Introduction
In addition to the recognized function of cytotoxic
T lymphocytes (CTLs) in immune response to cancer, it is now clear
that CD4+ T helper lymphocytes can also participate in
the generation of antitumour immunity. Tumour-infiltrating
CD4+ T cells recognize antigens presented through MHC
II, by conventional antigen-presenting cells (dendritic cells, B
lymphocytes and macrophages), able to capture, process and present
tumour antigens derived by cancer cells. Several published
evidences demonstrated that human solid cancers of
non-haematopoietic origin might express MHC II molecules and
stimulate tumour antigen-specific CD4+ T cells that
display direct cytotoxicity toward tumour cells expressing MHC II
(1–3).
The MHC II expression is regulated at
transcriptional level by a multi-protein enhanceosome complex that
binds the W-X-Y region of MHC II promoters, stabilized by the class
II transactivator CIITA (4). CIITA
exhibits a cell-type constitutive or IFN-γ inducible expression
profile. This cytokine activates the receptor-associated protein
tyrosine kinases Janus kinase 1 (JAK1) and 2 (JAK2) and leads to
the phosphorylation of signal transducer activator of transcription
1 (STAT1) that dimerizes or forms heterodimers with STAT3,
translocates to the nucleus and activates its target promoters.
p-STAT1 increases chromatin accessibility, through histone
acetylation of CIITA promoter and induces MHC II expression
(5,6).
Moreover, a post-transcriptional level of MHC II
regulation has been recently characterized through the discovery of
the ‘MHC II operon’ (7), a
functional unit that guarantees the coordinated transcription of
alpha and beta mRNAs, and the correct processing and balanced
surface expression for both HLA-DR and HLA-DQ isotypes.
Specifically, the MHC II operon is a ribonucleoprotein (RNP)
complex in which 5′UTR and 3′UTR of MHC II mRNAs are the binding
sites of a protein complex in which the RNA binding protein EBP1
(ErbB3 binding protein) has been identified (8). Several studies showed that p48, the
longer isoform of EBP1, is ubiquitously expressed, both in
malignant and non-malignant cells (9). Overexpression of EBP1 p48 in ErbB2/3
positive breast cancer cells and in prostate cancer cells inhibits
cell growth and results in a more differentiated phenotype
(10–13). Following heregulin treatment, EBP1
translocates in the nucleus where it inhibits the transcription of
E2F-regulated promoters, among which E2F1 transcription factor, D1
and E cyclins (14) and androgen
receptor (AR) (15). Many studies
have demonstrated the suppressor role of p48 in vivo, as it
inhibits the growth and invasion of adenoid cystic carcinoma
(16) and oral squamous cell
carcinoma (17). The
downregulation of the longer isoform of EBP1 is also associated
with hormone resistance in prostate cancer tissues (12), while in human bladder cancer
tissues (18) and hepatocellular
carcinoma (19) the decreased
expression was associated with advanced pathologic stage and poor
prognosis.
Other studies, performed in nerve growth factor
(NGF)-treated PC12 cells, focused on the expression of the two
isoforms of EBP1 and assigned them a different function: p48,
localized in both cytoplasm and nucleus, blocks cell
differentiation and promotes proliferation and survival, while the
shorter form p42, localized only in the cytoplasm, suppresses cell
proliferation and enhances differentiation (20). In human glioblastoma, p48 isoform
is highly expressed revealing an oncogene function and a
correlation with poor clinical outcome, while it facilitates
tumorigenesis and enhances tumour growth in mouse xenograft models
(21).
Concerning the function of RNA binding protein, it
has been demonstrated that EBP1 influences the stability of bcl2
(22), promotes decay of AR mRNAs and inhibits its translation
(23,24). Moreover, EBP1 is found within
pre-ribosomal ribonucleoprotein complexes where it is involved in
ribosome assembly and regulation of intermediate and late steps of
rRNA processing (25,26). In the present study, we aimed to
unravel the transcriptional and post-transcriptional mechanisms
used by EBP1 to modulate MHC II expression in non-hematopoietic
cancers. We hypothesize that the oncogene/suppressor role of p42
and p48 EBP1 isoforms, in different cells type, influences the
variable MHC II expression by solid tumours, necessary to stimulate
a tumor-specific immune-response of CD4+ T cells.
Materials and methods
Cell lines, reagents and
transfection
M14 human melanoma and U87 human glioblastoma cells
were cultured in RPMI-1640 medium, while MCF7 human breast-cancer
cell line was cultured in Dulbecco’s modified Eagle’s medium
(DMEM), supplemented with 10% fetal calf serum (FCS). The
authenticity of cell lines, was ensured by checking HLA class II
genomic asset; in particular, we carried out HLA-DRB1 and HLA-DQA1
genotypes by PCR using the AllSet Gold SSP HLA-DR Low-Resolution
kit, and DQA1 SSP UniTray kit, all purchased by Invitrogen (Life
Technologies Corp. Rome, Italy). The cDNA encoding p48 isoform of
EBP1 protein (10) was cloned in
the CMV10-3xFLAG vector (FLAG-p48). The cDNA encoding p42 isoform
of EBP1 (FLAG-p42) was amplified using p48 cDNAs template, and
using a forward primer starting at the third ATG
(ATGATTATGGAAGAAACAGG GAAA) and a reverse primer at the
transcription STOP coding sequence (TCAGTCCCCAGCTTCATTTTCT). M14,
U87 and MCF7 cell lines were transfected with FLAG-p48 and FLAG-p42
constructs and several clones, analysed by FLAG expression and
cells growth rate assessment, showed a comparable phenotype.
M14-p48, M14-p42, U87-p48, U87-p42, MCF7-p48 and MCF7-p42 single
clones were selected for the experiments described below.
The induction of MHC II expression in MCF7 was
obtained by adding 500 U/ml of recombinant IFNγ (PeproTech EC Ltd.,
London, UK) for 48 h; STAT1 protein phosphorylation was observed
after 15 min of IFNγ treatment. To measure the mRNA half-life,
actinomycin D (Sigma-Aldrich), 10 μg/ml, was added to cell cultures
for 2, 4 and 6 h.
MHC II phenotype, proliferation, cell
cycle and apoptosis analysis by flow cytometry
Determination of cell surface expression of MHC II
antigens was performed using FACSCanto II and DIVA software (BD
Biosciences, Franklin Lakes, NJ, USA). FITC mouse anti-human HLA-DR
and the appropriate isotype control were purchased from BD
Biosciences. For the growth rate measurements, 150,000 cells/well
were plated and counted every 24 h for 4 days in triplicate.
To perform cell cycle, dynamic proliferation
analysis and apoptosis, the cells were synchronized in medium
deprived of serum for 42 h.
To analyse the progression of the cell cycle, the
synchronized cells were released into the growth media containing
10% FBS for 8 h and then harvested, fixed and permeabilized with 1
ml of cold 70% ethanol. They were finally stained with propidium
iodide 50 μg/ml, in the presence of 100 μg/ml RNase A (Serva). The
distribution in cell cycle phases were analysed by FACSCanto II (BD
Biosciences).
To evaluate the dynamic proliferation, synchronized
cells were restarted into the growth media containing 10% FBS in
the presence of EdU (5-ethynyl-2-deoxyuridine) for 24 h. The
detection of EdU was performed by Click-iT EdU flow cytometry assay
kit (Molecular Probes, Eugene, OR, USA). Apoptotic cells, after 24
h into growth media containing 10% FBS, were identified by double
staining with Annexin V FITC kit (Miltenyi Biotec GmbH, Bergisch
Gladbach, Germany), according to the manufacturer’s
instructions.
RNA quantification
Total RNA, after lysis of cells in QIAzol lysis
reagent (Qiagen), was purified using phenol-chloroform extraction.
cDNA was synthetized using reverse transcriptase from Bio-Rad
Laboratories. The amount of specific transcripts was measured by
qRT-PCR using QuantiTect SYBR-Green PCR kit (Bio-Rad Laboratories)
in the presence of specific primers synthesized by PRIMM (Table I) through the DNA Engine Opticon
Real-Time PCR detection system (Bio-Rad Laboratories); each assay
was run in triplicate. The relative amount of specific transcripts
was calculated by the comparative cycle threshold method (27). The amount of GAPDH and β-actin
transcripts was measured to normalize specific RNA levels.
 | Table IPrimers used for qRT-PCR. |
Table I
Primers used for qRT-PCR.
| Gene | Primers | Sequences
5′→3′ | Annealing
temperature (°C) | Fragment size |
|---|
| β-actin | ACT-F |
TCATGAAGTGTGACGTTGACA | 58 | 285 nt |
| ACT-R |
CCTAGAAGCATTTGCGGTGCAC | | |
| GAPDH | G-F |
AACGGATTTGGTCGTATTGGC | 58 | 216 bp |
| G-R |
TCGCTCCTGGAAGATGGTGATG | | |
| HLA-DRA | DRA-F |
GGACAAAGCCAACCTGGAAA | 60 | 120 bp |
| DRA-R |
AGGACGTTGGCTCTCTCAG | | |
| HLA-DRB | DRB1-F |
CTCAGCATCTTGCTCTTGTGCAG | 60 | 228 bp |
| DRB1-R |
CAGCATTAAAGTCAGGTGGTTCC | | |
| HLA-DQA1 | DQA1-F |
GTGTAAACTTGTACCAGT | 58 | 263 bp |
| DQA1-R |
GAGACTTGGAAAACACT | | |
| HLA-A,B,C | MHCI-F |
AGTGGGCTACGTGGACGACA | 58 | 300 bp |
| MHCI-R |
ATGTAATCCTTGCCGTCGTA | | |
| CIITA | CIITA-F |
CCGACACAGACACCATCAAC | 58 | 222 bp |
| CIITA-R |
CTTTTCTGCCCAACTTCTGC | | |
The mRNA half-live was calculated by the equation
(t1/2=Ln(0.5)/slope). The oligonucleotides used for DRB
mRNA quantification were primers common to all DRB1 alleles
(7).
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed on M14 and M14-p48 as
previously described (7). The
pre-cleared chromatin was immune-precipitated with 5 μg of anti-RNA
Pol II CTD repeat P-S5 antibody and rabbit anti-IgG as control
(Abcam). One tenth of the immune-precipitated DNA and input DNA
were analysed by qRT-PCR, using DRA-c-F
(ATTTTTCTGATTGGCCAAAGAGTAATT) and DRA-c-R
(AAAAGAAAAGAGAATGTGGGGTGTAA) promoter primers.
Western blot analysis
Western blot analysis was carried out using cell
extract prepared using either RIPA buffer (10 mM Tris-HCl, pH 7.6,
150 mM NaCl, 1% NP-40) or PARIS kit (Ambion) in order to separate
nuclear from cytoplasmic extracts.
Clones overexpressing p42 and p48 isoforms were
screened using mouse monoclonal anti-FLAG (Sigma-Aldrich) and
rabbit polyclonal anti-EBP1 antibodies (Millipore). Monoclonal
anti-α-tubulin (Sigma-Aldrich) and anti-GAPDH (Abcam) were used to
normalize cytoplasmic extract and anti-Laminin A+C (Abcam) for
nuclear extract. STAT1 and pSTAT-1 were assessed by specific
antibodies (BD Bioscience).
All membranes were developed using the ECL kit
(Bio-Rad Laboratories) and exposed to X-ray film.
Statistical analysis
All experimental results are the mean of three
independent experiments. Statistical analysis was performed using
the unpaired Student’s t-test with two-tailed distribution and
two-sample equal variance parameter. In the figures, single
asterisk corresponds to P<0.05 and double asterisk corresponds
to P<0.01.
Results
Ectopic expression of EBP1 isoforms
reveals a different phenotype in glioblastoma and melanoma vs.
breast cancer
We performed a comparative analysis of the phenotype
induced by the overexpression of p48 and p42 EBP1 isoforms in MCF7
breast carcinoma, U87 glioblastoma and M14 melanoma cell lines.
Constructs FLAG-p48 and FLAG-p42 were used to obtain stable cells
lines and the clones expressing ectopic FLAG proteins were selected
by western blot analysis with anti-FLAG antibody. We confirmed
comparable transfection efficiency by qRT-PCR measurement of FLAG
mRNAs (data not shown).
First of all, we evaluated the effect of p48 and p42
overexpression on the proliferation of the selected clones. We
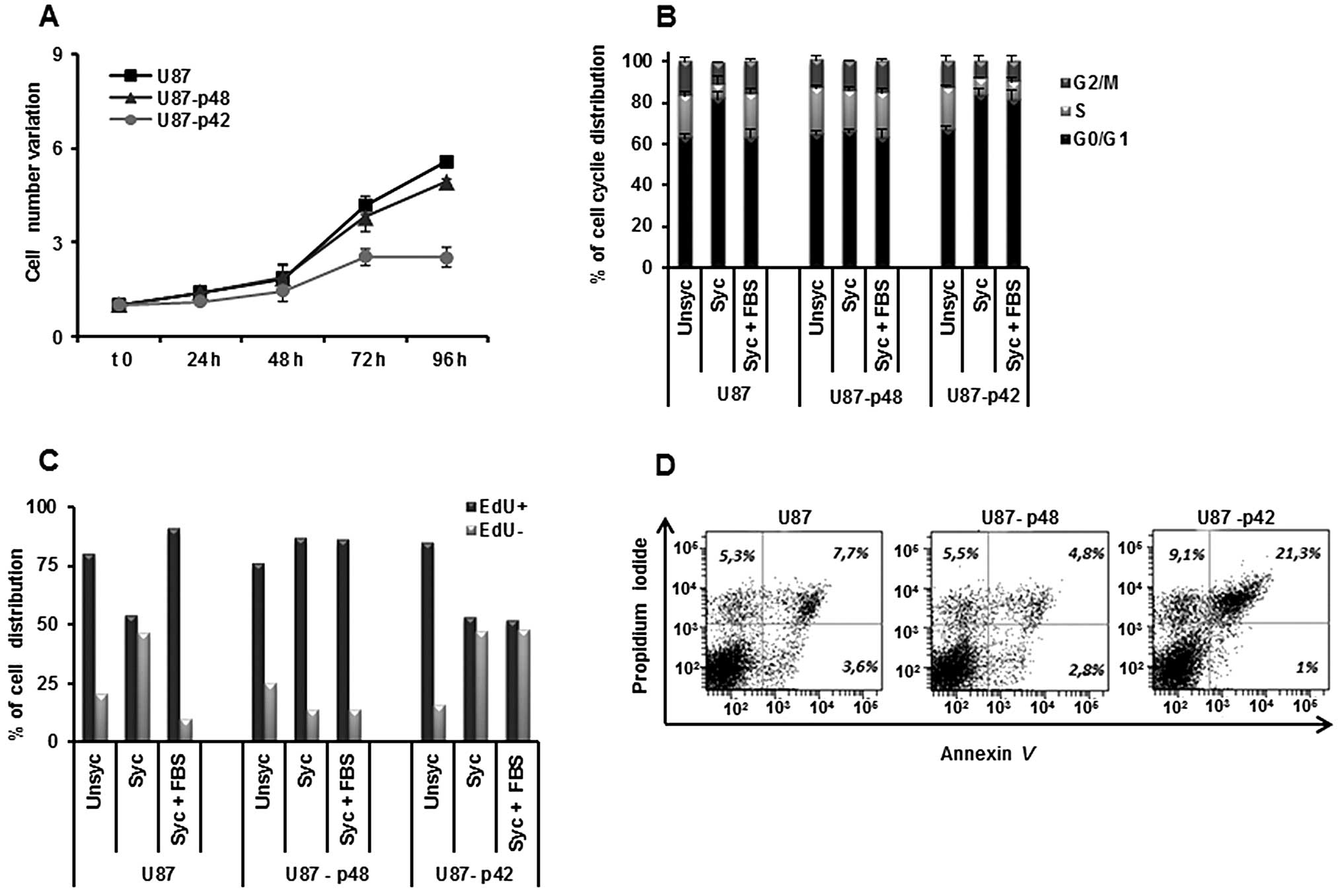
determined growth kinetics, by counting U87-p48 and U87-p42 cells
and we observed that p42 slows down the cell growth, while p48 has
no effect (Fig. 1A), consistent
with published data (21).
Examining the cell cycle progression of glioblastoma, we observed
that synchronization does not block proliferation, because U87-p48
cells persist in S-phase also in the absence of serum (Fig. 1B). In contrast, p42 overexpression
retains cells in G1 phase (81%) as compared to U87-p48 (63%) and to
the control (65%) also during the restart of the cycle (Fig. 1B).
In order to endorse the differences in the dynamic
proliferation between two stable cell lines, we performed EdU
staining which incorporation occurs in the cycling cells. We
observed that, during the synchronization phase, 85% of U87-p48
cells were EdU labelled. The percentage was unchanged in the
subsequent 24 h of cycle restart, indicating that the cells remain
in the S phase and continue to synthesize DNA (Fig. 1C). Otherwise, the number of U87-p42
cycling and not cycling are similar in synchronization and released
phases, suggesting a block in the G1/S transition. The double
staining with Annexin V and PI, 24 h after synchronization,
clarified the consequences of the proliferative block induced by
p42 and we observed 21.3% apoptotic U87-p42 cells (Fig. 1D), indicating that the block of
G1/S transition induced apoptosis.
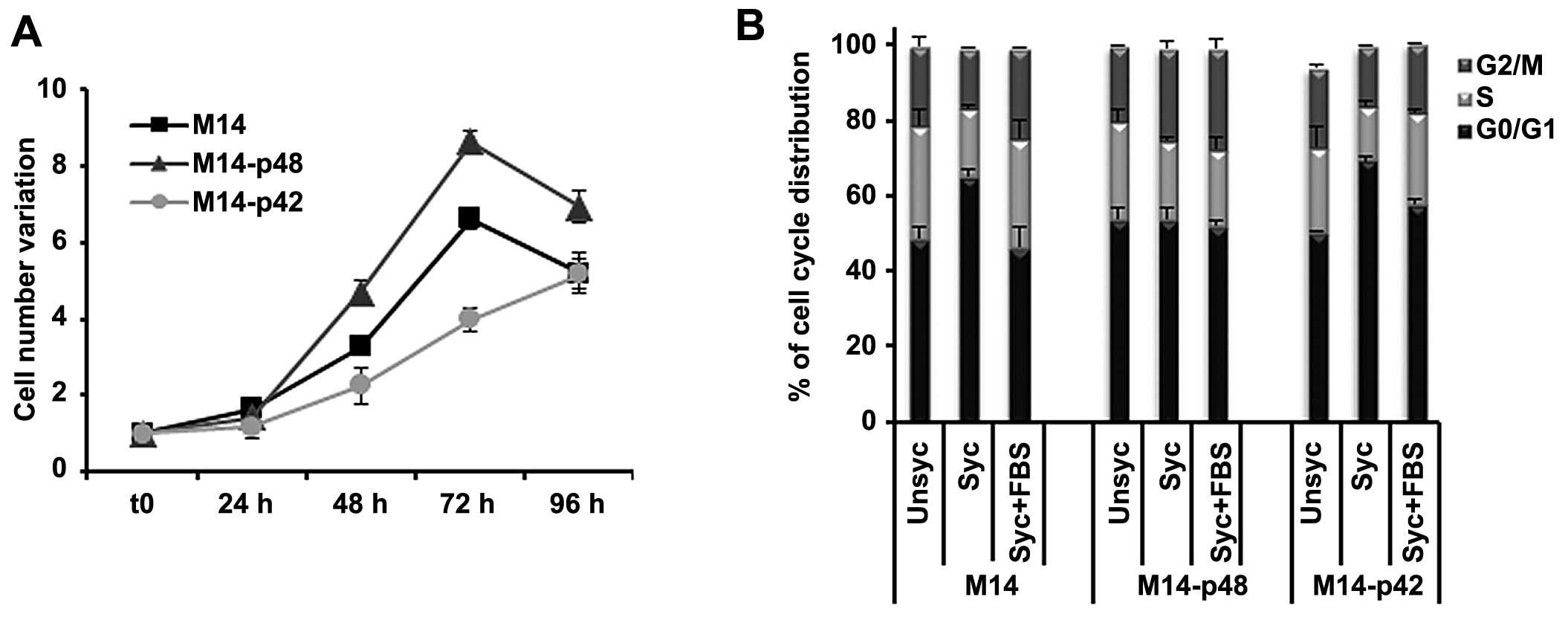
In parallel, we analysed the phenotype of M14
melanoma tumors overexpressing p42 and p48. M14-p48 cells showed an
increased cell growth rate (Fig.
2A), and a permanent distribution of cells in S phase of the
cell cycle, also during synchronization (Fig. 2B) with a phenotype similar to
U87-p48. Our data indicated that p48 isoform functions as oncogene
in glioblastoma and melanoma by increasing cell proliferation,
while p42 acts as a suppressor, by blocking proliferation and
favouring apoptosis.
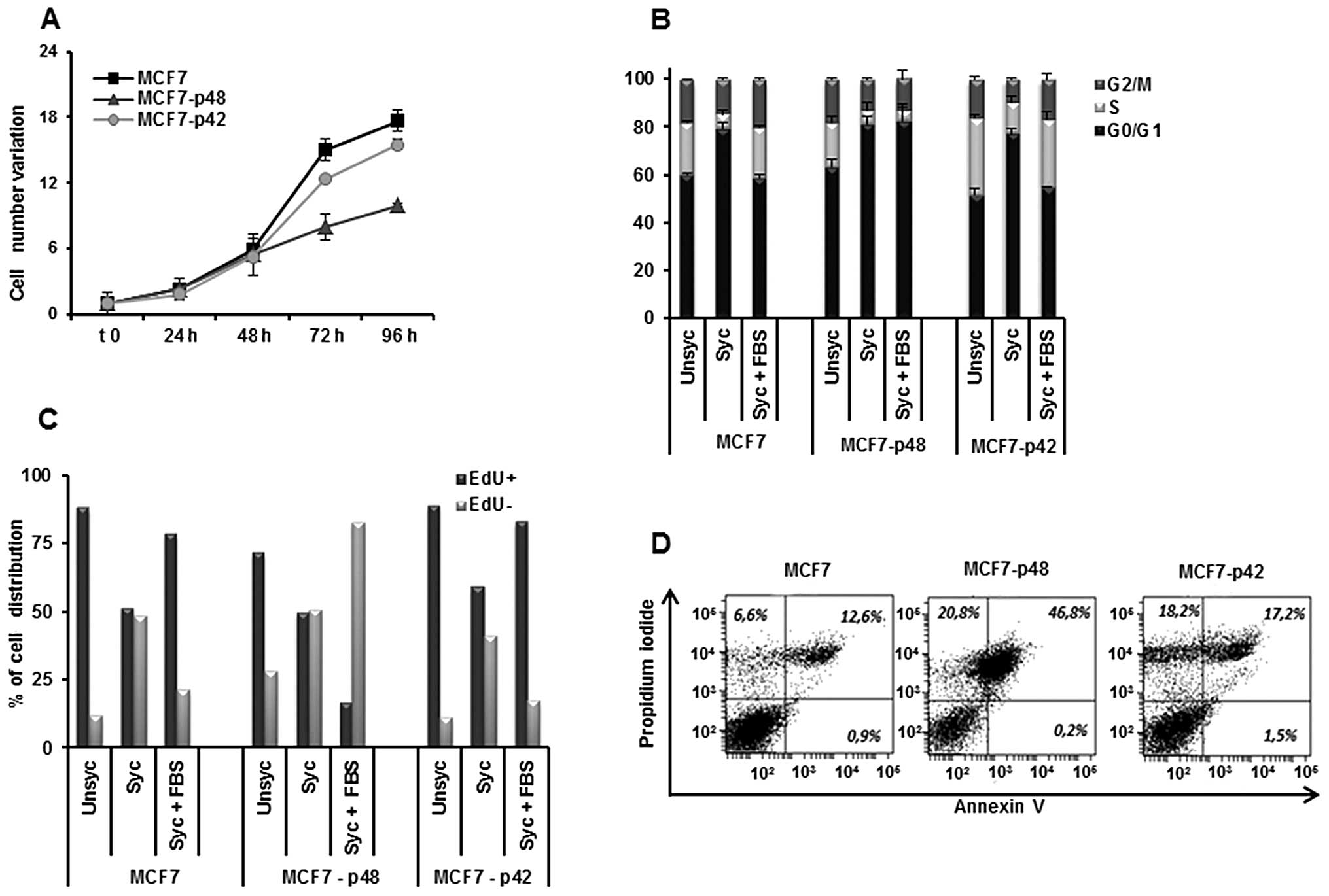
Otherwise, the overexpression of p48 in MCF7 cells
leads to an inhibition of cell growth rate as compared to MCF7-p42
(Fig. 3A), in agreement with
published data (11,28).
The analysis of the cell cycle (Fig. 3B) showed a significant increase of
MCF7-p48 cell distribution in G1 phase (82%) as compared to MCF7
and MCF7-p42 cells (55 and 59%, respectively), associated with a
drastic reduction of cells in S phase (4.7% in MCF7-p48 vs. 21% in
MCF7 and 28% in MCF7-p42). EdU staining confirmed these differences
and showed that, during the restart of the cycle, only 15% of
MCF7-p48 is represented by proliferating cells as compared to 80%
of MCF7-p42 (Fig. 3C). The block
of MCF7-p48 proliferation determined the induction of apoptosis
with 46.2% Annexin V-PI double positive (Fig. 3D) cells. In conclusion, p48
functions as tumor suppressor in breast carcinoma inducing
apoptosis, while p42 does not affect the cell phenotype.
EBP1 increases MHC class II
expression
We have previously demonstrated that the knock-out
of p48 EBP1 protein affects the expression of MHC II molecules
(8). To further investigate the
mechanism by which EBP1 influences MHC II regulation, we measured
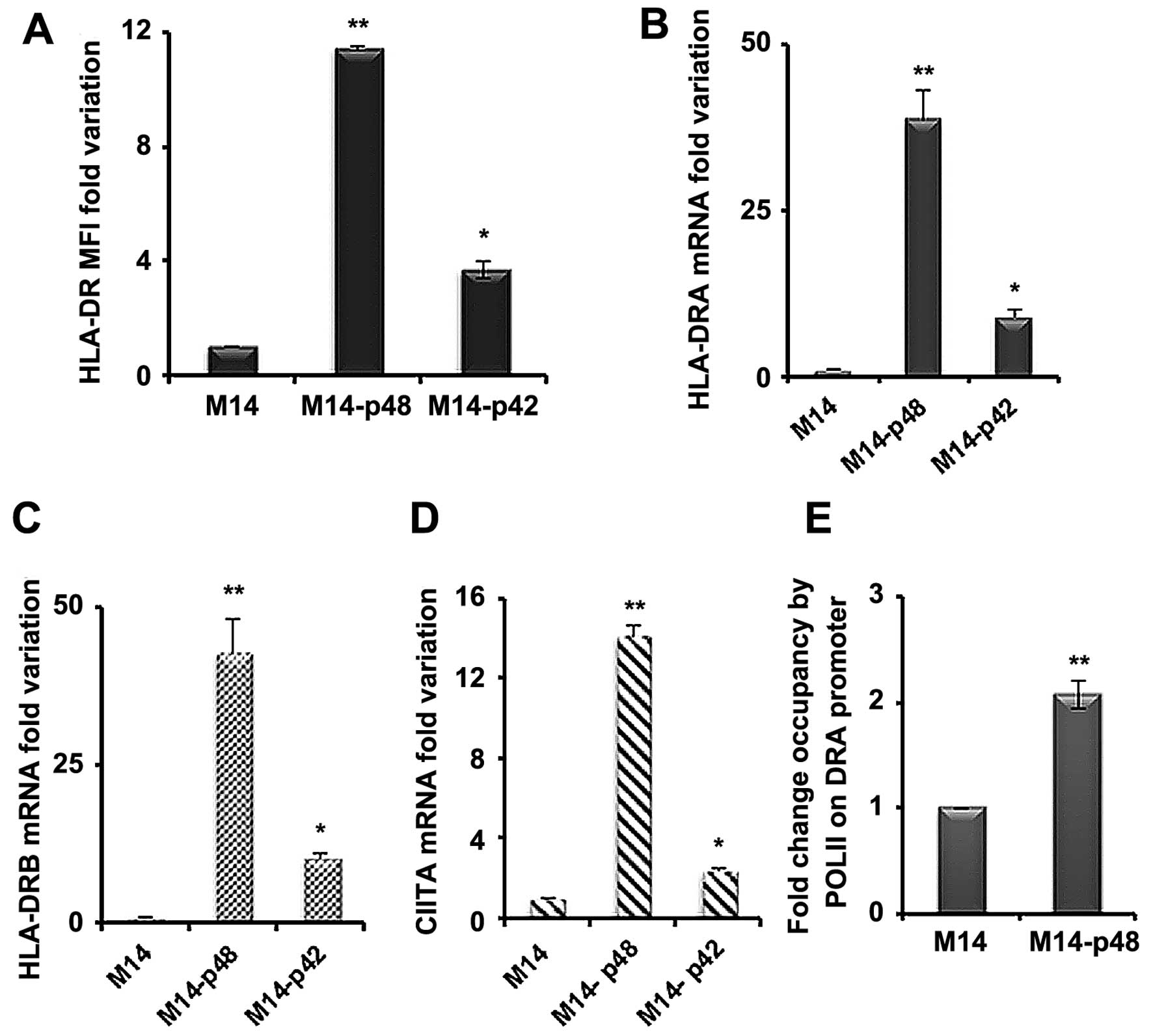
the HLA-DR surface level in M14 transfected cell lines. We showed
that HLA-DR MFI is 10- to 12-fold higher in M14-p48 and 4-fold
increased in M14-p42 cells compared to the control (Fig. 4A). To determine whether the surface
increase of DR molecules is supported by an equal mRNA rise, we
assessed the mRNA variations by qRT-PCR. We observed 40- to 50-fold
increase in the amount of HLA-DRA and HLA-DRB mRNAs (Fig. 4B and C) in M14-p48 and 9-to 11-fold
increase in M14-p42 cells. A significant increment was also
observed for HLA-DQA1 mRNA in the M14-p48 cells, while MHC class I
(MHC I) mRNA was unchanged (data not shown). In order to establish
whether the variation of MHC II molecules was determined at the
transcription level, we measured the expression of CIITA
transactivator mRNAs and Pol II activity on DRA promoter. We
observed a 14-fold increase of the total CIITA mRNA in M14-p48
cells compared to 3-fold rise in M14-p42 (Fig. 4D), indicating that MHC II
over-expression is caused by a transcriptional control involving
CIITA activation.
Moreover, it was confirmed that the CIITA increase
corresponds to the higher Pol II activity by ChIP assay. We
measured the endogenous promoter activity of DRA gene, encoding for
alpha mRNA, and we observed that the fold occupancy of DRA promoter
in M14-p48 cells by Pol II is double compared to M14 transfected
with empty vector, demonstrating that p48 isoform affects MHC II
transcription (Fig. 4E).
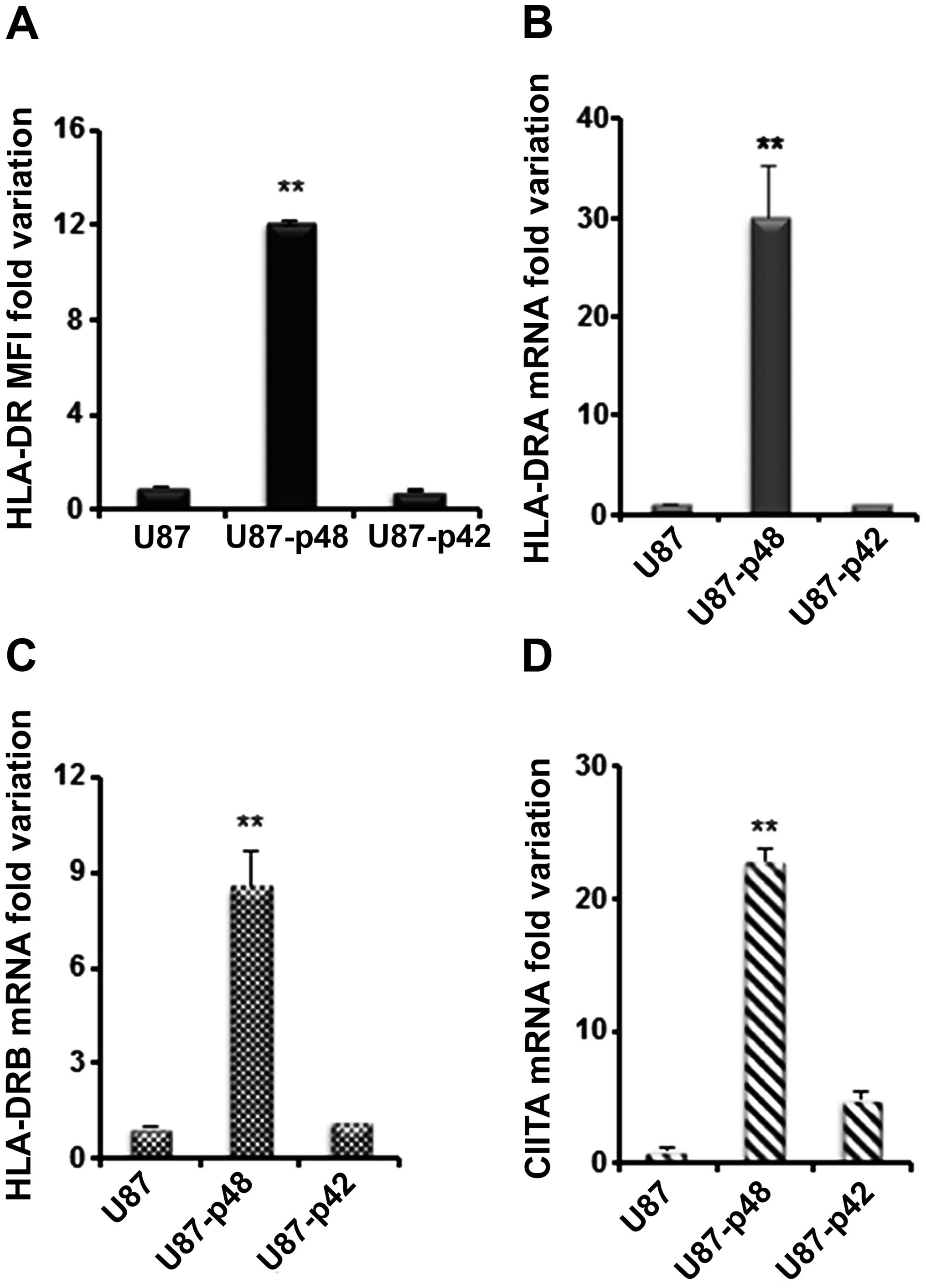
We then analysed the effect of p42 and p48 isoforms
overexpression on MHC II in glioblastoma. HLA-DR surface expression
in U87-p48 showed a 12-fold increase as compared to U87-p42 and to
control (Fig. 5A). The surface
variation corresponds to 30-fold increase of DRA mRNA (Fig. 5B) and 9-fold increase of DRB mRNA
(Fig. 5C) in U87-p48 cells, while
no variation was observed in the case of U87-p42. The measurement
of DQA1 and MHC I mRNAs (data not shown) confirmed data obtained in
M14. In addition, we quantified the CIITA mRNA amount and we found
23-fold increase in U87-p48 and 5-fold increase in U87-p42
(Fig. 5D). Similarly to the
results obtained with M14, we demonstrated that in glioblastoma
cell line p48 protein increases the transcription of MHC II genes
through CIITA activation.
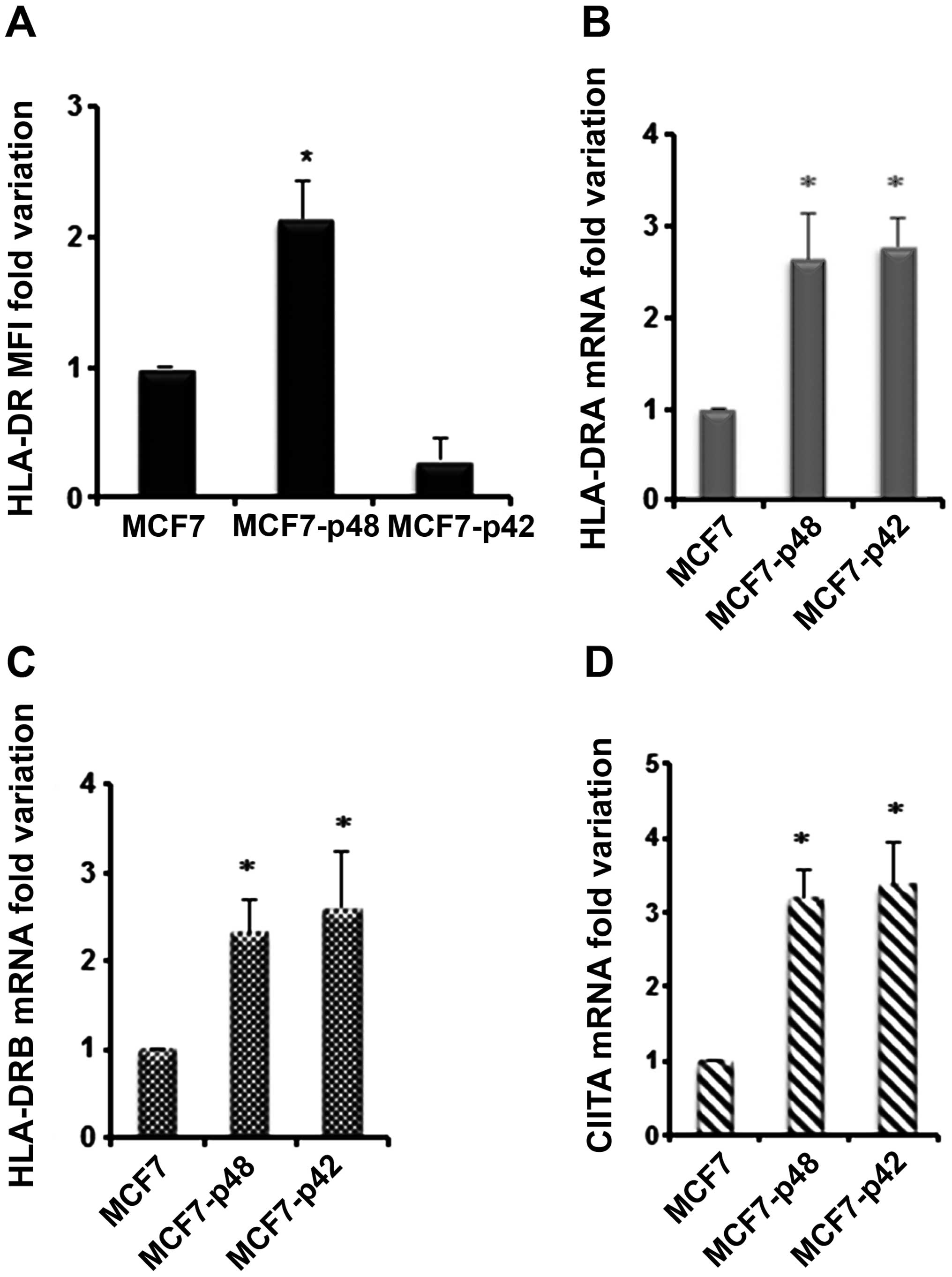
Finally, we used MCF7 cells which express MHC II
molecules only upon stimulation with IFNγ. Since we performed the
same treatment in the control, we assumed that any variation should
be attributed to the difference between the two stable cell lines.
We determined HLA-DR surface expression of MCF7 transfected cell
lines after IFNγ. As shown in Fig.
6A, we observed that HLA-DR MFI of MCF7-p48 shows 2.2-fold
increase compared to the cells transfected with empty vector, while
MCF7-p42 showed a downregulation of HLA-DR expression as compared
to the control. Surprisingly, we observed a 4-fold increase in the
amount of HLA-DRA and HLA-DRB not only in p48 but also in p42
transfectants (Fig. 6B and C),
which does not correspond with an increase in MHC II protein
(Fig. 6A). The expression of DQA1
is comparable to DR mRNAs while MHC I mRNA is unchanged (data not
shown). In order to explain the MHC II mRNA differences, we
analysed CIITA mRNA that showed 3-fold increase in both MCF7-p48
and MCF7-p42 (Fig. 6D). In
conclusion, both isoforms in carcinoma determine a comparable CIITA
activation and MHC II transcription, with only p42 isoform being
able to block MHC II protein synthesis through a mechanism which is
currently under investigation.
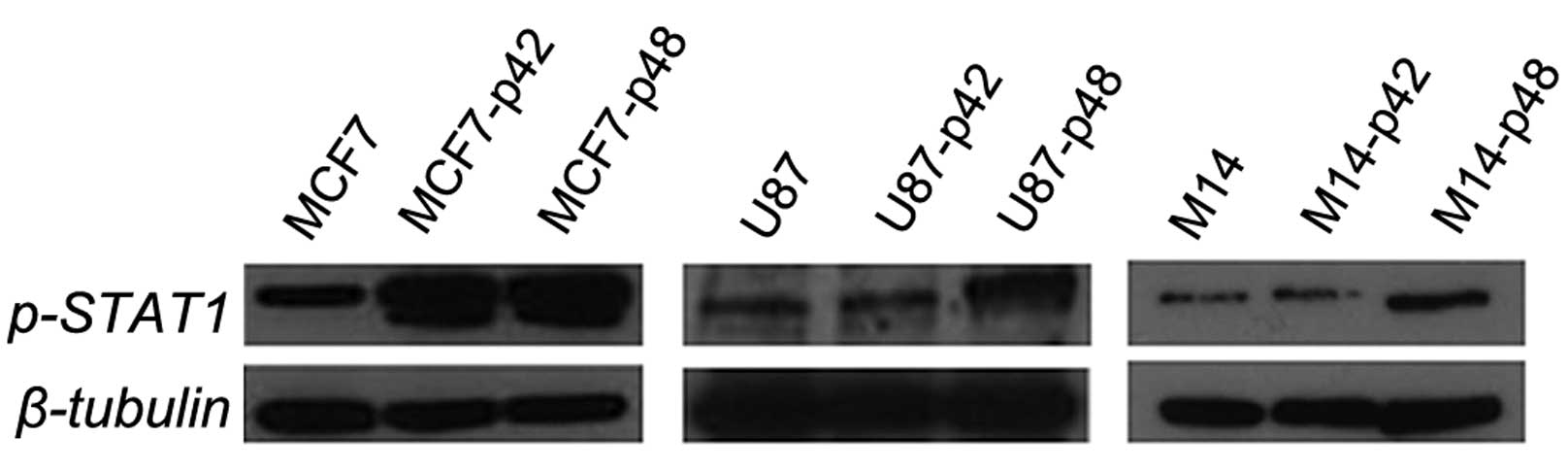
p48 oncogene influences the STAT-1
pathway
In order to unravel the protein pathway activation
upstream of CIITA, we evaluated the STAT1 phosphorylation in all
cell lines overexpressing EBP1 isoforms. We observed that both U87
and M14 cells, constitutively expressing MHC II, show upregulation
of p-STAT1 in the p48 overexpressing cells only and not in the p42
cells (Fig. 7). These results
clearly demonstrate that p48 induces STAT1 phosphorylation in the
absence of cytokine stimulation when it acts as an oncogene in U87
and M14. For MCF7-p42 and MCF7-p48 cells, we observed no
differences in the phosphorylation status of STAT1, and this in
agreement with the comparable CIITA increase (Fig. 6D).
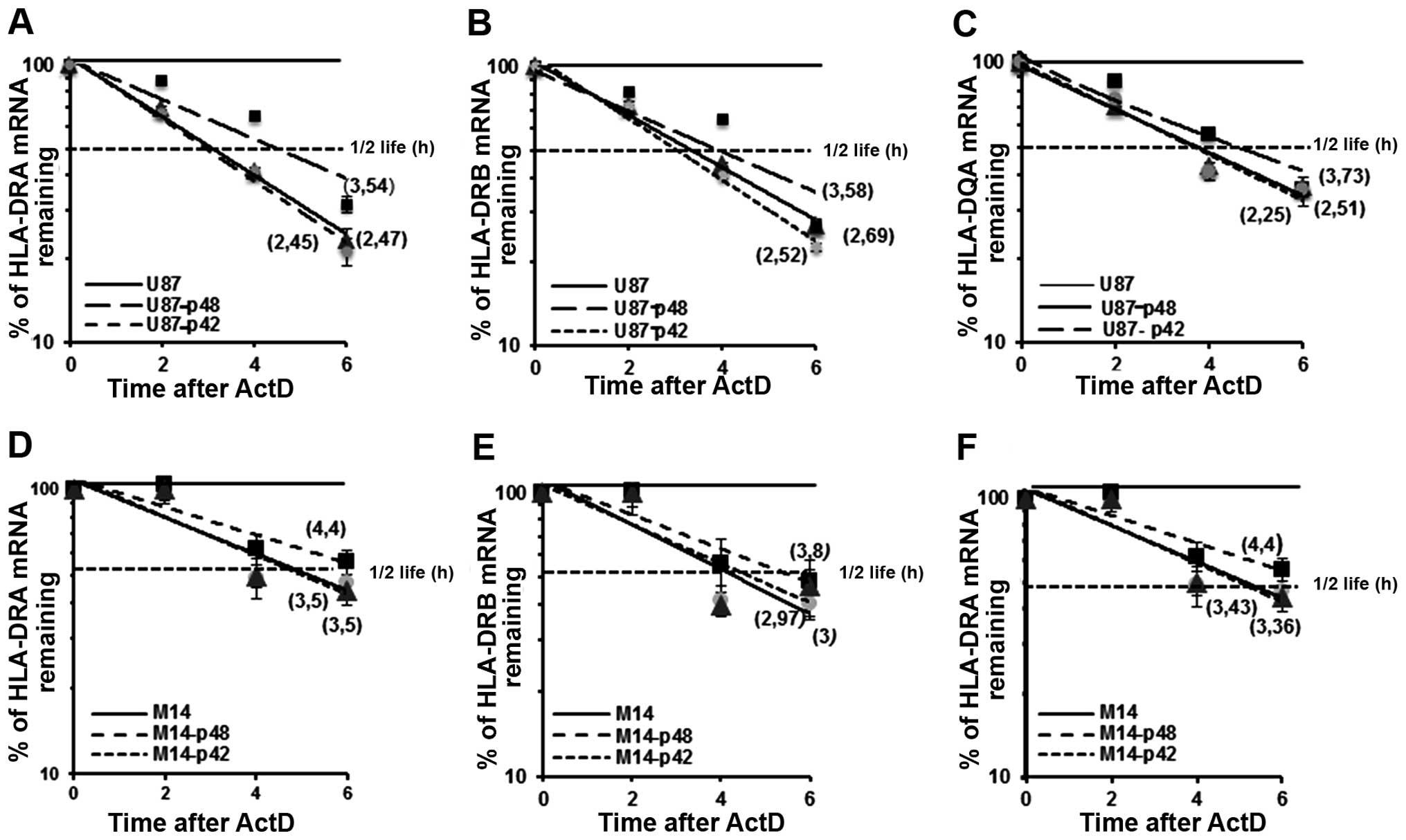
p48 isoform regulates MHC II
stability
We already demonstrated that p48 isoform influences
MHC II posttranscriptional regulation through the binding to UTRs
of the messengers (7,8). In the present study, we evaluated the
kinetics of mRNA decay in U87-p48 and U87-p42 cells after blocking
transcription at 2, 4 and 6 h with ActD. Total mRNA was analysed
for DRA, DRB and DQA1 expression by qRT-PCR, normalized by β-actin
mRNA level and expressed as percentage of maximum. When calculating
the half-lives of endogenous mRNAs in glioblastoma cells
transfected with p48, we found that DRA mRNA (t1/2=3.54
h; Fig. 8A) was more stable than
the DRA messenger from cells transfected with p42 or empty vector
(t1/2=2,45 h; Fig. 8A).
Also, the steady-state of DRB (Fig.
8B) and DQA1 (Fig. 8C) mRNA in
cells overexpressing p48 resulted in an increase of almost 1 h as
compared to mRNAs from U87-p42 cells. This increase was also
demonstrated in M14-p48 cells as shown in Fig. 8D–F. In conclusion, the
overexpression of p48 isoform, through the binding to 3′UTR of
transcripts, decreases endogenous MHC II mRNA decay, while p42
isoform is not able to interact with UTRs and influence their
stability.
Discussion
Recent studies have shown that CD4+ T
cells are critical for the generation and persistence of CTL
responses by providing help through multiple interactions with
antigen-presenting cells or by directly providing co-stimulatory
signals to the CTLs, which enhance their function and survival at
the tumour site. In addition, CD4+ T helper cells may
function as effector cells either by the local production of
cytokines (IFNγ and IL2) that curtail tumour growth or trigger the
release of cytotoxic mediators towards MHC II tumour cells
(29,30).
All these findings emphasize the interest for the
MHC II expression by tumour cells, which is variable and not
clearly related to the clinical outcome (31). Although the majority of solid
cancers do not constitutively express MHC II at significant levels
and cannot be directly recognized or killed in vitro by
CD4+ lymphocytes, many tumour cells can upregulate the
MHC II upon stimulation with IFNγ. Cytokine stimulation activates
CIITA expression as consequence of promoter IV epigenetic
de-repression (32). Moreover, no
data are available to correlate the function of specific oncogenes
with MHC II activation in solid tumours.
We have previously demonstrated that MHC II mRNAs
are regulated at post-transcriptional level by an RNP complex that
affects the processing and guarantees a coordinate expression of
mRNAs encoding two chains of MHC heterodimeric molecules. One of
the factors involved in the RNP complex is p48 isoform of EBP1, an
RNA binding protein that, interacting with UTRs of MHC II
messengers, affects MHC II post-transcriptional regulation
(7,8).
In the present study, we first studied the effect of
the p48 and p42 EBP1 isoforms on cell cycle in different tumour
cell lines of non-hematopoietic origin, and then analysed the role
of EBP1 isoforms on MHC II expression.
Many studies reported that p48 isoform represses
transcription of genes involved in cell cycle progression in the
nucleus of carcinoma, with consequent inhibition of proliferation
(10–12,15,28,33).
In these papers, it has been showed that p48 was able to interact
with retinoblastoma (Rb) protein through the binding of the
C-terminal region to form a repressor complex with Sin3A and HDAC2,
which tightly binds E2F family proteins preventing the
transcription of E2F regulated cell cycle genes (14,34,35).
Others authors have demonstrated that in glioblastoma p48 isoform
induces proliferation, in vitro and in vivo, because
it causes p53 poly-ubiquitination and degradation trough the
interaction with HDM2, while p42 reduces growth and promotes
differentiation (20,21,36).
In the present study, we have analysed the different
role of the two isoforms in tumorigenesis using three different
cell lines: glioblastoma (U87), melanoma (M14) and a breast
carcinoma (MCF7), overexpressing p48 or p42. We assessed in
parallel the effect of p48 and p42 on cell cycle and apoptosis, in
relationship with the cell type. In MCF7-p48, we observed a block
of cell proliferation and a strong induction of apoptosis, whereas,
the overexpression of p42, does not show differences in the cell
cycle progression as compared to the control. In this case, we
confirmed the anti-proliferative and apoptotic functions of p48,
already demonstrated in different types of carcinoma, such as
breast, prostate, bladder and hepatocellular carcinoma (11,18,19,28).
In contrast, we found that, in glioblastoma and melanoma, p48
overexpression increases proliferation by blocking cells in S
phase, thus confirming the phenotype already observed (36–38).
Furthermore, we demonstrated for the first time that p42 isoform
inhibits proliferation of glioblastoma, by blocking cells in G1
phase of the cell cycle and by inducing apoptosis. In conclusion,
our findings confirm that p48 acts as an oncogene in glioblastoma
and as an onco-suppressor in carcinoma.
We then performed the analysis of MHC II expression
pattern in cells overexpressing p48 and p42 and we found that the
overexpression of p48 increases the surface amount of HLA-DR
heterodimer in U87, MCF7 and M14 cell lines and this phenotype is
due to increased transcription and mRNA stability of DRA and DRB
genes. MHC II transcriptional activation is a consequence of the
CIITA transactivator activity, that occurs in normal and tumour
cells of hematopoietic origin, as well as in several tumours of
nonhematopoietic origin and in a consistent number of cell lines
(e.g., glioblastoma, pancreas adenocarcinoma, melanoma and bladder
carcinoma, ATLAS data base), in some cases after IFNγ stimulation.
In this study, the cell lines overexpressing p48 show a strong
increase of CIITA in M14 and U87, and a lower but significant
three-fold increase in MCF7, following IFNγ activation. Next, we
investigated the pathway upstream of CIITA transactivator,
especially the STAT1 protein, that activates CIITA transcription
through the binding to GAS (interferon-gamma-activated sequence)
element in the CIITA promoter, upon cytokine stimulation (5). We found higher level of STAT1
phosphorylation in both U87-p48 and M14-p48 cells indicating a
clear involvement of this transcription factor in the CIITA and MHC
II upregulation through p48 protein. U87 and M14 overexpressing p42
isoform did not show any variation of MHC II, CIITA or p-STAT1
protein levels with respect to the control. On the other hand, MCF7
shows a comparable level of STAT1 phosphorylation in p48 and p42
overexpressing cells, that explains the increase of CIITA mRNA but
not the difference in HLA-DR surface protein. These results
probably could be dependent on post-transcriptional regulation
which is currently under investigation.
In our previous study, we showed that p48 interacts
with UTRs of MHC class II mRNAs (8), here we further investigated its role
during RNA processing. We show a clear increase of DRA, DRB and
DQA1 mRNA half-lives in U87-p48 and M14-p48. No variation of mRNA
decay was observed in cells overexpressing p42 probably because
this isoform lacks of a specific RNA binding motif at the
N-terminus of the protein (39).
In conclusion, the present study explored the
different roles of two EBP1 isoforms, analysing them simultaneously
in overexpressing cell lines and confirming their influence on the
tumour phenotype in a cell-type specific manner. p48 showed an
anti-proliferative and apoptotic role in carcinoma versus a
proliferative function in glioblastoma and melanoma. In this
regard, we hypothesize that in glioblastoma the different function
of p48 may be regulated by HDM2 that is able to interact with both
tumour suppressors p53 and Rb (40). Conversely, p42 isoform shows a
clear function of tumour suppressor, inhibiting proliferation and
inducing apoptosis.
One hallmark of tumour cells is the acquisition of a
new profile of expressed proteins, determined by changes in gene
regulation. In this contest, the ability of tumour cells to express
MHC II could be a consequence of p48 EBP1 activity that upregulates
MHC II expression at transcriptional level via STAT1 activation and
by a post-transcriptional regulation, which increases mRNAs
stability.
We propose a scenario in which, when an oncogene
such as p48 affects the normal gene expression and induces tumor
progression, the increment of MHC II surface molecules can be
considered a strategy used by cells to activate a tumor-specific
immune response of CD4+ T cells.
Acknowledgements
The authors thank Professor A. Hamburger who kindly
provided the cDNA encoding p48 isoform of EBP1 protein, Dr Maria
Patrizia Stoppelli for critical revision of the manuscript. IGB
FACS facility for technical support. L.P. fellowship was funded by
MODO Project (POR-Campania 2007–2013).
References
|
1
|
Kobayashi H and Celis E: Peptide epitope
identification for tumor-reactive CD4 T cells. Curr Opin Immunol.
20:221–227. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Campoli M and Ferrone S: HLA antigen
changes in malignant cells: Epigenetic mechanisms and biologic
significance. Oncogene. 27:5869–5885. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haabeth OA, Tveita AA, Fauskanger M,
Schjesvold F, Lorvik KB, Hofgaard PO, Omholt H, Munthe LA, Dembic
Z, Corthay A, et al: How do CD4+ T cells detect and
eliminate tumor cells that either lack or express MHC class II
molecules? Front Immunol. 5:1742014. View Article : Google Scholar
|
|
4
|
Reith W, LeibundGut-Landmann S and
Waldburger JM: Regulation of MHC class II gene expression by the
class II transactivator. Nat Rev Immunol. 5:793–806. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pisapia L, Pozzo GD, Barba P, Citro A,
Harris PE and Maffei A: Contrasting effects of IFNα on MHC class II
expression in professional vs. nonprofessional APCs: Role of CIITA
type IV promoter. Results Immunol. 2:174–183. 2012. View Article : Google Scholar
|
|
6
|
Ting JP and Trowsdale J: Genetic control
of MHC class II expression. Cell. 109(Suppl): S21–S33. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pisapia L, Cicatiello V, Barba P, Malanga
D, Maffei A, Hamilton RS and Del Pozzo G: Co-regulated expression
of alpha and beta mRNAs encoding HLA-DR surface heterodimers is
mediated by the MHCII RNA operon. Nucleic Acids Res. 41:3772–3786.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Corso C, Pisapia L, Citro A, Cicatiello V,
Barba P, Cigliano L, Abrescia P, Maffei A, Manco G and Del Pozzo G:
EBP1 and DRBP76/NF90 binding proteins are included in the major
histocompatibility complex class II RNA operon. Nucleic Acids Res.
39:7263–7275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia X, Lessor TJ, Zhang Y, Woodford N and
Hamburger AW: Analysis of the expression pattern of Ebp1, an
ErbB-3-binding protein. Biochem Biophys Res Commun. 289:240–244.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lessor TJ, Yoo JY, Xia X, Woodford N and
Hamburger AW: Ectopic expression of the ErbB-3 binding protein ebp1
inhibits growth and induces differentiation of human breast cancer
cell lines. J Cell Physiol. 183:321–329. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Wang XW, Jelovac D, Nakanishi T,
Yu MH, Akinmade D, Goloubeva O, Ross DD, Brodie A and Hamburger AW:
The ErbB3-binding protein Ebp1 suppresses androgen
receptor-mediated gene transcription and tumorigenesis of prostate
cancer cells. Proc Natl Acad Sci USA. 102:9890–9895. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Linn D, Liu Z, Melamed J, Tavora
F, Young CY, Burger AM and Hamburger AW: EBP1, an ErbB3-binding
protein, is decreased in prostate cancer and implicated in hormone
resistance. Mol Cancer Ther. 7:3176–3186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Ali TZ, Zhou H, D’Souza DR, Lu Y,
Jaffe J, Liu Z, Passaniti A and Hamburger AW: ErbB3 binding protein
1 represses metastasis-promoting gene anterior gradient protein 2
in prostate cancer. Cancer Res. 70:240–248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Woodford N, Xia X and Hamburger
AW: Repression of E2F1-mediated transcription by the ErbB3 binding
protein Ebp1 involves histone deacetylases. Nucleic Acids Res.
31:2168–2177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y and Hamburger AW: Specificity and
heregulin regulation of Ebp1 (ErbB3 binding protein 1) mediated
repression of androgen receptor signalling. Br J Cancer.
92:140–146. 2005. View Article : Google Scholar
|
|
16
|
Sun J, Luo Y, Tian Z, Gu L, Xia SC and Yu
Y: Expression of ERBB3 binding protein 1 (EBP1) in salivary adenoid
cystic carcinoma and its clinicopathological relevance. BMC Cancer.
12:4992012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou X, Chen W, Zhang Y, Sun J, Wang Q and
Yu Y: Potential therapeutic strategy for oral squamous cell
carcinoma by ErbB3-binding protein 1 gene transfer. J Cancer Res
Clin Oncol. 136:891–896. 2010. View Article : Google Scholar
|
|
18
|
He HC, Ling XH, Zhu JG, Fu X, Han ZD,
Liang YX, Deng YH, Lin ZY, Chen G, Chen YF, et al: Down-regulation
of the ErbB3 binding protein 1 in human bladder cancer promotes
tumor progression and cell proliferation. Mol Biol Rep.
40:3799–3805. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu B, Xiong Y, Ni R, Wei L, Jiang D, Wang
G, Wu D, Xu T, Zhao F, Zhu M, et al: The downregulation of ErbB3
binding protein 1 (EBP1) is associated with poor prognosis and
enhanced cell proliferation in hepatocellular carcinoma. Mol Cell
Biochem. 396:175–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Z, Ahn JY, Liu X and Ye K: Ebp1
isoforms distinctively regulate cell survival and differentiation.
Proc Natl Acad Sci USA. 103:10917–10922. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim CK, Nguyen TL, Joo KM, Nam DH, Park J,
Lee KH, Cho SW and Ahn JY: Negative regulation of p53 by the long
isoform of ErbB3 binding protein Ebp1 in brain tumors. Cancer Res.
70:9730–9741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bose SK, Sengupta TK, Bandyopadhyay S and
Spicer EK: Identification of Ebp1 as a component of cytoplasmic
bcl-2 mRNP (messenger ribonucleoprotein particle) complexes.
Biochem J. 396:99–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou H, Mazan-Mamczarz K, Martindale JL,
Barker A, Liu Z, Gorospe M, Leedman PJ, Gartenhaus RB, Hamburger AW
and Zhang Y: Post-transcriptional regulation of androgen receptor
mRNA by an ErbB3 binding protein 1 in prostate cancer. Nucleic
Acids Res. 38:3619–3631. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou H, Zhang Y and Hamburger AW: EBP1
inhibits translation of androgen receptor mRNA in castration
resistant prostate cancer cells. Anticancer Res. 31:3129–3135.
2011.PubMed/NCBI
|
|
25
|
Squatrito M, Mancino M, Donzelli M, Areces
LB and Draetta GF: EBP1 is a nucleolar growth-regulating protein
that is part of pre-ribosomal ribonucleoprotein complexes.
Oncogene. 23:4454–4465. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Squatrito M, Mancino M, Sala L and Draetta
GF: Ebp1 is a dsRNA-binding protein associated with ribosomes that
modulates eIF2alpha phosphorylation. Biochem Biophys Res Commun.
344:859–868. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Zhang Y, Akinmade D and Hamburger AW:
Inhibition of heregulin mediated MCF-7 breast cancer cell growth by
the ErbB3 binding protein EBP1. Cancer Lett. 265:298–306. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie Y, Akpinarli A, Maris C, Hipkiss EL,
Lane M, Kwon EK, Muranski P, Restifo NP and Antony PA: Naive
tumor-specific CD4+ T cells differentiated in vivo
eradicate established melanoma. J Exp Med. 207:651–667. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Quezada SA, Simpson TR, Peggs KS, Merghoub
T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et
al: Tumor-reactive CD4+ T cells develop cytotoxic
activity and eradicate large established melanoma after transfer
into lymphopenic hosts. J Exp Med. 207:637–650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thibodeau J, Bourgeois-Daigneault MC and
Lapointe R: Targeting the MHC class II antigen presentation pathway
in cancer immunotherapy. Oncoimmunology. 1:908–916. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Holling TM, van Eggermond MC, Jager MJ and
van den Elsen PJ: Epigenetic silencing of MHC2TA transcription in
cancer. Biochem Pharmacol. 72:1570–1576. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Y, Fondell JD, Wang Q, Xia X, Cheng
A, Lu ML and Hamburger AW: Repression of androgen receptor mediated
transcription by the ErbB-3 binding protein, Ebp1. Oncogene.
21:5609–5618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Akinmade D and Hamburger AW: The
ErbB3 binding protein Ebp1 interacts with Sin3A to repress E2F1 and
AR-mediated transcription. Nucleic Acids Res. 33:6024–6033. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia X, Cheng A, Lessor T, Zhang Y and
Hamburger AW: Ebp1, an ErbB-3 binding protein, interacts with Rb
and affects Rb transcriptional regulation. J Cell Physiol.
187:209–217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim CK, Lee SB, Nguyen TL, Lee KH, Um SH,
Kim J and Ahn JY: Long isoform of ErbB3 binding protein, p48,
mediates protein kinase B/Akt-dependent HDM2 stabilization and
nuclear localization. Exp Cell Res. 318:136–143. 2012. View Article : Google Scholar
|
|
37
|
Liu Z, Oh SM, Okada M, Liu X, Cheng D,
Peng J, Brat DJ, Sun SY, Zhou W, Gu W, et al: Human BRE1 is an E3
ubiquitin ligase for Ebp1 tumor suppressor. Mol Biol Cell.
20:757–768. 2009. View Article : Google Scholar :
|
|
38
|
Kwon IS and Ahn JY: p48 Ebp1 acts as a
downstream mediator of Trk signaling in neurons, contributing
neuronal differentiation. Neurochem Int. 58:215–223. 2011.
View Article : Google Scholar
|
|
39
|
Monie TP, Perrin AJ, Birtley JR, Sweeney
TR, Karakasiliotis I, Chaudhry Y, Roberts LO, Matthews S,
Goodfellow IG and Curry S: Structural insights into the
transcriptional and translational roles of Ebp1. EMBO J.
26:3936–3944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yap DB, Hsieh JK, Chan FS and Lu X: mdm2:
A bridge over the two tumour suppressors, p53 and Rb. Oncogene.
18:7681–7689. 1999. View Article : Google Scholar
|