Introduction
In the present scenario lung cancer is the leading
cause of cancer-related deaths worldwide and the incidence of lung
cancer has almost reached epidemic proportions in both developing
and developed countries (1).
Despite decades of research, the available treatment options for
lung cancer patients remain inadequate, either to offer a cure or
even a substantial survival advantage owing to its inherent
resistance to chemotherapy. Moreover, the clinical efficacy and
usefulness of chemotherapy is still limited because of its
dose-limiting toxicity (2) which
has considerably shifted the focus towards complementary and
alternative medicine (CAM), that are low in toxic side effects
(3). Among different CAM regimens,
homeopathy a nearly 200-year-old system of medicine has been shown
to decrease side effects of chemotherapy in cancer patients and
possess antitumorigenic property (4–8).
Homeopaths have described observations that tumors recede from the
use of homeopathic treatment and have, from time to time,
documented long-term recoveries from cancer in response to
homeopathic treatment (4–6). Unfortunately, scientific studies
corroborating these clinical observations are very few. There are
only few reports on the mechanism of action of homeopathic drugs in
experimental cancers and cell cultures (9–14).
In the present study, we investigated the basic
molecular mechanism of antitumorigenic effect of sulphur, a
promising homeopathic remedy, on non-small cell lung cancer cells.
Sulphur, the most ancient archetype in the history of our planet,
is used by homeopaths for treating inflammation and cancer and also
in treating other skin diseases (15–18).
Potent chemo-preventive effects have been demonstrated in various
in vivo and in vitro models for sulphur-containing
compounds found in naturally occurring products, such as, onions
and garlic (19,20). Protective effect of sulphur has
also been reported against cytotoxicity in neuroblastoma cells
(21). In vitro treatment
with sulphur significantly increased apoptosis in neuroblastoma
cells (22). Reports have stated
growth inhibitory and apoptosis-related effects of sulphur on
immortalized human oral keratinocytes and on oral cancer cells
(23). Taken together, these
findings indicate a promising anticancer potential of sulphur.
However, the underlying mode of action for its professed
antitumorigenic effect in highly resistant non-small cell lung
carcinoma (NSCLC) is still unidentified and requires further study.
To the best of our knowledge, therefore, this is the first report
delineating the detailed mechanism of sulphur on NSCLC cells.
It is well established that development and growth
of tumor cells are controlled by complex signalling pathways
involved in the regulation of cell death, survival and
proliferation. In mammalian cells, the regulatory contribution of
NFκB and p53 to cancer development and progression is well
documented where inactivation of p53 and hyper-activation of NFκB
are the common occurrences (24–27).
It has been acknowledged that NFκB pathway activation renders
inherent resistance to chemotherapy to NSCLC cells apparently via
induction of survival and anti-apoptotic proteins (25,28).
Therefore, targeting NFκB pathway may serve as a novel approach to
regress NSCLC cells. Conversely, tumor suppressor p53, the
‘guardian of genome’ translates stress signals into cell cycle
arrest or apoptosis, depending on the balance between pro-apoptotic
and anti-proliferative genes (29–31).
Thus, drugs reviving tumor suppressor functions of p53 will be
proficient for targeted cancer therapy. Considering the
deregulation of NFκB and p53 pathways in numerous cancers,
including NSCLC cells, it is not surprising that extensive
cross-talk between these pathways exists at various levels. In
fact, NFκB activation was shown to play a role in neoplastic
transformation by inhibiting p53 gene expression (32,33).
Moreover, reports have shown that NFκB by inducing the E3 ubiquitin
ligase MDM2 attenuated p53 protein stability (34). Furthermore, the NFκB gene promoter
is activated by p53 mutants, and p52 subunit of NFκB can modulate
the promoter activity of p53 target genes (34). Both NFκB and p53 compete for
co-activators, for example, the histone acetyltransferases p300 and
CBP (35,36). An ideal therapeutic approach
should, therefore, involve tailoring NFκB-governed survival pathway
in favor of p53-regulated apoptotic pathway to regress otherwise
drug-resistant NSCLC cells.
The present study investigated the molecular
mechanism underlying the antitumorigenic potential of homeopathic
remedy sulphur, commonly known as a healing mineral. Our findings
revealed that sulphur preferentially induces apoptosis in NSCLC
cells sparing normal cells. Our exploration for the detailed
molecular mechanism revealed that sulphur inhibits NFκB-induced
Bcl-2 mediated survival pathway while triggering p53-induced Bax
mediated apoptosis in NSCLC cells. In NSCLC cells, the
constitutively active NFκB associates with p300 and this NFκB-p300
complex binds to the promoter region of Bcl-2 thereby leading to
transcriptional upregulation of Bcl2 that in turn endorses
activation of survival pathway. On the contrary, upon sulphur
treatment, pro-apoptotic gene p53 gets activated and occupies p300
to form p53–p300 complex. NFκB-p300 complex formation, therefore,
gets hampered and the newly formed p53–p300 complex binds to the
promoter region of p53 target gene, Bax thereby leading to the
transcriptional upregulation of Bax that consecutively directs
activation of apoptotic pathway. Sulphur thus plays an essential
role in dictating pro and anti-apoptotic permutation to create an
environment conducive for induction of apoptosis in NSCLC
cells.
Materials and methods
Cell culture
Human non-small cell carcinoma cell line, A549 was
obtained from NCCS, India. Peripheral blood collected from healthy
human volunteers with informed consent (Institutional Review Board
1382) was centrifuged over Ficoll-Hypaque density gradient
(Amersham Pharmacia, Uppsala, Sweden) to obtain total peripheral
blood mono-nuclear cells. Cells were routinely maintained in DMEM
supplemented with 10% heat inactivated fetal bovine serum (Lonza,
NH, USA), L-glutamine (2 mM), sodium pyruvate (100 μg/ml),
non-essential amino acids (100 μM), streptomycin (100 μg/ml),
penicillin (50 U/ml; Invitogen, CA, USA) at 37°C in a humidified 5%
CO2 incubator. Cells were maintained in an exponential
growth phase for all experiments. Viable cell numbers were
determined by trypan-blue exclusion test.
Treatment of cells
Placebo and sulphur 6C, 30C or 200C were procured
from Hahnemann Publishing Co., India. Cells were treated with
sulphur/placebo of potencies 6, 30 or 200C exposure at the
different concentration (10, 15, 20 and 30 μl/ml) for different
time-points (6, 12, 24, 36 and 48 h) to select the optimum time
required for cell killing. To understand the sequence of events
leading to apoptosis, cancer cells were treated with mitochondrial
pore inhibitor CsA (25 μM; Merck, Germany) for 1 h prior to
incubation with sulphur.
Flow cytometry
For the determination of cell death, cells were
stained with 7AAD and Annexin V-FITC and analyzed on flow cytometry
(FACS Verse, BD Biosciences). Electronic compensation of the
instrument was done to exclude overlapping of the emission spectra.
A total of 10,000 events were acquired for analysis using CellQuest
software. For the assessment of mitochondrial transmembrane
potential, cells were stained with potentially-sensitive dye
Dihexyloxacarbonicao cyanine (DiOC6, Merck, Germany)
during the last 30 min of treatment at 37°C in the dark.
Fluorescence of retained DiOC6 was determined flow
cytometrically using logarithmic amplification by CellQuest
software (BD Biosciences) (31).
Fluorescence imaging
Chromatin condensation and nuclear fragmentation was
analyzed using the standard protocol. Briefly, cells were grown on
coverslips, fixed with 3% p-formaldehyde for 10 min and then
permeabilized with 0.1% Triton X-100 for 5 min. Cells were then
incubated with 4′6-diamidino-2-phenyl-indole (DAPI; BD Pharmingen,
CA, USA). The morphology of the cell nuclei was visualized using a
fluorescence microscope (Leitz microscope fitted with
epifluorescence illuminator through a 60× aperture oil immersion
lens, Carl Zeiss, Germany). For fluorescence imaging, cells growing
on a cover slip were fixed with 3% p-formaldehyde and were stained
with anti-p53 and anti-p65NFκB antibodies (SantaCruz, CA, USA),
after permeabilization with Triton X-100, followed by FITC and
TRITC conjugated secondary antibodies, respectively and visualized
with confocal microscope (Carl Zeiss, Germany) (13,37).
Plasmids, siRNA and transfections
The expression constructs pcDNA3.0/HA-tagged
IκBα-32A/36A [IκBα super-repressor (IκBα-SR), a kind gift from Dr
J. Didonato, The Cleveland Clinic Foundation],
pcDNA3.1-p65NFκB/p53/Bcl-2 overexpression plasmids (2 μg/million
cells) were introduced into exponentially growing cancer cells
using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according
to the protocol provided by the manufacturer. In a similar manner,
p53 expression was knocked down in A549 cells using p53-shRNA
(Santa Cruz) and Lipofectamine 2000 (Invitrogen). Stably expressing
clones were isolated by limiting dilution and selected with G418
sulphate (Cellgro) at a concentration of 400 μg/ml and cells
surviving this treatment were cloned and screened by western blot
analysis with specific antibody. A549 cells were transfected with
300 pmol of Bcl-2-/Bax-/control-ds-siRNA (Santa Cruz) and
Lipofectamine 2000 separately for 12 h. The levels of respective
proteins were estimated by western blotting (37).
Western blotting
To obtain whole cell lysates, cells were homogenized
in lysis buffer (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM
MgCl2, 1 mM Na-EDTA, 1 mM Na-EGTA and 1 mM DTT)
supplemented with protease and phosphatase inhibitor cocktails.
Mitochondrial and cytosolic fractions were prepared according to
Lahiry et al (31). For
direct western blot analysis, a total of 50 μg of protein was
resolved using SDS-PAGE and transferred to nitrocellulose membrane
for western blotting using required antibodies e.g.,
anti-caspase-9, anti-caspase-3, anti-p53 (DO-1), anti-p65NFκB,
anti-IκBα, anti-Bcl-2, anti-Bax (N-20), anti-cytochrome c
and anti-p300. Thereafter proteins of interest were visualized by
chemiluminiscence. Equivalent protein loading in cytosolic, nuclear
and mitochondrial fractions were verified using anti-α-actin/
histone H1/MnSOD antibodies (Santa Cruz, CA, USA) respectively.
Co-immunoprecipitation
For the determination of direct interaction between
two proteins, co-immunoprecipitation technique was employed
(38,39). p53–p300 and p65NFκB-p300
interaction was determined by co-immunoprecipitation. Samples (300
μg of protein from the total lysate) were incubated at 4°C
overnight with anti-p300/-IgG antibody and then incubated for 2 h
at 4°C with protein A-Sepharose. Immunocomplexes were washed of
unbound proteins with cold TBS with protease inhibitors, and
pelleted beads were boiled for 5 min in SDS-PAGE sample buffer. The
immunoprecipitated proteins were resolved on SDS-PAGE and analyzed
by western blotting for detection of associated proteins. Equal
protein loading was confirmed using anti-histone H1 antibody.
Reverse transcriptase-PCR
Total RNA (2 μg) each from untreated,
placebo-/sulphur-treated NSCLC cells was extracted by TRIzol
(Invitrogen) and was reverse transcribed and then subjected to PCR
with enzymes and reagents of the RTplusPCR System (Eppendorf,
Hamburg, Germany) using GeneAmpPCR 2720 (Applied Biosystems, Foster
City, CA, USA) (40). The cDNAs
were amplified with primers specific for Bax
(5′-GGAATTCCAAGAAGCTGAGCGAGTGT-3′/
5′-GGAATTCTTCTTCCAGATGGTGAGCGAG-3′), Bcl-2
(5′-CCTGTGCCACCATGTGTCCATC-3′/5′-GCTGAGAACA GGGTCTTCAGAGAC-3′) and
GAPDH (internal control: 5′-TGATGACATCAAGAAGGTGGTGAAG-3′/5′-TCCTTGG
AGGCCATGTAGGCCAT-3′).
Chromatin immunoprecipitation (ChIP)
ChIP assays were carried out for identification of
p53 and p65NFκB binding region on Bax and Bcl-2-promoters
respectively, using a ChIP assay kit (Millipore) according to the
manufacturer’s instructions. PCR assay was performed using primer
sets as follows: Bcl-2 forward primer 5′-GATTCCTGCGGATTGACA
TTTC-3′, Bcl-2 reverse primer 5′-CATCAATCTTCAGCACTC TCC-3′; Bax
forward primer 5′-TCAGCACAGATTAGTTT CTG-3′, Bax reverse primer
5′-GGGATTACAGGCATGAG CTA-3′. Extracted DNA (2 μl) was used for 45
cycles of amplification in 5 μl of reaction mixture under the
following conditions: 95°C for 30 sec, 56°C for 30 sec and 72°C for
60 sec. The PCR products were analysed by 2% agarose gel
electrophoresis (41,42).
Statistical analysis
Values are shown as standard error of mean, except
when otherwise indicated. Data were analyzed and, significance
(P<0.05) of the differences between mean values was determined
by a Student’s t-test.
Results
Sulphur induces non-small cell carcinoma
apoptosis
The effect of sulphur, a homeopathic drug, on the
viability of human NSCLC cell line (A549) and normal peripheral
blood mononuclear cell (PBMC) was examined at different potencies
of sulphur, i.e., 6C, 30C, 200C, where for each potency a
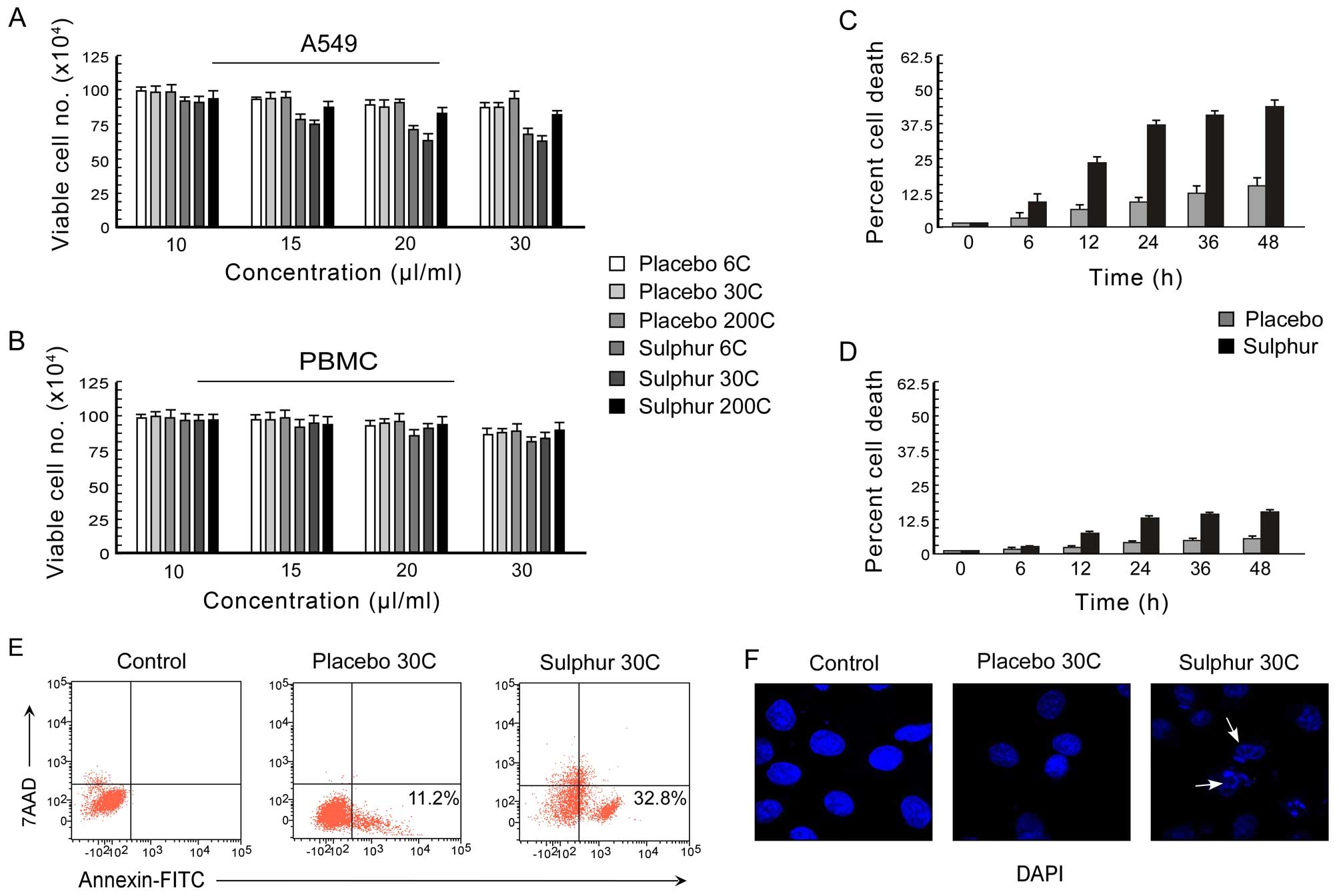
differential dose of 0–30 μl/ml was applied (Fig. 1A and B). The percent cell death was
scored by trypan-blue dye-exclusion assay. It was observed that
among all the potencies of sulphur, 30C potency resulted in most
significant decrease (p<0.001) in cell viability (Fig. 1A). Moreover, at 20 μl/ml dose of
30C, the percent A549 cell death reached its optimum (Fig. 1A) while under the same conditions
the PBMC viability was found to be >90% (Fig. 1B). These results indicating the
better efficacy of 20 μl/ml dose of 30C sulphur in A549 cell
killing with minimum toxicity led us to perform all further
experiments using this particular potency and dose of sulphur. The
time-dependent effect of sulphur 30C (20 μl/ml), in comparison to
placebo, on A549 and PBMC cells was examined at different time
intervals (0–48 h) and percentage cell death was assessed. Sulphur
30C, at a concentration of 20 μl/ml, exerted time-dependent
significant death in A549 cells (Fig.
1C). However, significant cell death in PBMCs was noted from 24
h onwards following the treatment (Fig. 1D).
Next, to confirm the nature of cell death as
apoptosis, we utilized double labelling techniques using Annexin
V-FITC/7-AAD to distinguish between apoptotic and necrotic cells.
Our flow cytometric data demonstrated that in comparison to
placebo-treated A549 cells, sulphur-treated unfixed A549 cells
showed Annexin V-FITC binding with minimum 7-AAD binding (Fig. 1E) indicating that the mode of cell
death was apoptosis but not necrosis. These findings were
re-confirmed by the development of nuclear blebbing as evidenced by
DAPI-stained fluorescent images of sulphur-treated A549 cells
(Fig. 1F). These data together
supported the notion that sulphur 30C asserts apoptogenic effect in
the NSCLC A549 cells.
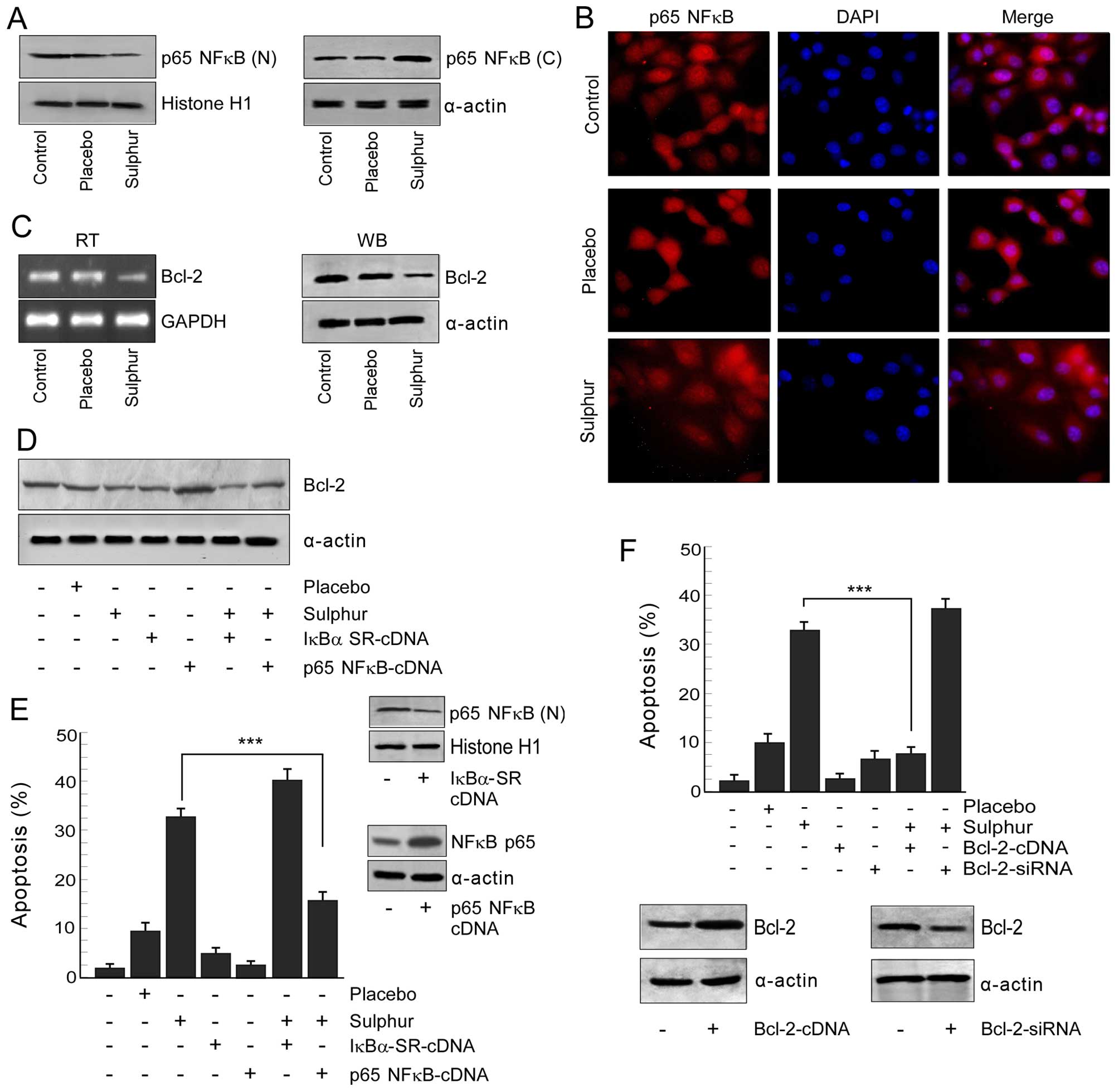
Sulphur decreases the survival advantage
of NSCLC cells by perturbing nuclear translocation of p65NFκB
Since p65NFκB has been reported to be globally
involved in survival of cancer cells (27,28,43),
we examined whether sulphur suppresses this pathway to combat
NFκB-mediated survival to induce apoptosis in drug-resistant NSCLC
cells. Our search revealed that sulphur treatment efficiently
blocked nuclear translocation of NFκB in NSCLC cells as observed by
both western blotting (Fig. 2A)
and confocal imaging experiments (Fig.
2B). In addition, the mRNA and protein levels of NFκB-target
gene, Bcl-2, was found to be downregulated by sulphur treatment in
NSCLC cells (Fig. 2C). We further
observed that transfecting NSCLC cells with super repressor
IκBα-SR-cDNA decreased Bcl-2 followed by significant apoptosis in
response to sulphur (Figs. 2D and
E). On the other hand NSCLC cells expressing p65NFκB-cDNA
manifested enhanced Bcl-2 with significant resistance upon sulphur
exposure (Fig. 2D and E). The
anti-apoptotic role of NFκB-dependent Bcl-2 upregulation was
re-confirmed by evaluating response of Bcl-2-engineered cells
towards sulphur treatment. Overexpression of Bcl-2 in NSCLC cells
bestowed them with survival advantage upon sulphur treatment,
whereas transfection with Bcl-2-siRNA efficiently enhanced
sulphur-induced apoptosis (Fig.
2F). Collectively, these results confirmed that p65NFκB
activation and subsequent Bcl-2 upregulation were primarily
involved in survival of NSCLC cells which upon inhibition by
sulphur induced apoptosis in these cells.
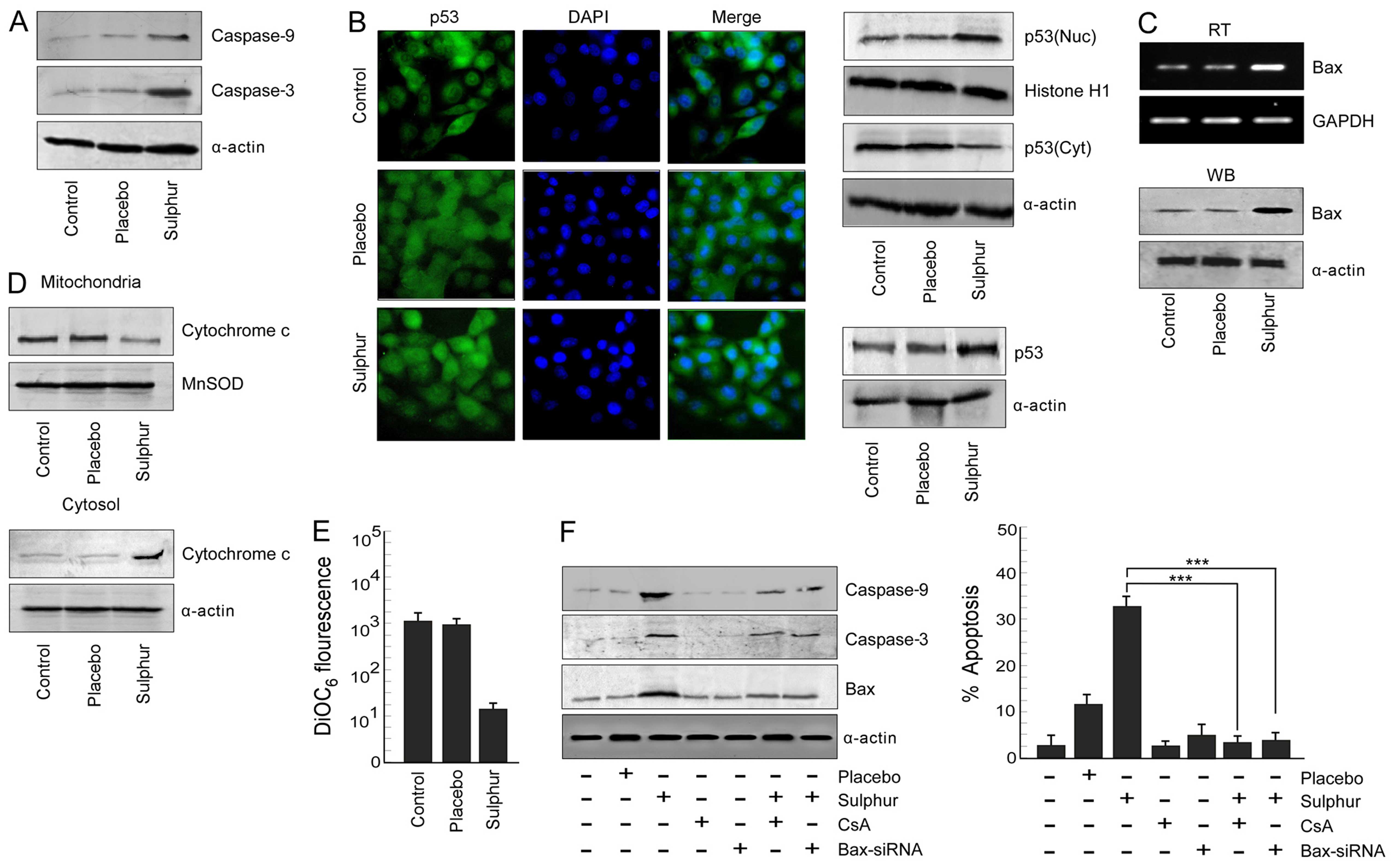
Sulphur treatment triggers the
p53-mediated mitochondria-dependent apoptotic pathway in NSCLC
cells
Since inhibition of p65NFκB activity in NSCLC cells
induced a powerful apoptotic response, we predicted the involvement
of the cellular apoptotic proteins during sulphur-induced
apoptosis. We evaluated the status of apoptotic proteases, i.e.,
caspase-9 and caspase-3, respectively, in response to sulphur
treatment. It was noted that sulphur treatment significantly
upregulated levels of cleaved caspase-9 and caspase-3 in A549 cells
(Fig. 3A). Since tumor suppressor
protein p53 plays an important role in canonical apoptotic pathway,
the above results tempted us to compare the p53 activation status
upon sulphur exposure in NSCLC cells. Results of Fig. 3B left panel revealed that sulphur
induced p53 expression in A549 cells when compared to untreated
cells. These results were further confirmed by confocal microscopy,
the results of which not only demonstrated accumulation of p53
protein but also its translocation from cytosol to nucleus of
sulphur-exposed A549 cells (Fig.
3B, middle panel). Furthermore, findings of western blot
analysis verified greater translocation of p53 from cytosol to
nucleus upon sulphur exposure in A549 cells (Fig. 3B right panel). It is acknowledged
that p53 when localized in nucleus transactivates its downstream
target genes (31). We, therefore,
next assessed the status of its trans-activated gene product, i.e.,
Bax, by western blot and RT-PCR analyses in sulphur-treated and
untreated A549 cells. Our results re-established that in comparison
to un-/placebo-treated tumor cells, sulphur-exposed cells showed
increase in the levels of p53 trans-activated gene product Bax
(Fig. 3C) Downstream of Bax,
increase in cytosolic cytochrome c with its concomitant
decrease in mitochondria (Fig. 3D)
were observed and also significant mitochondrial transmembrane
potential (MTP) loss is observed in sulphur-exposed A549 cells as
compared to control and placebo-treated A549 cells (Fig. 3E).
Next, A549 cells were transfected with Bax-siRNA or
pre-treated with cyclosporine A (CsA), mitochondrial pore formation
blocker, prior to sulphur treatment for validation of the
involvement of Bax in mitochondria cascade-mediated NSCLC
apoptosis. Silencing Bax, or CsA pre-treatment significantly
decreased percent apoptosis of NSCLC cells and also down-modulated
the expression levels of caspase-9 and caspase-3 (Fig. 3F). Taken together these findings
validated the contribution of Bax in sulphur-induced A549 cell
apoptosis via mitochondrial death cascade.
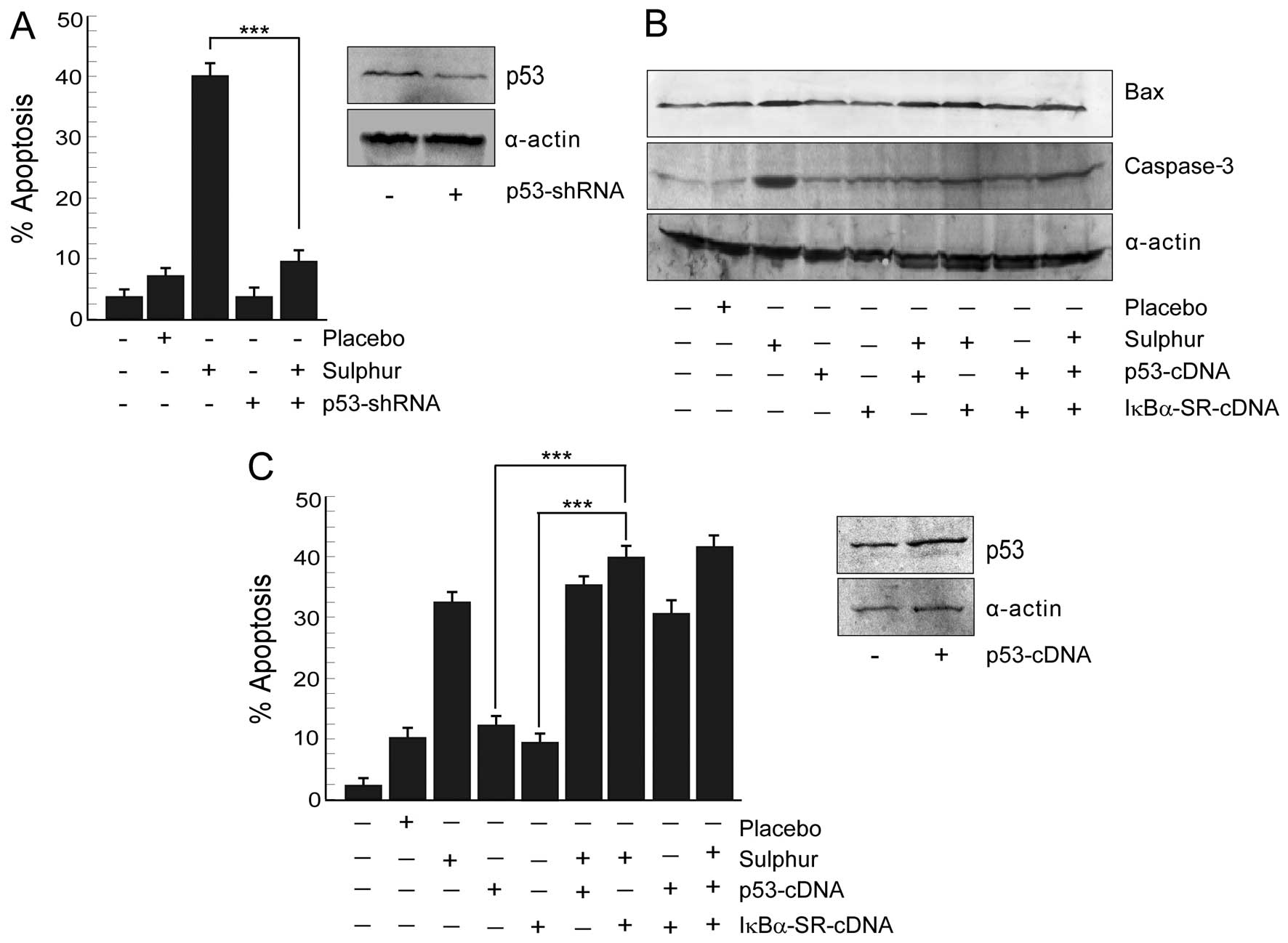
Inhibition of p65NFκB by sulphur triggers
p53-mediated apoptosis in NSCLC cells
Next, we verified whether sulphur-induced cancer
cell death is p53-dependent or not. To this end, human cancer cell
lines with differential p53 status, e.g., wild-type p53-expressing
and p53-shRNA-transfected A549 cells were tested for
sulphur-dependent apoptosis by scoring the number of Annexin
V-positive cells flow cytometrically (Fig. 4A). Interestingly, sulphur 30C at 20
μl/ml dose significantly (p<0.001) induced apoptosis in
wild-type p53-expressing A549 cells. The apoptogenic insult
asserted by sulphur after minimization of placebo effect in A549
cells were 31%, while p53-knockdown cells resisted such insult
(Fig. 4A). These results indicated
the contribution of functional p53 in sulphur-induced cancer cell
apoptosis.
In parallel experiment when A549 cells were
transfected with p53-cDNA, p53 expression though increased, Bax
expression level failed to reach that of sulphur-treated A549 cells
(Fig. 4B). These findings revealed
that increasing p53 levels alone in NSCLC cells failed to restore
p53 transcriptional functions and to induce apoptosis (Fig. 4C). This raised the possibility of
the involvement of p53 transcriptional ‘inhibitor(s)’ in NSCLC
cells that somehow opposed p53-dependent transcription of apoptotic
genes. Sulphur, on the other hand, by restraining this inhibitor
might have activated the p53-transcriptional program. Since our
previous results (Fig. 2A and B)
have demonstrated sulphur-induced inhibition of NFκB activation, we
hypothesized that activated NFκB might be blocking p53-dependent
apoptotic program in NSCLC cells. To confirm this hypothesis we
utilized IκBα-SR-cDNA transfected cells and checked p53-dependent
execution of apoptosis in these cells upon transfection with
p53-cDNA. Indeed these transfectants displayed robust p53 induction
along with upregulation of Bax (Fig.
4B). Activation of caspase-3 in these cells (Fig. 4B) finally confirmed that NFκB
intervened the functioning of p53-dependent apoptotic program.
All these results together signified that sulphur by
inhibiting p65NFκB-governed survival signalling skewed the cellular
microenvironment in favor of p53-transcriptional activation to
result in apoptosis.
Sulphur rescues p300 from p65NFκB to
establish p53–p300 collaboration in NSCLC cells
We next attempted to unveil the detail mechanisms
underlying NFκB-mediated inhibition of p53 transcription functions.
Recent studies indicate that the transcriptional activity of p53 is
regulated by its interaction with the transcriptional co-activator
p300 (27,31). To verify the effects of sulphur on
p53–p300 cross-talk, if any, we immunoprecipitated nuclear p300 in
NSCLC cells treated with placebo, or sulphur and verified its
interaction with p53 by western blotting. It was observed that in
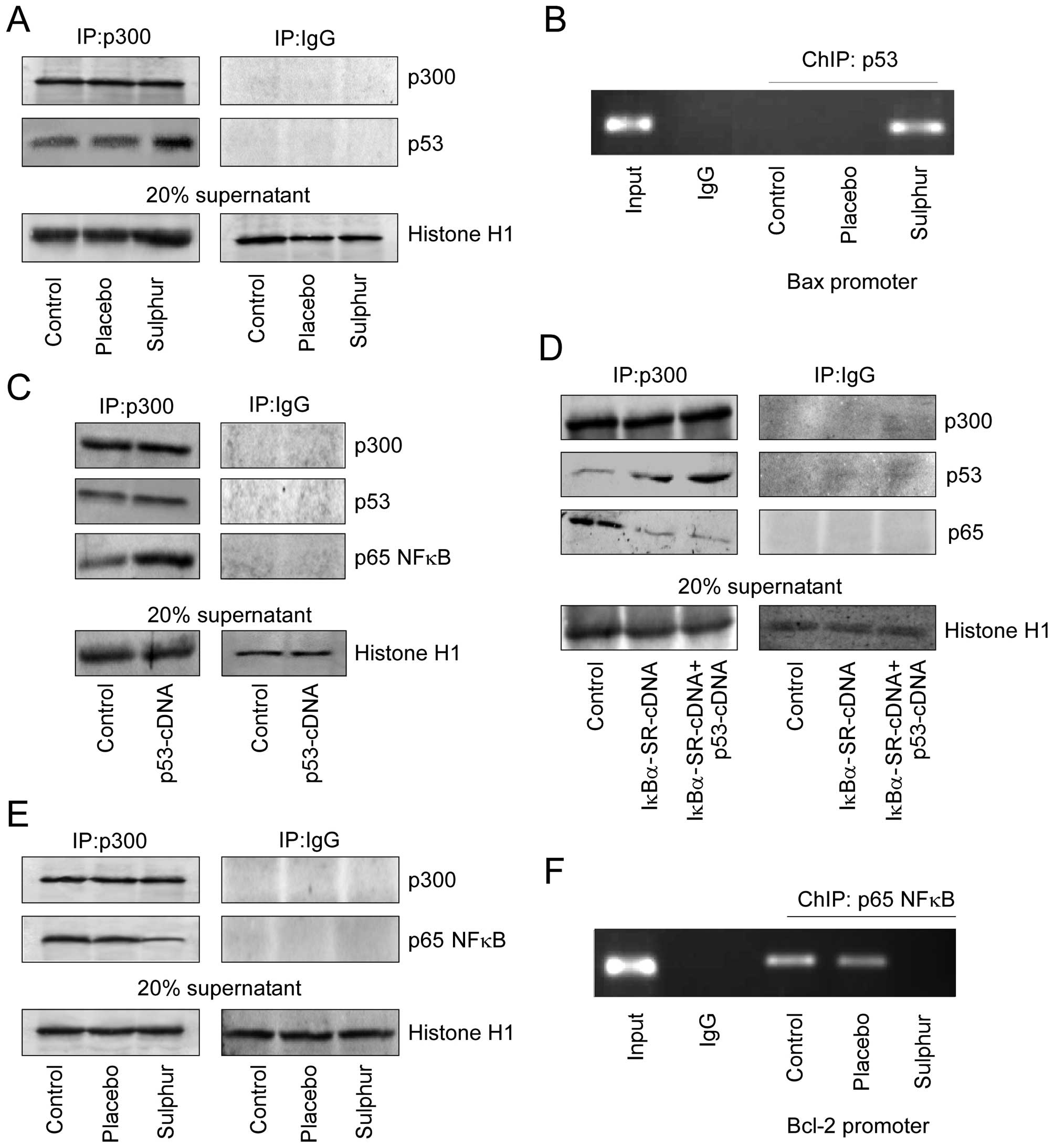
contrast to placebo, sulphur induced p53–p300 interaction (Fig. 5A). Consistently, it was further
observed by ChIP analysis that sulphur treatment in NSCLC cells
enhanced p53 binding on Bax promoter, which is a pre-requisite for
transcriptional activation of Bax (Fig. 5B). This subsequently enabled
p53-dependent apoptosis as observed earlier (Fig. 3A). Since, sulphur triggered
p53–p300 interaction in cells where p65NFκB activation was not
taking place, we proposed that nuclear translocation of p65NFκB in
untreated p53-cDNA transfected A549 cells might have sequestered
p300 thereby abridging p53–p300 cross-talk. As anticipated, these
untreated p53-cDNA transfected A549 cells manifested significant
p65NFκB-bound p300 in their nuclear lysates (Fig. 5C). Furthermore, upon genetically
perturbing nuclear translocation of p65NFκB in these p53-cDNA
trasfected A549 cells, significant increase in p53–p300 interaction
was observed with concomitant decrease in p65NFκB-bound p300
(Fig. 5D). Interestingly sulphur
treatment inhibited p65NFκB-p300 cross-talk (Fig. 5E) thereby preventing p65NFκB
binding on Bcl-2 promoter to initiate p65NFκB-mediated Bcl-2
transcription (Fig. 5F). These
results indicate a competition between NFκB and p53 for availing
p300, and depending on the relative availability, the winner, and
the fate of the cells are decided.
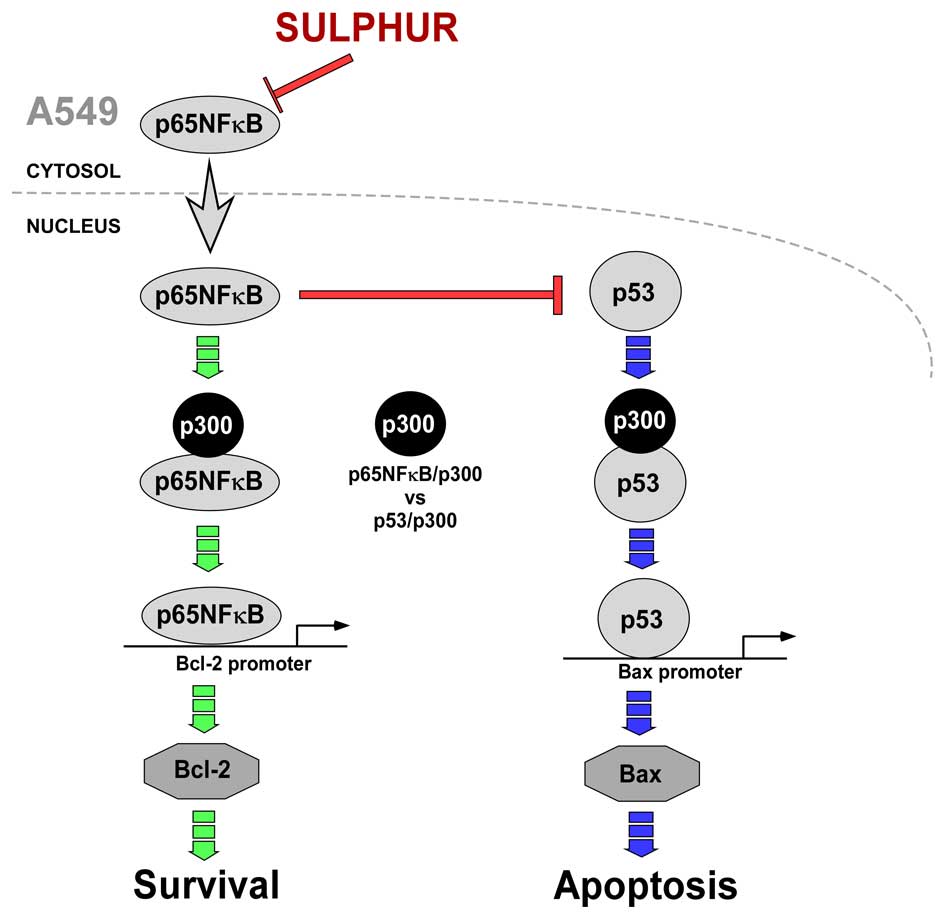
In summary, the above results conclude that in
drug-resistant NSCLC cells, p65NFκB competes with p53 for the
transcriptional co-activator p300 thereby inhibiting the apoptotic
program and upregulates the survival-machinery of the cell. On the
other hand, by inhibiting p65NFκB, sulphur censored the survival
pathway thereby making p300 available for p53 interaction to ensure
the transcription of pro-apoptotic protein Bax for effective
induction of apoptosis in otherwise resistant NSCLC cells (Fig. 6).
Discussion
Sulphur has been widely accredited for its antitumor
potential (15–18). Although few reports have also
verified the anticancer effect of this remedy (17,19),
detailed report elucidating the molecular mechanisms underlying the
anticancer effect of sulphur is still warranted. The present study
demonstrated that the antitumor effect of sulphur on non-small cell
lung carcinoma cells was not a ‘placebo effect’ as
placebo-(potentized hydro-alcoholic solution) treated cells failed
to show significant death when compared to control cells. This
study further revealed that sulphur asserted its effects by
re-orienting the molecular choreography of cancer cells.
Importantly, preferential induction of cytotoxic effects in
drug-resistant NSCLC cells, as compared to normal cells, raised the
exciting possibility for a window of safe and non-toxic therapeutic
opportunity.
Here we report that sulphur induces apoptosis in
NSCLC cells by inhibiting p65NFκB-mediated survival pathway and
activating p53-apoptotic signaling. In fact, inactivation of NFκB
pathway by sulphur rescued p300 from p65NFκB and launched p53–p300
collaboration to induce p53-dependent Bax-transactivation and
instigation of downstream mitochondria-dependent death cascade in
NSCLC cells. The regulatory contribution of NFκB and p53 to cancer
development and progression is well documented where inactivation
of p53 and hyper-activation of NFκB are the common occurrences
(25–27,44).
In agreement with such complex regulation of NFκB and p53 at
several steps, these transcription factors can functionally
antagonize, cooperate or exhibit independence (45–48).
Likewise in our study we observed that p65NFκB, being activated in
NSCLC cells, interfered with p53 functions by p300 sequestration.
Inhibition of p65NFκB by sulphur or IκBα super repressor rescued
p300 from p65NFκB-clutch to restrain the resistance pathway.
Consistently there are studies reporting that p65NFκB has a
high-affinity for p300 that may lead to its sequestration thereby
making it unavailable to other transcription factors (35). In line with these studies we
observed that upon sulphur treatment, inhibition of p65NFκB rescued
p300 making it available to other transcription factor/s like p53
in the present case, thereby allowing p53-dependent transactivation
of apoptotic proteins.
Our findings were consistent with those of Webster
and Perkins (48) who first
reported that the RelA (p65) subunit of NFκB antagonized p53
transactivation through sequestration of the p300 and CBP
co-activators. It is acknowledged that p300 and CBP participate at
various stages of the p53 response, functioning as essential
co-activators in p53-dependent trans-activation of target genes
(49). They promote transcription
of specific p53 targets by two mechanisms. First, p300 and CBP are
recruited by p53 to target gene promoters where they acetylate
histones. Secondly, p53 acetylation, secondary to DNA damage,
stabilizes the p53-DNA complex at target gene promoters (49). Similarly acetylation of p65NFκB is
important for p65NFκB-DNA binding activity and p300 activation is
known to enhance p65NFκB acetylation (50). The N- and C-terminal domains of
both CBP/p300 functionally interact with a region of p65NFκB
containing the transcriptional activation domain and thereby
promote the trans-activating functions of p65NFκB transcription
factors (50). Therefore, our
results along with others, suggest that p65NFκB and p53 compete for
transcriptional co-activator p300 and depending upon whether
p65NFκB or p53 hires p300, execution of downstream effector
pathways oscillates between survival and apoptotic responses
(Fig. 6).
In conclusion, our study for the first time
indicated an apoptosis-inducing capability of sulphur in otherwise
drug-resistant NSCLC cells by shifting the cellular milieu from
NFκB-mediated survival environment towards p53-mediated apoptosis.
Even though further investigations and clinical trials are needed,
overall these findings provide evidence for a molecular signature
of the apoptotic effects of sulphur on NSCLC cells.
Acknowledgements
The authors are thankful to Mr. Uttam Ghosh and Mr.
Ranjan Dutta for their technical help. This study was supported by
the grants from Central Council for Research in Homeopathy (CCRH),
Government of India.
References
|
1
|
Dela Cruz CS, Tanoue LT and Matthay RA:
Lung cancer: Epidemiology, etiology, and prevention. Clin Chest
Med. 32:605–644. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peters BG: An overview of chemotherapy
toxicities. Top Hosp Pharm Manage. 14:59–88. 1994.PubMed/NCBI
|
|
3
|
Cassileth BR and Vickers AJ: Complementary
and alternative therapies. Urol Clin North Am. 30:369–376. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barnes PM, Bloom B and Nahin RL:
Complementary and alternative medicine use among adults and
children: United States, 2007. Natl Health Stat Rep. 12:1–23.
2008.
|
|
5
|
Johannessen H, von Bornemann Hjelmborg J,
Pasquarelli E, Fiorentini G, Di Costanzos F and Miccinesi G:
Prevalence in the use of complementary medicine among cancer
patients in Tuscany, Italy. Tumori. 94:406–410. 2008.PubMed/NCBI
|
|
6
|
Rossi E, Vita A, Baccetti S, Di Stefano M,
Voller F and Zanobini A: Complementary and alternative medicine for
cancer patients: Results of the EPAAC survey on integrative
oncology centres in Europe. Support Care Cancer. 23:1795–1806.
2015. View Article : Google Scholar
|
|
7
|
Flinn JE: Bromium in acute lymphatic
leukemia. J Am Inst Homeopath. 58:213–214. 1965.PubMed/NCBI
|
|
8
|
Gruchmann W: Arsenic: Destroyer and
healer; a contribution to the management of carcinoma. Hippokrates.
27:444–445. 1956.(In German). PubMed/NCBI
|
|
9
|
Saha S, Hossain DM, Mukherjee S, Mohanty
S, Mazumdar M, Mukherjee S, Ghosh UK, Nayek C, Raveendar C, Khurana
A, et al: Calcarea carbonica induces apoptosis in cancer cells in
p53-dependent manner via an immuno-modulatory circuit. BMC
Complement Altern Med. 13:2302013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
MacLaughlin BW, Gutsmuths B, Pretner E,
Jonas WB, Ives J, Kulawardane DV and Amri H: Effects of homeopathic
preparations on human prostate cancer growth in cellular and animal
models. Integr Cancer Ther. 5:362–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pathak S, Kumar Das J, Jyoti Biswas S and
Khuda-Bukhsh AR: Protective potentials of a potentized homeopathic
drug, Lycopodium-30, in ameliorating azo dye induced
hepatocarcinogenesis in mice. Mol Cell Biochem. 285:121–131. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frenkel M, Mishra BM, Sen S, Yang P,
Pawlus A, Vence L, Leblanc A, Cohen L and Banerji P and Banerji P:
Cytotoxic effects of ultra-diluted remedies on breast cancer cells.
Int J Oncol. 36:395–403. 2010.PubMed/NCBI
|
|
13
|
Saha S, Bhattacharjee P, Mukherjee S,
Mazumdar M, Chakraborty S, Khurana A, Nayak D, Manchanda R,
Chakrabarty R, Das T, et al: Contribution of the ROS-p53 feedback
loop in thuja-induced apoptosis of mammary epithelial carcinoma
cells. Oncol Rep. 31:1589–1598. 2014.PubMed/NCBI
|
|
14
|
Sikdar S, Kumar Saha S and Rahman
Khuda-Bukhsh A: Relative apoptosis-inducing potential of
homeopathic condurango 6C and 30C in H460 lung cancer cells in
vitro: Apoptosis-induction by homeopathic Condurango in H460 cells.
J Pharmacopuncture. 17:59–69. 2014. View Article : Google Scholar
|
|
15
|
Parcell S: Sulfur in human nutrition and
applications in medicine. Altern Med Rev. 7:22–44. 2002.PubMed/NCBI
|
|
16
|
Sobolewska D, Podolak I and Makowska-Wąs
J: Allium ursinum: Botanical, phytochemical and pharmacological
overview. Phytochem Rev. 14:81–97. 2015. View Article : Google Scholar :
|
|
17
|
Melino S, Sabelli R and Paci M: Allyl
sulfur compounds and cellular detoxification system: Effects and
perspectives in cancer therapy. Amino Acids. 41:103–112. 2011.
View Article : Google Scholar
|
|
18
|
|
|
19
|
Milner JA: Mechanisms by which garlic and
allyl sulfur compounds suppress carcinogen bioactivation. Garlic
and carcinogenesis. Adv Exp Med Biol. 492:69–81. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mikaili P, Maadirad S, Moloudizargari M,
Aghajanshakeri S and Sarahroodi S: Therapeutic uses and
pharmacological properties of garlic, shallot, and their
biologically active compounds. Iran J Basic Med Sci. 16:1031–1048.
2013.
|
|
21
|
Koike S, Ogasawara Y, Shibuya N, Kimura H
and Ishii K: Polysulfide exerts a protective effect against
cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling
in neuroblastoma cells. FEBS Lett. 587:3548–3555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matias AC, Manieri TM, Cipriano SS,
Carioni VM, Nomura CS, Machado CM and Cerchiaro G:
Diethyldithiocarbamate induces apoptosis in neuroblastoma cells by
raising the intracellular copper level, triggering cytochrome c
release and caspase activation. Toxicol In Vitro. 27:349–357. 2013.
View Article : Google Scholar
|
|
23
|
Lee J, Lee HJ, Park JD, Lee SK, Lee SI,
Lim HD, Lee YM, Yun YG, Jeon BH, Ree IS, et al: Anti-cancer
activity of highly purified sulfur in immortalized and malignant
human oral keratinocytes. Toxicol In Vitro. 22:87–95. 2008.
View Article : Google Scholar
|
|
24
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryan KM: p53 and autophagy in cancer:
Guardian of the genome meets guardian of the proteome. Eur J
Cancer. 47:44–50. 2011. View Article : Google Scholar
|
|
27
|
Sen GS, Mohanty S, Hossain DMS,
Bhattacharyya S, Banerjee S, Chakraborty J, Saha S, Ray P,
Bhattacharjee P, Mandal D, et al: Curcumin enhances the efficacy of
chemotherapy by tailoring p65NFκB-p300 cross-talk in favor of
p53–p300 in breast cancer. J Biol Chem. 286:42232–42247. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mohanty S, Saha S, Md S Hossain D,
Adhikary A, Mukherjee S, Manna A, Chakraborty S, Mazumdar M, Ray P,
Das K, et al: ROS-PIASγ cross talk channelizes ATM signaling from
resistance to apoptosis during chemosensitization of resistant
tumors. Cell Death Dis. 5:e10212014. View Article : Google Scholar
|
|
29
|
Brown CJ, Lain S, Verma CS, Fersht AR and
Lane DP: Awakening guardian angels: Drugging the p53 pathway. Nat
Rev Cancer. 9:862–873. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saha B, Adhikary A, Ray P, Saha S,
Chakraborty S, Mohanty S, Das K, Mukherjee S, Mazumdar M, Lahiri L,
et al: Restoration of tumor suppressor p53 by differentially
regulating pro- and anti-p53 networks in HPV-18-infected cervical
cancer cells. Oncogene. 31:173–186. 2012. View Article : Google Scholar
|
|
31
|
Lahiry L, Saha B, Chak raborty J,
Bhattacharyya S, Chattopadhyay S, Banerjee S, Choudhuri T, Mandal
D, Bhattacharyya A, Sa G, et al: Contribution of p53-mediated Bax
transactivation in theaflavin-induced mammary epithelial carcinoma
cell apoptosis. Apoptosis. 13:771–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim DS, Park SS, Nam BH, Kim IH and Kim
SY: Reversal of drug resistance in breast cancer cells by
transglutaminase 2 inhibition and nuclear factor-kappaB
inactivation. Cancer Res. 66:10936–10943. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bassères DS and Baldwin AS: Nuclear
factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar
|
|
35
|
Schneider G, Henrich A, Greiner G, Wolf V,
Lovas A, Wieczorek M, Wagner T, Reichardt S, von Werder A, Schmid
RM, et al: Cross talk between stimulated NF-kappaB and the tumor
suppressor p53. Oncogene. 29:2795–2806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Adhikary A, Chakraborty S, Mazumdar M,
Ghosh S, Mukherjee S, Manna A, Mohanty S, Nakka KK, Joshi S, De A,
et al: Inhibition of epithelial to mesenchymal transition by
E-cadherin up-regulation via repression of slug transcription and
inhibition of E-cadherin degradation: Dual role of scaffold/ matrix
attachment region-binding protein 1 (SMAR1) in breast cancer cells.
J Biol Chem. 289:25431–25444. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chakraborty S, Das K, Saha S, Mazumdar M,
Manna A, Chakraborty S, Mukherjee S, Khan P, Adhikary A, Mohanty S,
et al: Nuclear matrix protein SMAR1 represses c-Fos-mediated HPV18
E6 transcription through alteration of chromatin histone
deacetylation. J Biol Chem. 289:29074–29085. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chakraborty J, Banerjee S, Ray P, Hossain
DM, Bhattacharyya S, Adhikary A, Chattopadhyay S, Das T and Sa G:
Gain of cellular adaptation due to prolonged p53 impairment leads
to functional switchover from p53 to p73 during DNA damage in acute
myeloid leukemia cells. J Biol Chem. 285:33104–33112. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mazumdar M, Adhikary A, Chakraborty S,
Mukherjee S, Manna A, Saha S, Mohanty S, Dutta A, Bhattacharjee P,
Ray P, et al: Targeting RET to induce medullary thyroid cancer cell
apoptosis: An antagonistic interplay between PI3K/Akt and
p38MAPK/caspase-8 pathways. Apoptosis. 18:589–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mukherjee S, Mazumdar M, Chakraborty S,
Manna A, Saha S, Khan P, Bhattacharjee P, Guha D, Adhikary A,
Mukhjerjee S, et al: Curcumin inhibits breast cancer stem cell
migration by amplifying the E-cadherin/β-catenin negative feedback
loop. Stem Cell Res Ther. 5:1162014. View Article : Google Scholar
|
|
41
|
Saha S, Mukherjee S, Mazumdar M, Manna A,
Khan P, Adhikary A, Kajal K, Jana D, Sa G, Mukherjee S, et al:
Mithramycin A sensitizes therapy-resistant breast cancer stem cells
toward genotoxic drug doxorubicin. Transl Res. 165:558–577. 2015.
View Article : Google Scholar
|
|
42
|
Hossain DM, Panda AK, Manna A, Mohanty S,
Bhattacharjee P, Bhattacharyya S, Saha T, Chakraborty S, Kar RK,
Das T, et al: FoxP3 acts as a cotranscription factor with STAT3 in
tumor-induced regulatory T cells. Immunity. 39:1057–1069. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saha S, Adhikary A, Bhattacharyya P, Das T
and Sa G: Death by design: Where curcumin sensitizes drug-resistant
tumours. Anticancer Res. 32:2567–2584. 2012.PubMed/NCBI
|
|
44
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Scian MJ, Stagliano KE, Anderson MA,
Hassan S, Bowman M, Miles MF, Deb SP and Deb S: Tumor-derived p53
mutants induce NF-kappaB2 gene expression. Mol Cell Biol.
25:10097–10110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Schumm K, Rocha S, Caamano J and Perkins
ND: Regulation of p53 tumour suppressor target gene expression by
the p52 NF-kappaB subunit. EMBO J. 25:4820–4832. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Furia B, Deng L, Wu K, Baylor S, Kehn K,
Li H, Donnelly R, Coleman T and Kashanchi F: Enhancement of nuclear
factor-kappa B acetylation by coactivator p300 and HIV-1 Tat
proteins. J Biol Chem. 277:4973–4980. 2002. View Article : Google Scholar
|
|
48
|
Webster GA and Perkins ND: Transcriptional
cross talk between NF-kappaB and p53. Mol Cell Biol. 19:3485–3495.
1999.PubMed/NCBI
|
|
49
|
Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE,
Cariati M, Brindle K, Aparicio S and Caldas C: p300 regulates
p53-dependent apoptosis after DNA damage in colorectal cancer cells
by modulation of PUMA/p21 levels. Proc Natl Acad Sci USA.
101:7386–7391. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhong H, Voll RE and Ghosh S:
Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional
activity by promoting a novel bivalent interaction with the
coactivator CBP/p300. Mol Cell. 1:661–671. 1998. View Article : Google Scholar : PubMed/NCBI
|