Introduction
Endometrial carcinoma is the most common malignancy
of the female reproductive tract. Although the majority of patients
with early-stage disease are cured with surgery and/or radiation
therapy, advanced endometrial carcinoma continues to portend a poor
prognosis. Chemotherapy is an important treatment option for women
with advanced and recurrent endometrial cancer or women in early
stage with high-risk factor. Enhancing the sensitivity of
endometrial carcinoma cells to chemotherapy agents could lead to an
improved clinical outcome.
The combination of paclitaxel and carboplatin is
currently regarded as the standard regimen of endometrial carcinoma
based on its favourable toxicity profile and good response rates
(1–4). Paclitaxel is a potent mitotic
inhibitor and inducer of apoptosis. Paclitaxel binds to β-tubulin
thereby inhibiting microtubule depolymerization, which ultimately
results in cell cycle arrest in G2/M phase and subsequent
apoptosis. Furthermore, previous studies have shown that paclitaxel
can induce autophagy in many cancer cell lines. Autophagy is a
tightly regulated process in which a cell engulfs cytoplasmic
constituents within a double-membrane vacuole known as an
autophagosome, and delivers them to the lysosome for degradation.
Degradation of the contents within the autophagosome generates free
amino and fatty acids that can be recycled intrinsically or
delivered systemically to distant sites within the organism.
Therefore, the process of autophagy promotes cellular homeostasis
and viability by way of maintaining quality control of both
proteins and organelles.
Early studies demonstrate that a variety of chemical
insults can activate autophagy in vitro as well as in
vivo in certain types of cancers (5). Autophagy induced by certain cancer
therapeutics may cause the undesired effect of protecting cancer
cells from apoptosis (6,7). As increasing evidence suggests that
autophagy promotes cancer cell survival after chemotherapy and that
the inhibition of autophagy resensitizes resistant cancer cells to
anticancer agents, it has been proposed that suppression of the
autophagic pathway may be combined with conventional or
experimental antitumor regimens to achieve increased efficacy,
thereby allowing for lower dosage and limited side effects.
Conversely, some cytotoxic drugs have been discovered to trigger
progressive autophagy that ultimately leads to autophagic cell
death, particularly in apoptosis-defective cells. As such,
induction of autophagic cell death through over-stimulation of
autophagy has also been considered a potential therapeutic strategy
to eradicate cancer cells.
Several studies have demonstrated that the
pro-apoptotic drug paclitaxel can elicit an autophagic response
that actually plays a protective role, ultimately impeding cell
death (8–10). Upregulation of the autophagic
pathway has also been shown to be associated with paclitaxel
resistance in breast cancer cells (11). However, Eum and Lee (12) found that both autophagy and
apoptosis act as cooperative partners to induce cell death in
paclitaxel treated v-Ha-ras-transformed NIH3T3 cells. Veldhoen
et al (13) reported that
in non-mitotic paclitaxel-treated MCF-7 cells, autophagosomes were
generated to sensitize cells to paclitaxel toxicity. Nonetheless,
paclitaxel-induced autophagy and its effect on
paclitaxel-associated cytotoxity in endometrial carcinoma cells
have not been described. In this study, we focused on the effects
of autophagy on paclitaxel-insensitive endometrial carcinoma cell
lines. We found that paclitaxel induced reactive oxygen species
(ROS)-mediated cytoprotective autophagy in endometrial carcinoma
cells. Furthermore, we demonstrated that the suppression of
autophagy using chloroquine (CQ) increased paclitaxel-induced cell
death, thereby suggesting a new approach for enhancing the
chemotherapeutic efficacy of paclitaxel in endometrial
carcinoma.
Materials and methods
Cell culture
HEC-1A human endometrial carcinoma cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA), AN3CA human endometrial carcinoma cells from the Shanghai
Cell Collection, Chinese Academy of Sciences (Shanghai, China) and
JEC human endometrial carcinoma cells from the China Center for
Type Culture Collection (Wuhan, China). Ishikawa human endometrial
carcinoma cells were kindly provided by Professor Zehua Wang
(Department of Obstetrics and Gynecology, Union Hospital, Tongji
Medical College, Huazhong University of Science and Technology,
Wuhan, China). Cells were cultured in Dulbecco’s modified Eagle’s
medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal
bovine serum (Hyclone, South Logan, UT, USA) with varying
concentrations of paclitaxel (Bristol-Myers Squibb, New York, NY,
USA), and/or chloroquine (CQ) (Sigma-Aldrich, St. Louis, MO, USA),
and/or N-acetyl-L-cysteine (NAC) (Sigma-Aldrich) at 37°C in a
humidified chamber with 5% carbon dioxide.
Cell viability assay
Cell viability were measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (MP, CA, USA). Cells of different cell lines were seeded in
96-well plates at varied density
(4×103-1.1×104 per well) and grown to 70%
confluence. Paclitaxel was added to culture medium at the indicated
concentrations. After treatment, cells were cultured for an
additional 24 h. Twenty microliters of MTT (5 mg/ml) was added to
each well, and cells were subsequently incubated at 37°C for an
additional 4 h. Crystals were dissolved in 150 μl of DMSO.
Absorbance was measured at a wavelength of 490 nm using a
microplate reader (Bio-Tek, ELx800, USA).
Confocal microscopy
Cells were grown on glass coverslips and transfected
with YFP-LC3. Twenty-four hours after transfection, cells were
treated with paclitaxel and analyzed after an additional 24 h.
Cells were fixed with 4% paraformaldehyde in PBS for 30 min at room
temperature, then mounted on slides in anti-fading solution and
stored at 4°C. Slides were subsequently examined under a
laser-scanning confocal microscope (Olympus, FV-1000, Japan).
Immunoblot analysis
Cells were seeded in 6-well plates and grown to 70%
confluence. Cells were harvested 24 h after the indicated
treatment. Lysates were prepared by dissolving cell pellets in 100
μl lysis buffer [20 mM Na2PO4 (pH 7.4), 150
mM NaCl, 1% Triton X-100, 1% aprotinin, 1 mM phenymethysulfonyl
fluoride, 10 mg/ml leupeptin, 100 mM NaF and 2 mM
Na3VO4]. Cell lysates were centrifuged for 10
min at 14,000 g. Protein concentrations in the supernatant were
determined by BCA protein assay. Total protein (25 μg) was
separated by SDS-PAGE, then transferred to PVDF membranes.
Membranes were blocked with non-fat dry milk (5%) in TBST (10 mM
Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and incubated with
primary antibodies for 1 h at 4°C. Primary antibodies included
those to detect microtubule associated protein 1 light chain 3
(LC3) (Novus Biologicals Inc., Littleton, CO, USA), P62/SQSTM1
(Santa Cruz Biotechnlogy, Inc., Santa Cruz, CA, USA) and Beclin 1
(Cell Signaling, Beverly, MA, USA). After primary antibody
incubation, membranes were washed three times with TBST for 30 min,
then incubated with secondary antibody conjugated with horseradish
peroxidase for 2 h at room temperature. After washing out the
secondary antibody, the bound antibody complex was detected using
an ECL chemiluminescence reagent.
RNA interference
Protein depletion through RNA-mediated interference
(RNAi) was mediated using the GV246 shRNA system. The target
sequences for beclin 1-specific shRNA were
5′-UGGAAUGGAAUGAGAUUAATT-3′ and 5′-GCUCAGUAUCAGAGAGAAUTT-3′
(GeneBank accession no. NM003766.2). Lentiviruses were generated by
co-transfection of GV246-shRNA plasmids with lentivirus plasmids
PIK into 293T cells by lipidosome. Lentiviruses were collected in
high-serum media at 72 and 96 h following transfection. HEC-1A
cells were transduced with lentiviruses and 8 μg/ml polybrene
(hexadimethrine bromide, Sigma-Aldrich) followed by incubation with
virus at 37°C for 4–6 h. shRNA-transduced cells were selected with
1 μg/ml puromycin for 72 h. Knockdown was assessed by western
blotting at an appropriate time-point each time the experiment was
replicated.
Measurement of intracellular ROS
Reactive Oxygen Species Assay kit was purchased from
Beyotime (Beijing, China). Intracellular ROS levels were measured
by detecting the conversion of cell permeable 2,
7-dichlorofluorescein diacetate (DCFH-DA) to fluorescent
dichlorofluorescein (DCF). Cells were seeded in 12-well plates and
treated with the experimental reagents indicated for 24 h. After
washing several times with PBS, cells were incubated with 5 μM 2′,
7′-dichlorofluorescein diacetate (DCFH-DA) for 1 h at 37°C. After a
second wash step with PBS, positively stained cells were analyzed
at an excitation wavelength of 488 nm and an emission wavelength of
525 nm by BD FACScan flow cytometry (BD LSRII, USA).
Results
Paclitaxel induces autophagy in
insensitive endometrial carcinoma cells
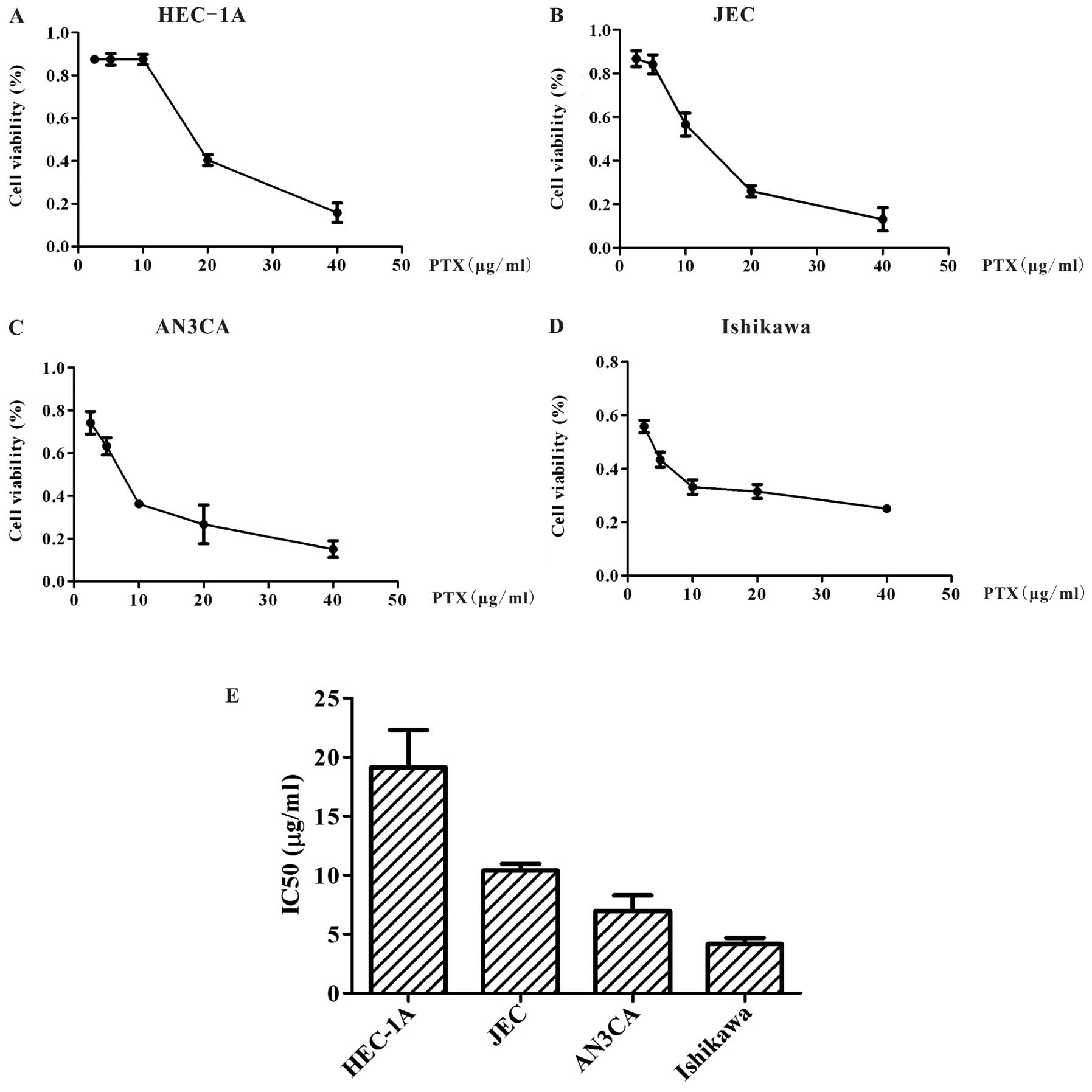
Four endometrial carcinoma cell lines including
HEC-1A, JEC, AN3CA and Ishikawa cells were chosen to investigate
paclitaxel resistance in endometrial carcinoma. MTT assay was used
to detect the IC50 values of paclitaxel on each
endometrial carcinoma cell line. As shown in Fig. 1, upon exposure to paclitaxel for 24
h, the IC50 of HEC-1A, JEC, AN3CA and Ishikawa cells was
19.14±3.16 μg/ml (Fig. 1A),
10.39±0.55 μg/ml (Fig. 1B),
6.95±1.33 μg/ml (Fig. 1C) and
4.15±0.52 μg/ml (Fig. 1D),
respectively. HEC-1A proved to be the least sensitive to paclitaxel
as compared to other cell lines, due to its relatively high
IC50 value (Fig. 1E).
Therefore, we chose HEC-1A as the paclitaxel-resistant endometrial
carcinoma cell line for subsequent studies. We sought to determine
whether paclitaxel could induce autophagy in HEC-1A cells. LC3 and
P62 were used to detect autophagy. LC3 is an important autophagy
marker recruited to the autophagosomal membrane. LC3 has two
isoforms, LC3-I and LC3-II. During autophagy, LC3-I is conjugated
to autophagic membrane-associated phosphatidylethanolamine and
converted to LC3-II. Since LC3-II is localized in autophagosomal
membranes throughout the autophagy process, LC3-II expression is
regarded as a good marker for autophagosome formation. Increased
LC3-II expression, especially an increased LC3-II/LC3-I ratio, may
indicate the occurrence of autophagy (14,15).
P62/SQSTM1 is a highly conserved scaffolding protein involved in
the transportation of ubiquitinylated proteins destined for
proteosomal degradation (15,16).
A decrease in p62 expression could indicate an early autophagic
degradation, which makes it an alternative method for detecting
autophagic flux (17). Using
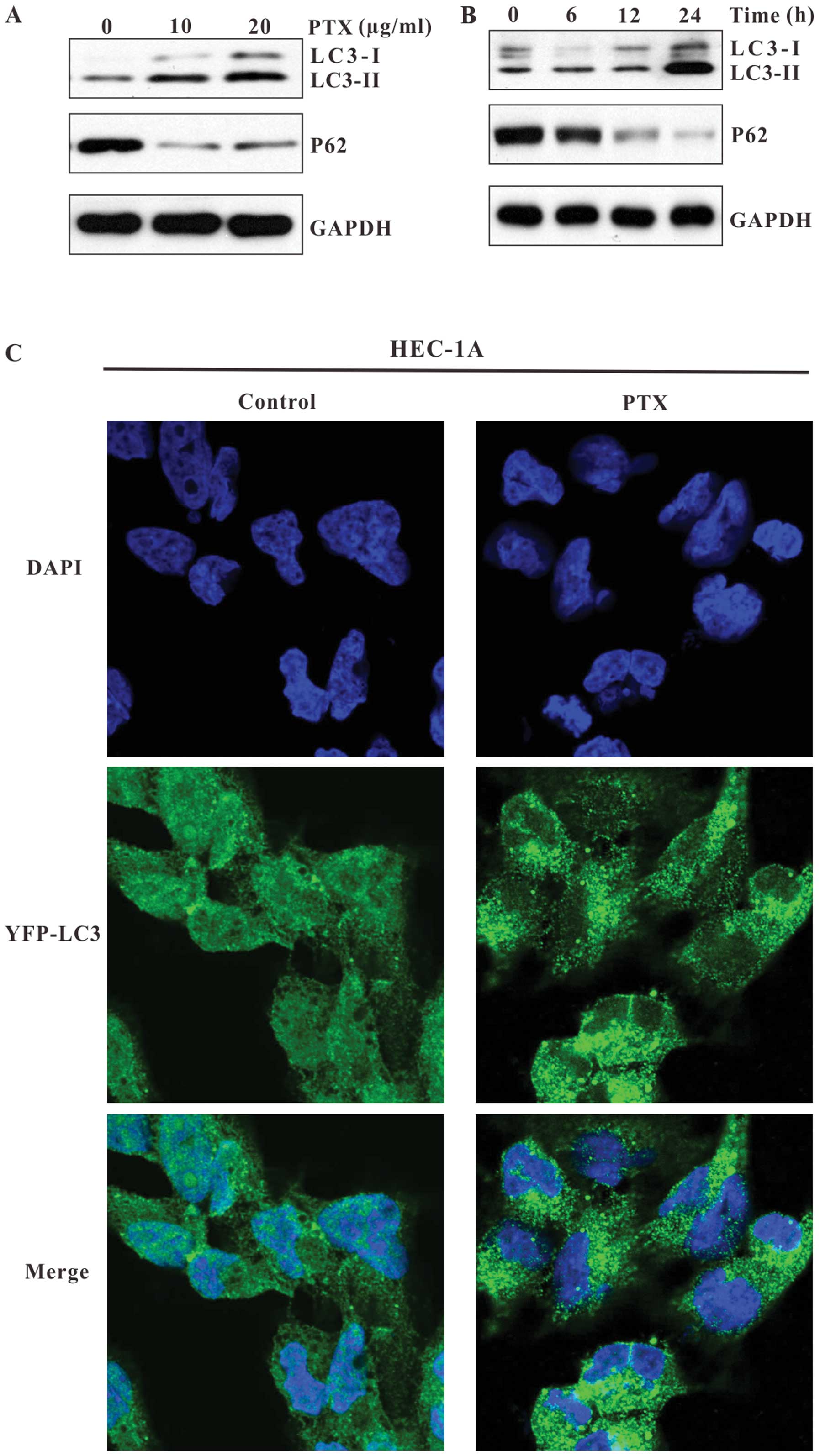
immunoblot analysis, we observed that paclitaxel induced a
significant conversion of LC3I to LC3II, as well as an alteration
of P62 expression in HEC-1A cells. As shown in Fig. 2A and B, in cells treated with
paclitaxel, the expression of LC3-II increased in a dose- and
time-dependent manner, and the expression of P62 dramatically
decreased, as compared to control cells. These results demonstrate
that paclitaxel induces autophagy in HEC-1A cells.
To confirm the induction of autophagy in HEC-1A
cells, we examined the formation of autophagosomes after paclitaxel
treatment using YFP-LC3, an LC3 expression construct fused to a
yellow fluorescent protein. HEC-1A cells were transfected with
YFP-LC3 and exposed to paclitaxel. As shown in Fig. 2C, in control cells, YFP-LC3 was
evenly distributed throughout the entire cytoplasm. However, in
paclitaxel treated cells, the punctate dots of YFP-LC3 were
detectable in the cytosol, indicating the association of YFP-LC3
with autophagosomal membranes, which suggests the induction of
autophagy.
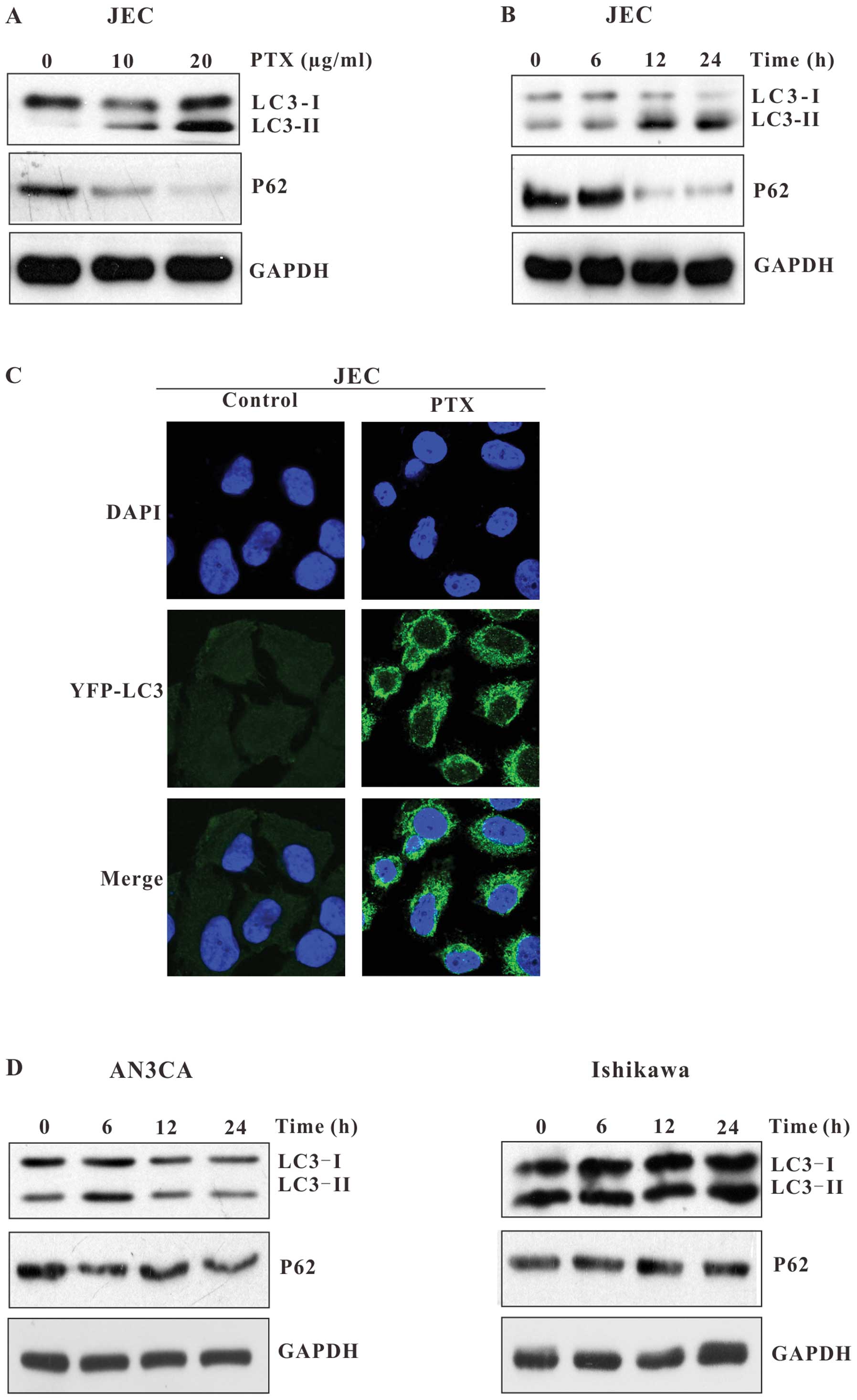
We extended our studies to test whether autophagy
was induced in the other endometrial carcinoma cell lines. As shown
in Fig. 3, the paclitaxel-induced
increase in LC3-II and decrease in P62 were also exhibited in JEC
cells (Fig. 3A and B), but not in
AN3CA or Ishikawa cells (Fig. 3D).
YFP-LC3 punctate dots were also detectable in the cytoplasm of
paclitaxel-treated JEC cells (Fig.
3C). Because autophagy was only induced in HEC-1A and JEC cells
which exhibited high IC50 values, we inferred that
autophagy may be involved in the resistance of endometrial
carcinoma cells to paclitaxel.
Knockdown of Beclin 1 by shRNA increases
the cytotoxic sensitivity of endometrial carcinoma cells to
paclitaxel
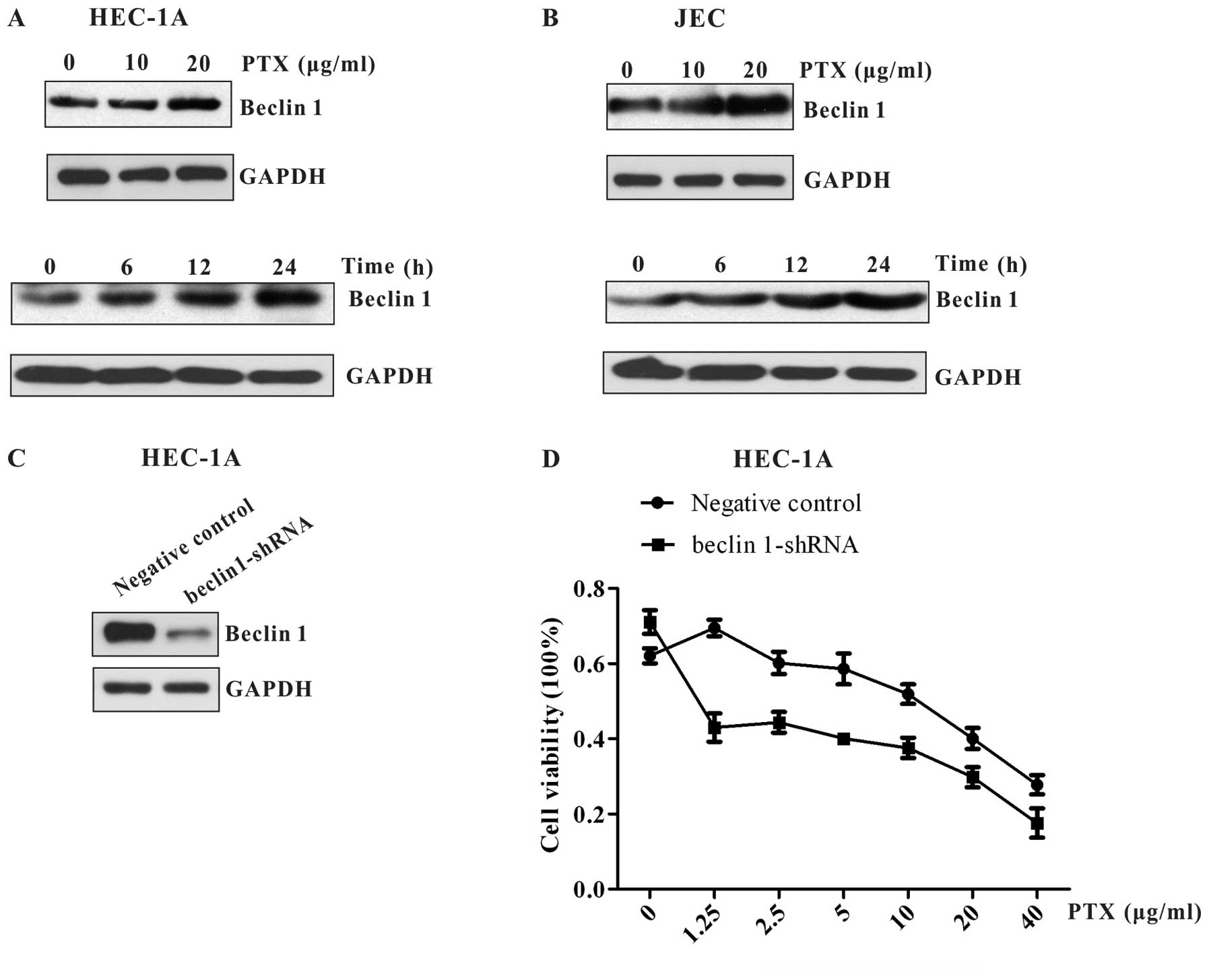
It has been shown that Beclin 1 and its binding
partner class III phosphoinositide 3-kinase (PI3K), also named
Vps34, are required for the initiation of the formation of the
autophagosome. As such, we detected the expression of Beclin 1 in
HEC-1A and JEC cells. We found that Beclin 1 expression increased
in a time- and dose-dependent manner similarly to LC3 and P62 in
these two cell lines (Fig. 4A and
B). The result indicates that paclitaxel-induced autophagy in
endometrial carcinoma cells may be regulated by Beclin 1
complex.
To exploit whether Beclin 1 plays an intrinsic role
in autophagy induced by paclitaxel, the effect of Beclin 1
knockdown on paclitaxel-mediated cell death was investigated. We
used shRNA to knock down Beclin 1 expression in HEC-1A cells
(Fig. 4C). As shown in Fig. 4D, when Beclin 1 expression was
markedly suppressed by beclin 1 shRNA, HEC-1A cells were more
sensitive to paclitaxel. These data suggest that Beclin 1 is
upregulated to initiate cytoprotective autophagy in endometrial
carcinoma cells following paclitaxel treatment.
Inhibition of autophagy increases
paclitaxel-induced cell death
To investigate the impact of autophagy on
paclitaxel-mediated cell death, chloroquine (CQ), an autophagy
inhibitor, was used in combination with paclitaxel in endometrial
carcinoma cell lines. CQ is a weak base that can be trapped in
acidic vesicles, elevate intralysosomal pH (18), and thus block autophagosome
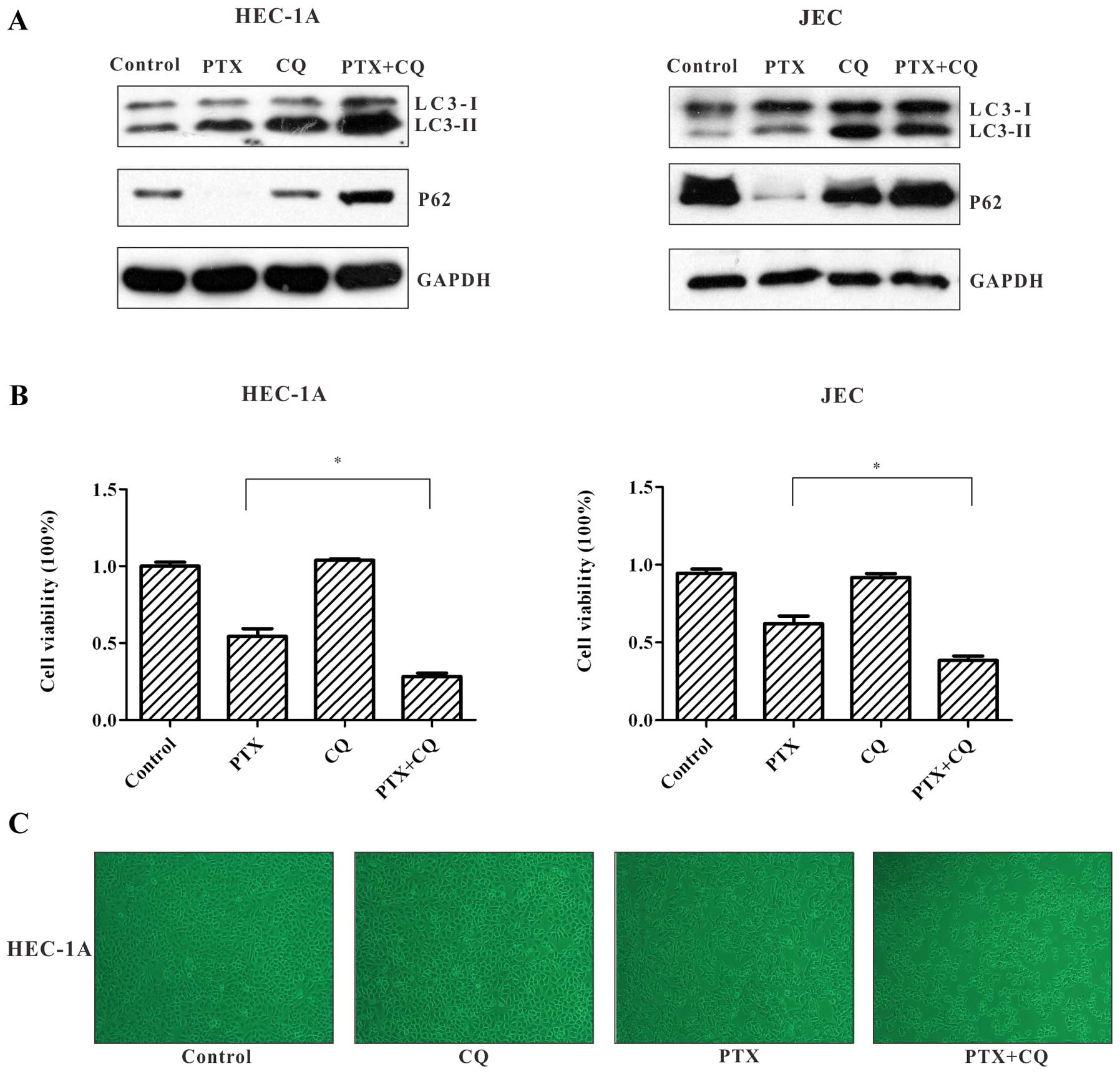
degradation (19,20). As depicted in Fig. 5A, in immunoblot analysis,
paclitaxel increased the production of LC3-II in HEC-1A and JEC
cells, which was in accordance with the aforementioned results.
Compared to paclitaxel treatment alone, co-treatment of paclitaxel
and CQ led to accumulation of LC3-II and impaired p62 degradation.
These results suggest that autophagic flux induced by paclitaxel
could be inhibited by CQ in the autophagic degradation stage.
We then examined the impact of autophagy inhibition
on the survival of HEC-1A and JEC cells exposed to paclitaxel. MTT
assay showed that the cell viability after exposure of HEC-1A and
JEC cells to paclitaxel for 24 h was significantly decreased in the
presence of CQ. (Fig. 5B). We also
observed that in the paclitaxel + CQ group, HEC-1A cells appeared
to exhibit severe shrinkage, and the number of cells was further
reduced compared to the paclitaxel alone treatment group (Fig. 5C).
These results confirm that autophagy is induced to
counteract the cytotoxity of paclitaxel in HEC-1A cells and JEC
cells, and that the inhibition of autophagy enhances sensitivity to
paclitaxel.
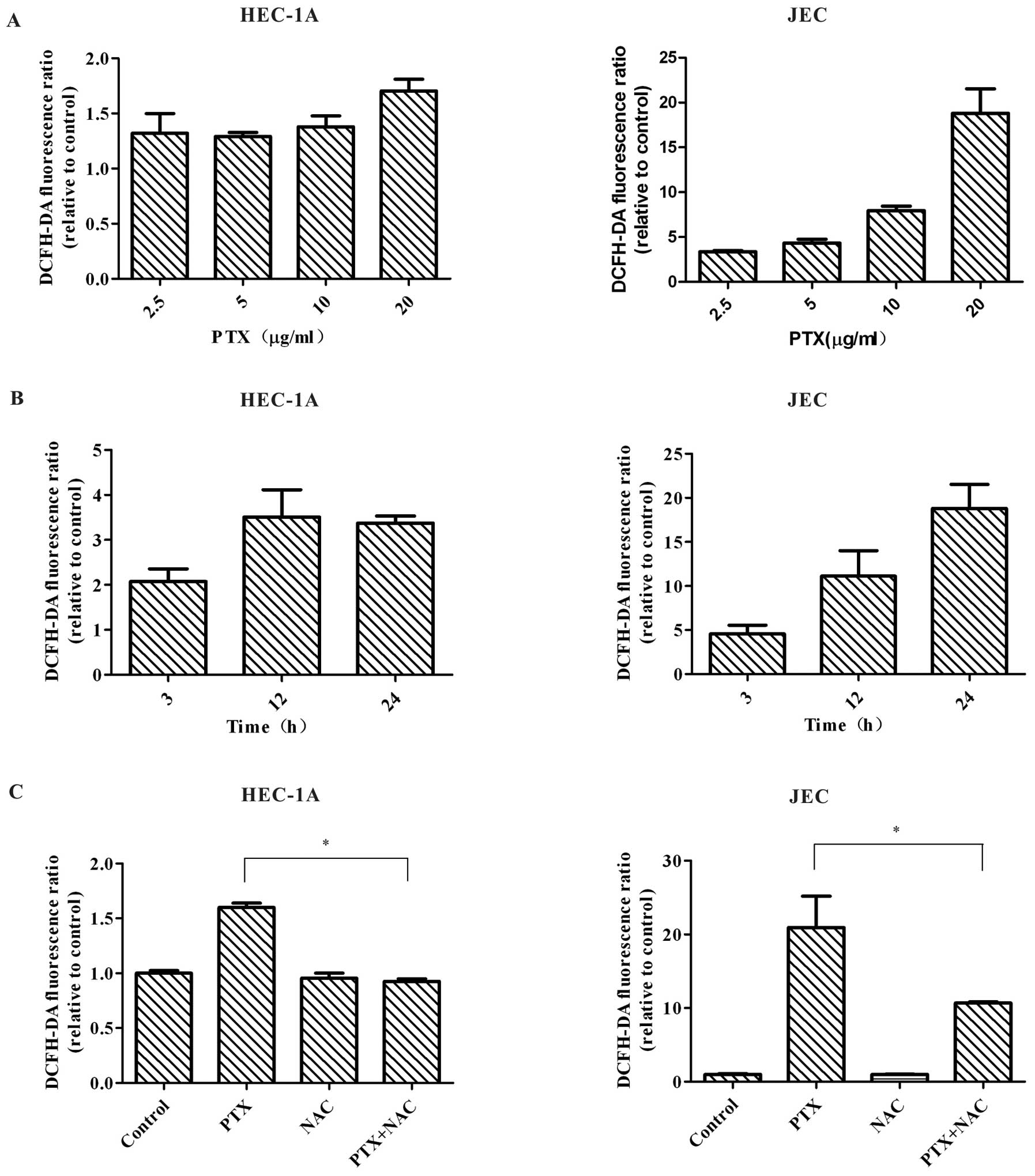
Paclitaxel treatment generates reactive
oxygen species
As paclitaxel can trigger the generation of
intracellular ROS in many cancer cell lines (21–23),
we next investigated whether ROS was elevated in HEC-1A and JEC
cells following paclitaxel exposure. Since DCFH-DA was often used
to assess paclitaxel-induced ROS in previous studies (21,23),
we used DCFH-DA to measure the intracellular levels of ROS in our
study. DCFH-DA can diffuse into cells through the cell membrane,
and subsequently become hydrolyzed to non-fluorescent DCFH. ROS
leads to oxidation of DCFH to DCF, which resulting in the emission
of a fluorescent signal. Our study showed that treatment with
paclitaxel significantly increased ROS level (indicated by
increased green fluorescence) in HEC-1A and JEC cells. The increase
of ROS level was time- and dose-dependent (Fig. 6A and B). We then used NAC, a ROS
scavenger, to treat HEC-1A and JEC cells together with paclitaxel.
Co-treatment of NAC and paclitaxel led to attenuation of ROS level
(Fig. 6C). All the results
indicated that paclitaxel promotes ROS generation in HEC-1A and JEC
cells.
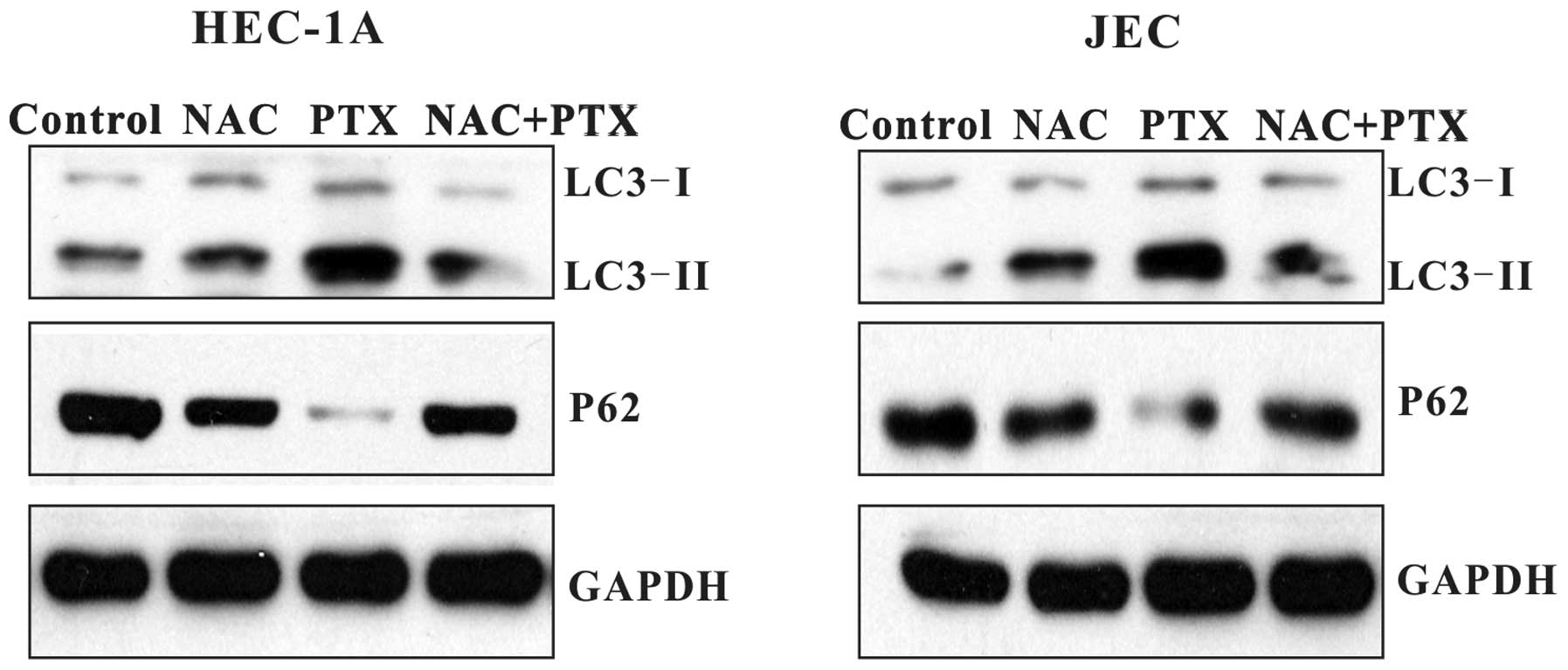
Paclitaxel induces ROS-mediated
autophagy
To examine the role of ROS in paclitaxel-induced
autophagy, HEC-1A and JEC cells were co-treated with paclitaxel and
NAC. As shown in Fig. 7, when
co-treated with NAC, paclitaxel-induced LC3 conversion was
decreased in HEC-1A and JEC cells. P62 level was elevated. This
data demonstrated that when ROS was decreased, autophagy flux was
blocked in the early stage. The result indicates that
paclitaxel-induced autophagy in endometrial carcinoma cells is
mediated by ROS.
Discussion
In the present study, we found that paclitaxel
induced the generation of reactive oxygen species (ROS), which
resulted in cytoprotective autophagy in insensitive endometrial
carcinoma cells. Notably, we demonstrated that the suppression of
autophagy using chloroquine (CQ) increased paclitaxel-induced cell
death, thereby suggesting a novel approach for enhancing the
chemotherapeutic effect of paclitaxel in the treatment of
endometrial carcinoma.
Autophagy has been shown to be involved in mediating
the resistance of cancer cells to anticancer therapy. In our study,
autophagy-related properties including increased LC3-II/LC3-I
ratio, decrease in p62 abundance, and punctate dots of YFP-LC3 in
cytosol, were obvious in paclitaxel-insensitive endometrial
carcinoma cells HEC-1A and JEC but not in the two
paclitaxel-responsive endometrial carcinoma cell lines, AN3Ca and
Ishikawa. Subsequent experiments demonstrated that inhibiting
autophagy resulted in an increase in paclitaxel-induced cell death,
thereby confirming a protective role of autophagy induced by
paclitaxel in certain types of endometrial carcinoma. Based on
these findings, it can be hypothesized that autophagy is activated
in response to paclitaxel-induced damage to various cellular
structures, and that this autophagic process has the unintended
result of maintaining the survival of cancer cells and paclitaxel
resistance. Previous studies have shown that paclitaxel can induce
cytoprotective autophagy in multiple cancer cell lines (8–10),
suggesting that the cytoprotective effect of autophagy induced by
paclitaxel is not cell-type specific, and may be associated with
paclitaxel resistance.
Reactive oxygen species (ROS) are
chemically-reactive molecules containing oxygen, which form as a
natural byproduct of the normal metabolism of oxygen. When produced
in moderate amounts, ROS are thought to operate as signaling
molecules in signal transduction pathways regulating cell growth,
differentiation, survival, inflammation and the immune response.
When produced in excessive amounts, ROS may inflict oxidative
damage to vital biological molecules, such as DNA, lipids and
proteins, which alters their functionality and causes impairment of
cellular integrity. Recently, it has been demonstrated that ROS can
induce autophagy (24). Early
studies have shown that paclitaxel can induce the generation of ROS
in several cancer cell lines, and that this induction of ROS is
involved in paclitaxel-induced apoptosis (21–23).
Our study also demonstrated that paclitaxel induced ROS generation
in a time- and dose-dependent manner. Intriguingly, we found that
when ROS levels were decreased using NAC, a general ROS scavenger,
autophagy was blocked, indicating that ROS generation mediates
autophagy upon paclitaxel treatment.
The generation of ROS by anticancer agents has been
reported to mediate autophagy in previous studies (25–29).
ROS have been shown to induce autophagy by several processes:
activating ERK and JNK, inhibiting mTOR signaling by activating
AMP-activated protein kinase (AMPK), and promoting the
translocation of oxidative HMGB1 from nucleus to cytoplasm. In
addition, arsenic trioxide, a chemical known for inducing
autophagic cell death by induction of oxidation stress in several
cancer cell lines, was found to stimulate autophagy through
upregulating BNIP3, which displaces Bcl-2/Bcl-XL from its complex
with Beclin 1 (30,31). Our study found that Beclin 1
expression increased with autophagy, indicating Beclin 1 complex
may be involved in the regulation of paclitaxel induced autophagy
in endometrial carcinoma cells. It remains to be further validated
whether paclitaxel-induced ROS production promotes autophagy by the
activation of multiple pro-autophagic signal pathways or by
liberating Beclin 1.
As mounting reports have provided data suggesting
that autophagy can facilitate the survival of cancer cells in
response to anticancer treatment, autophagy has been proposed as a
potential therapeutic target for anticancer drug resistance in the
future. To circumvent cytoprotective autophagy, autophagy
inhibitors are employed to overcome anticancer therapy resistance,
including pharmacological autophagy inhibitor 3-methyladenine
(3-MA), bafilomycin A1 (BafA), CQ and its derivatives, as well as
siRNA that target essential modulators of autophagic machinery.
Among them, CQ and its derivatives have gained increasing attention
due to their favorable pharmacological properties and physiological
tolerance as evidenced by their long history of use as
anti-malarial agents and in diseases such as rheumatoid arthritis.
CQ inhibits lysosomal acidification, which blocks the terminal
stage of autophagy and has indeed been shown to potentiate the
anticancer effects of various chemotherapeutic drugs both in
vitro and in vivo (32–35).
Currently, multiple clinical trials using CQ as a sensitizing
reagent, in combination with standard cancer therapies, are under
evaluation in different tumor types, including lung cancer,
glioblastoma multiforme, multiple myeloma, breast cancer, melanoma,
colon cancer, prostate cancer, and advanced solid tumors
unresponsive to chemotherapy (http://clinicaltrials.gov). In the present study,
co-treatment of CQ and paclitaxel in paclitaxel-insensitive HEC-1A
and JEC cells led to autophagy blockage and increased cell death.
This result suggests that CQ can enhance the responsiveness of
chemotherapeutic-resistant endometrial carcinoma cells to
paclitaxel. Thus, treating cancer cells with CQ and its derivatives
may provide an important technique to make endometrial carcinoma
responsive to paclitaxel chemotherapy.
In conclusion, the results of this study revealed
that paclitaxel can increase autophagic activity in
paclitaxel-insensitive endometrial carcinoma cells, and that
suppressing autophagy could enhance paclitaxel-induced cell death.
Taken together, this suggests that autophagy-inhibitor therapy
could be an effective and potent strategy to improve paclitaxel
treatment outcomes in the treatment of endometrial carcinoma.
Acknowledgements
This study was supported by grants from the National
Nature Science Foundation of China (30772332).
Abbreviations:
|
LC3
|
microtubule associated protein 1 light
chain 3
|
|
YFP
|
yellow fluorescent protein
|
|
CQ
|
chloroquine
|
|
ROS
|
reactive oxygen species
|
|
NAC
|
N-acetyl-L-cysteine
|
References
|
1
|
Mazgani M, Le N and Hoskins PJ; British
Columbia Cancer Agency. Reuse of carboplatin and paclitaxel in
patients with relapsed endometrial cancer - the British Columbia
Cancer Agency experience. Gynecol Oncol. 111:474–477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pectasides D, Xiros N, Papaxoinis G,
Pectasides E, Sykiotis C, Koumarianou A, Psyrri A, Gaglia A,
Kassanos D, Gouveris P, et al: Carboplatin and paclitaxel in
advanced or metastatic endometrial cancer. Gynecol Oncol.
109:250–254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sorbe B, Andersson H, Boman K, Rosenberg P
and Kalling M: Treatment of primary advanced and recurrent
endometrial carcinoma with a combination of carboplatin and
paclitaxel-long-term follow-up. Int J Gynecol Cancer. 18:803–808.
2008. View Article : Google Scholar
|
|
4
|
Vandenput I, Vergote I, Leunen K,
Berteloot P, Neven P and Amant F: Leuven dose-dense
paclitaxel/carboplatin regimen in patients with primary advanced or
recurrent endometrial carcinoma. Int J Gynecol Cancer.
19:1147–1151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lefranc F, Facchini V and Kiss R:
Proautophagic drugs: A novel means to combat apoptosis-resistant
cancers, with a special emphasis on glioblastomas. Oncologist.
12:1395–1403. 2007. View Article : Google Scholar
|
|
6
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morselli E, Galluzzi L, Kepp O, Vicencio
JM, Criollo A, Maiuri MC and Kroemer G: Anti- and pro-tumor
functions of autophagy. Biochim Biophys Acta. 1793:1524–1532. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Q, Si S, Schoen S, Chen J, Jin XB
and Wu G: Suppression of autophagy enhances preferential toxicity
of paclitaxel to folliculin-deficient renal cancer cells. J Exp
Clin Cancer Res. 32:992013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim HJ, Lee SG, Kim YJ, Park JE, Lee KY,
Yoo YH and Kim JM: Cytoprotective role of autophagy during
paclitaxel-induced apoptosis in Saos-2 osteosarcoma cells. Int J
Oncol. 42:1985–1992. 2013.PubMed/NCBI
|
|
10
|
Xi G, Hu X, Wu B, Jiang H, Young CY, Pang
Y and Yuan H: Autophagy inhibition promotes paclitaxel-induced
apoptosis in cancer cells. Cancer Lett. 307:141–148. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ajabnoor GM, Crook T and Coley HM:
Paclitaxel resistance is associated with switch from apoptotic to
autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis.
3:e2602012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eum KH and Lee M: Crosstalk between
autophagy and apoptosis in the regulation of paclitaxel-induced
cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem.
348:61–68. 2011. View Article : Google Scholar
|
|
13
|
Veldhoen RA, Banman SL, Hemmerling DR,
Odsen R, Simmen T, Simmonds AJ, Underhill DA and Goping IS: The
chemotherapeutic agent paclitaxel inhibits autophagy through two
distinct mechanisms that regulate apoptosis. Oncogene. 32:736–746.
2013. View Article : Google Scholar
|
|
14
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Overvatn A, Bjorkoy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ichimura Y and Komatsu M: Selective
degradation of p62 by autophagy. Semin Immunopathol. 32:431–436.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luiken JJ, Aerts JM and Meijer AJ: The
role of the intralysosomal pH in the control of autophagic
proteolytic flux in rat hepatocytes. Eur J Biochem. 235:564–573.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maclean KH, Dorsey FC, Cleveland JL and
Kastan MB: Targeting lysosomal degradation induces p53-dependent
cell death and prevents cancer in mouse models of lymphomagenesis.
J Clin Invest. 118:79–88. 2008. View
Article : Google Scholar
|
|
21
|
Alexandre J, Batteux F, Nicco C, Chéreau
C, Laurent A, Guillevin L, Weill B and Goldwasser F: Accumulation
of hydrogen peroxide is an early and crucial step for
paclitaxel-induced cancer cell death both in vitro and in vivo. Int
J Cancer. 119:41–48. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramanathan B, Jan KY, Chen CH, Hour TC, Yu
HJ and Pu YS: Resistance to paclitaxel is proportional to cellular
total antioxidant capacity. Cancer Res. 65:8455–8460. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lyle PA, Mitsopoulos P and Suntres ZE:
N-acetyl cysteine modulates the cytotoxic effects of Paclitaxel.
Chemotherapy. 57:298–304. 2011. View Article : Google Scholar
|
|
24
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miki H, Uehara N, Kimura A, Sasaki T, Yuri
T, Yoshizawa K and Tsubura A: Resveratrol induces apoptosis via
ROS-triggered autophagy in human colon cancer cells. Int J Oncol.
40:1020–1028. 2012.PubMed/NCBI
|
|
26
|
Kim JY, Cho TJ, Woo BH, Choi KU, Lee CH,
Ryu MH and Park HR: Curcumin-induced autophagy contributes to the
decreased survival of oral cancer cells. Arch Oral Biol.
57:1018–1025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang L, Yang M, Zhang H, Wang Z, Yu Y, Xie
M, Zhao M, Liu L and Cao L: S100A8-targeting siRNA enhances arsenic
trioxide-induced myeloid leukemia cell death by down-regulating
autophagy. Int J Mol Med. 29:65–72. 2012.
|
|
28
|
Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH,
Lin YF and Shih CM: Resveratrol enhances the therapeutic effect of
temozolomide against malignant glioma in vitro and in vivo by
inhibiting autophagy. Free Radic Biol Med. 52:377–391. 2012.
View Article : Google Scholar
|
|
29
|
Cheng P, Ni Z, Dai X, Wang B, Ding W, Rae
Smith A, Xu L, Wu D, He F and Lian J: The novel BH-3 mimetic
apogossypolone induces Beclin-1- and ROS-mediated autophagy in
human hepatocellular carcinoma [corrected] cells. Cell Death Dis.
4:e4892013. View Article : Google Scholar
|
|
30
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar
|
|
31
|
Chinnadurai G, Vijayalingam S and Gibson
SB: BNIP3 subfamily BH3-only proteins: Mitochondrial stress sensors
in normal and pathological functions. Oncogene. 27(Suppl 1):
S114–S127. 2008. View Article : Google Scholar
|
|
32
|
Bellodi C, Lidonnici MR, Hamilton A,
Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi
T, Yacobi R, et al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ni Z, Wang B, Dai X, Ding W, Yang T, Li X,
Lewin S, Xu L, Lian J and He F: HCC cells with high levels of Bcl-2
are resistant to ABT-737 via activation of the ROS-JNK-autophagy
pathway. Free Radic Biol Med. 70:194–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun K, Xie X, Liu Y, Han Z, Zhao X, Cai N,
Zhang S, Song J and Wei L: Autophagy lessens ischemic liver injury
by reducing oxidative damage. Cell Biosci. 3:262013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo XL, Li D, Hu F, Song JR, Zhang SS,
Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar : PubMed/NCBI
|