 445:589–596. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Straub BK, Boda J, Kuhn C, et al: A novel
cell-cell junction system: the cortex adhaerens mosaic of lens
fiber cells. J Cell Sci. 116:4985–4995. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith KD, Fu MA and Brown EJ: Tim-Tipin
dysfunction creates an indispensible reliance on the ATR-Chk1
pathway for continued DNA synthesis. J Cell Biol. 187:15–23. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mao Y, Fu A, Leaderer D, Zheng T, Chen K
and Zhu Y: Potential cancer-related role of circadian gene TIMELESS
suggested by expression profiling and in vitro analyses. BMC
Cancer. 13:4982013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin YM, Chang JH, Yeh KT, et al:
Disturbance of circadian gene expression in hepatocellular
carcinoma. Mol Carcinog. 47:925–933. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leman AR, Noguchi C, Lee CY and Noguchi E:
Human Timeless and Tipin stabilize replication forks and facilitate
sister-chromatid cohesion. J Cell Sci. 123:660–670. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scaggiante B, Dapas B, Bonin S, et al:
Dissecting the expression of EEF1A1/2 genes in human prostate
cancer cells: the potential of EEF1A2 as a hallmark for prostate
transformation and progression. Br J Cancer. 106:166–173. 2012.
View Article : Google Scholar :
|
|
30
|
Pinke DE and Lee JM: The lipid kinase
PI4KIIIbeta and the eEF1A2 oncogene co-operate to disrupt
three-dimensional in vitro acinar morphogenesis. Exp Cell Res.
317:2503–2511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pinke DE, Kalloger SE, Francetic T,
Huntsman DG and Lee JM: The prognostic significance of elongation
factor eEF1A2 in ovarian cancer. Gynecol Oncol. 108:561–568. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gross SR and Kinzy TG: Improper
organization of the actin cytoskeleton affects protein synthesis at
initiation. Mol Cell Biol. 27:1974–1989. 2007. View Article : Google Scholar :
|
|
33
|
Gross SR and Kinzy TG: Translation
elongation factor 1A is essential for regulation of the actin
cytoskeleton and cell morphology. Nat Struct Mol Biol. 12:772–778.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Amiri A, Noei F, Jeganathan S, Kulkarni G,
Pinke DE and Lee JM: eEF1A2 activates Akt and stimulates
Akt-dependent actin remodeling, invasion and migration. Oncogene.
26:3027–3040. 2007. View Article : Google Scholar
|
|
35
|
Jeganathan S and Lee JM: Binding of
elongation factor eEF1A2 to phosphatidylinositol 4-kinase beta
stimulates lipid kinase activity and phosphatidylinositol
4-phosphate generation. J Biol Chem. 282:372–380. 2007. View Article : Google Scholar
|
|
36
|
Khacho M, Mekhail K, Pilon-Larose K, Pause
A, Cote J and Lee S: eEF1A is a novel component of the mammalian
nuclear protein export machinery. Mol Biol Cell. 19:5296–5308.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li D, Wei T, Abbott CM and Harrich D: The
unexpected roles of eukaryotic translation elongation factors in
RNA virus replication and pathogenesis. Microbiol Mol Biol Rev.
77:253–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chuang SM, Chen L, Lambertson D, Anand M,
Kinzy TG and Madura K: Proteasome-mediated degradation of
cotranslationally damaged proteins involves translation elongation
factor 1A. Mol Cell Biol. 25:403–413. 2005. View Article : Google Scholar :
|