Introduction
Approximately one million people were newly
diagnosed as gastric cancer patients and 738,000 new deaths were
related to this malignancy in 2012 worldwide. The highest morbidity
and mortality of gastric cancer are reported in countries of
Eastern Asia, especially China (1,2).
Several high-risk etiologic factors, such as Helicobacter
pylori infection, heavy salt consumption, smoking, and genetic
alterations, have been proved to be associated with gastric cancer
(3,4). In addition, epigenetic alterations,
including DNA methylation, histone modification and microRNAs, are
also involved in the initiation and progression of gastric cancer
(5,6). Of note, promoter methylation has been
considered as a common mechanism leading to the silence of tumor
suppressor genes (TSGs) (7,8).
Since the precise mechanisms underlying the gastric tumor are still
unclear, the identification of TSGs inactivated by promoter
hypermethylation may provide new insights into the initiation and
development of gastric cancer.
BLU was first identified by Lerman and Minna in
2000, it is also named ZMYND10 (zinc finger, MYND-type containing
10) and located at human chromosome 3p21.3 (9). The BLU protein is ~50 kDa and
contains a MYND zinc finger DNA binding domain at its C-terminus,
which was similar to the domains found in transcription repressors
such as ETO (MTG8) and BS69 (10–12).
As reported in previous studies, BLU served as a tumor suppressor
in human nasopharyngeal carcinoma and ovarian carcinomas via
inhibiting proliferation, inducing apoptosis and anti-angiogenic
(13–16). Moreover, BLU was found frequently
inactivated by the promoter hypermethylation in various human
cancers, which could be restored by demethylating agent 5-Aza
treatment (17–21). However, the expression and
biological functions of BLU in gastric cancer remain unknown.
DNA methylation was reported to epigenetically
inhibit gene expression via blocking the binding of transcription
factor (TF) such as Sp1 to hypermethylated CpG dinucleotides within
TF-binding sites (22–25). In this study, our in silico
prediction has shown that two putative Sp1-binding sites are
harbored in TATA-less BLU promoter, which are relative to the
transcription start site of BLU. Moreover, given the fact that Sp1
is a transcription activator and Sp1-binding sites are crucial for
the basal transcription of genes with TATA-less promoter (26,27),
it is possible that the methylation of CpG within Sp1-binding site
in the BLU promoter can lead to inactivation of BLU. Furthermore,
Sp1 has been recognized to play an essential role in tumorigenesis
(28). Therefore, the
identification of Sp1-regulated expression of BLU may provide new
clues to pathogenesis of malignancies, including gastric
cancer.
Our present study supported the idea that BLU was
inactivated due to its promoter hypermethylation in human cancers.
However, the mechanisms by which hypermethylation results in the
silencing of BLU gene have not yet been reported. In the present
study, we suggest that BLU promoter hypermethylation is able to
prevent Sp1 from binding to BLU promoter in gastric cancer, and
this mechanism may provide an explanation for silencing of the BLU
gene.
Materials and methods
Cell culture
Six human gastric cancer cell lines (SGC7901, HGC27,
BGC823, MKN45, MGC803 and AGS) and human gastric epithelial cell
line GES-1 were obtained from the Cell Bank of the Chinese Academy
of Sciences (Shanghai, China). SGC7901, MKN45, MGC803, and AGS were
maintained in the RPMI-1640 medium (Hyclone, South Logan, UT, USA)
containing 10% fetal bovine serum (FBS, Invitrogen, Carisbad, CA,
USA). HGC27, BGC823 and GES-1 were cultured in the DMEM medium
(Invitrogen) supplemented with 10% FBS. All cells were incubated at
37°C in a humid atmosphere of 5% CO2.
Tissues samples
Fifty-two paired gastric cancer tissues and adjacent
non-cancerous gastric tissues were obtained after informed consents
from patients at the First Affiliated Hospital of Soochow
University. Tissue samples were collected, immediately snap-frozen
in liquid nitrogen and stored in −80°C until analysis. None of the
patients had received chemotherapy or radiotherapy prior to
surgery. This research was approved by the Academic Advisory Board
of Soochow University.
Demethylating agent 5-Aza treatment
5-Aza was purchased from Sigma-Aldrich Corp. (St.
Louis, MO, USA) and dissolved in sterile double distilled water.
Cells were plated at a density of 1×105 cells per well
in the 6-well plates and incubated overnight. On days 1, 3 and 5,
the medium was changed to serum-free RPMI-1640 containing 10 μM
5-Aza. On days 2 and 4, the medium was changed to 5-Aza free
RPMI-1640 with 3% FBS. The cells were harvested for further study
on day 5.
Quantitative real-time reverse
transcription PCR (qRT-PCR)
Total RNA was extracted from cells using RNAiso Plus
(Takara Biotech Co., Ltd., Dalian, China) according to the
manufacturer’s protocol. Total mRNA (1 μg) was reverse-transcribed
by the M-MLV reverse transcriptase (Invitrogen). Subsequently,
aliquots of cDNA were subjected to PCR by using FastStart Universal
SYBR Green Master (ROX) (Roche, Basel, Switzerland) on a
LightCycler® 96 Real-Time PCR System (Roche). Primers
for mRNA detection of BLU, Sp1 and β-actin (internal control) were
as follows: for BLU, 5′-AAC CAGCAGCATGAGAACCT-3′ (forward) and
5′-AGTTTGCG GTGGCAATAGTC-3′ (reverse); for Sp1, 5′-CCTCCAGACC
ATTAACCTCAG-3′ (forward) and 5′-TCCACCTGCTGTGT CATCAT-3′ (reverse);
for β-actin, 5′-CACAGAGCCTCGCCTT TGCC-3′ (forward) and
5′-CACATGCCGGAGCCGTTGTC-3′ (reverse). The amplification conditions
were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for
10 sec and 60°C for 30 sec. Relative fold-change in gene expression
was determined using the ΔΔCt method normalized to β-actin.
Bisulfite sequencing
Genomic DNA was extracted from cells using DNeasy
Blood and Tissue kit (Qiagen, Hilden, Germany) and then bisulfited
using EpiTect Bisulfite kit (Qiagen) according to the
manufacturer’s instructions. The sodium bisulfite-modified genomic
DNA was subjected to PCR amplification with primers which specially
designed to analysis the methylation status of 22 CpG sites within
a 244-bp region of the BLU promoter (−181 to +63). The primers
were: 5′-ATAAG GATTTGGAGTTTAGGAGAG-3′ (forward) and 5′-CCAAAA
TCTAAAACAAAACAATTAC-3′ (reverse). PCR products were purified by Gel
Extraction kit (Omega, USA) and cloned into pMD19-T vector (Takara)
for DNA sequencing. At least ten clones from each sample were
randomly selected and then sequenced.
Western blot assay
Cells were lysed in RIPA buffer (Cell Signaling
Technology) with the protease inhibitor (Sigma-Aldrich Corp.).
Equal amounts of total protein were separated on SDS-PAGE and
transferred to the nitrocellulose membranes (Millipore, Billerica,
MA, USA). The membranes were blocked with 3% BSA-TBST buffer for 1
h at room temperature and then incubated with appropriate primary
antibodies overnight at 4°C. Next, the membranes were washed with
TBST buffer three times and incubated with the corresponding
HRP-conjugated secondary antibodies for 2 h at room temperature.
The specific protein band detection was performed using a
commercial ECL kit (Pierce, Rockford, IL, USA). β-actin was used as
internal control. The following antibodies were used for western
blot analysis: rabbit anti-BLU polyclonal antibody (ab96700,
1:2,000, Abcam, Cambridge, UK); rabbit anti-Sp1 polyclonal antibody
(#9389S, 1:1,000, Cell Signaling Technology, Danvers, MA, USA);
rabbit anti-β-actin polyclonal antibody (sc-130656,1:1,000, Santa
Cruz Biotechnology, Santa Cruz, CA, USA).
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using EZ-ChIP™
Chromatin Immunoprecipitation kit (Millipore). Immunoprecipitation
was performed by the addition of anti-Sp1 antibody, and rabbit IgG
antibody was used as a negative control. A fragment of the BLU
promoter (−80/+108), which contains putative Sp1-binding sites
(−43/−35) was amplified by semi-quantitative PCR with primers as
follows: 5′-TAGAGACCCGCCCGGAT TTA-3′ (forward) and
5′-GGGTGGGGATGCTGTCACAT-3′ (reverse). PCR products were analyzed by
electrophoresis on a 2% agarose gel and stained with ethidium
bromide.
Electrophoresis mobility shift assay
(EMSA)
Nuclear extracts were isolated from cells using
Nuclear Protein Extraction kit (Solarbio, Beijing, China). The EMSA
assay was performed by LightShift Chmiluminescent EMSA kit (Pierce)
following the manufacturer’s instructions. The 5′ biotin-labeled
and double-stranded oligo probes containing putative Sp1 binding
sites (−43/−35) were chemically synthesized by Invitrogen. The
sequences of probes were as follows: −39C (unmethylated)
5′-CCTCCAGGGGCGGGGCCCAGTTG-3′ and −39 5mC
(methylated) 5′-CCTCCAGGGG5mCGGGGCCCAGTTG-3′; unlabeled
probes 5′-CCTCCAGGGGCGGGGCCCAGTTG-3′ were synthesized
as cold competitor. Electrophoresis mobility gel shift assay was
performed as described previously (29).
Plasmid construction, transfection and
luciferase report assays
The pGL3-basic dual luciferase vector (Promega,
Madison, WI, USA) was used to produce a series of constructs
containing various truncated regions of BLU promoter. Two fragments
of interest BLU promoter regions containing the unmethylated and
methylated −39 CpG (5′-CCTCCAGGGG CGGGGCCCAG-3′ and
5′-CCTCCAGGGG5mCGGGGCC CAG-3′, respectively) were directly
chemically synthesized and massively inserted into the luciferase
pGL3-basic vector, and then purified by using Universal DNA
purification kit (Tiangen, Beijing, China). To assess promoter
activity, cells were transiently co-transfected with BLU promoter
constructs and RenillaphRL-TK plasmid (Promega) using
Lipofectamine 2000 (Invitrogen). Twenty-four hours later, cells
were harvested, and luciferase activities were measured by the
Dual-Luciferase Reporter assay kit (Promega) on a TD20/20
Luminometer (Turner Designs, Sunnyvale, CA, USA). Results are
expressed as relative luciferase activities, which are normalized
to Renilla luciferase activities.
RNA interference experiments
Small interfering RNA (siRNA) sequences (Table I), which target Sp1 and BLU, were
chemically synthesized (GenePharma, Shanghai, China). Scrambled
siRNA was used as a negative control (NC). Cells were transiently
transfected with 100 pmol of siRNA sequences using Lipofectamine
2000 (Invitrogen). After 72-h transfection, the cells were
harvested for further experiments.
 | Table IsiRNA sequences used for silencing of
Sp1 and BLU. |
Table I
siRNA sequences used for silencing of
Sp1 and BLU.
| Name | Sequences
(5′-3′) |
|---|
| si-Sp1-1 | Sense:
5′-GAUCACUCCAUGGAUGAAATT-3′ |
| Antisense:
5′-UUUCAUCCAUGGAGUGAUCTT-3′ |
| si-Sp1-2 | Sense:
5′-GCAACAUGGGAAUUAUGAATT-3′ |
| Antisense:
5′-UUCAUAAUUCCCAUGUUGCTT-3′ |
| si-BLU-1 | Sense:
5′-CCUCCAUCAUCAACCUCUUTT-3′ |
| Antisense:
5′-UUAGAAGCCUCUGCACUGCTT-3′ |
| si-BLU-2 | Sense:
5′-GCAGUGCAGAGGCUUCUAATT-3′ |
| Antisense:
5′-UUAGAAGCCUCUGCACUGCTT-3′ |
| si-BLU-3 | Sense:
5′-CCCUCUCAGUACUACGCUATT-3′ |
| Antisense:
5′-UAGCGUAGUACUGAGAGGGTT-3′ |
Cell proliferation
Cell viability was measured by a cell Counting Kit-8
(CCK-8) (Yeasen, Shanghai, China). After being transfected with or
without siRNA as described above, 100 μl SGC7901 and AGS cell
suspensions (2×104 cells/ml) were plated into each well
of 96-well plates. Then 10 μl CCK-8 solution was added to each well
and incubated for 2 h. The absorbance was measured at 450 nm on a
microplate reader (MRX, Dynex Technologies, West Sussex, UK).
Colony formation assay
Two thousand cells were plated in 60-mm culture
dishes and incubated at 37°C for 2 weeks; visible cell colonies
were fixed with methanol for 15 min, then stained with Giemsa for 1
h, washed with water and colonies containing ≥50 cells were counted
under a microscope.
Statistical analysis
Statistical analysis was performed using the SPSS
19.0 software (SPSS Inc, Chicago, IL, USA). All quantitative data
were obtained from at least three independent experiments and
represented as means ± SD or SEM. Difference between the means of
two groups was compared using Student’s t-tests. Comparisons
between clinicopathologic characteristics and expression levels of
mRNA in gastric cancer samples were performed with non-parametric
tests (Mann-Whitney U test for 2 groups, Kruskall-Wallis test for
≥3 groups). P<0.05 was considered to be statistically
significant.
Results
BLU expression is reduced in gastric
cancer cells and tissues
To verify whether inactivation of BLU contributes to
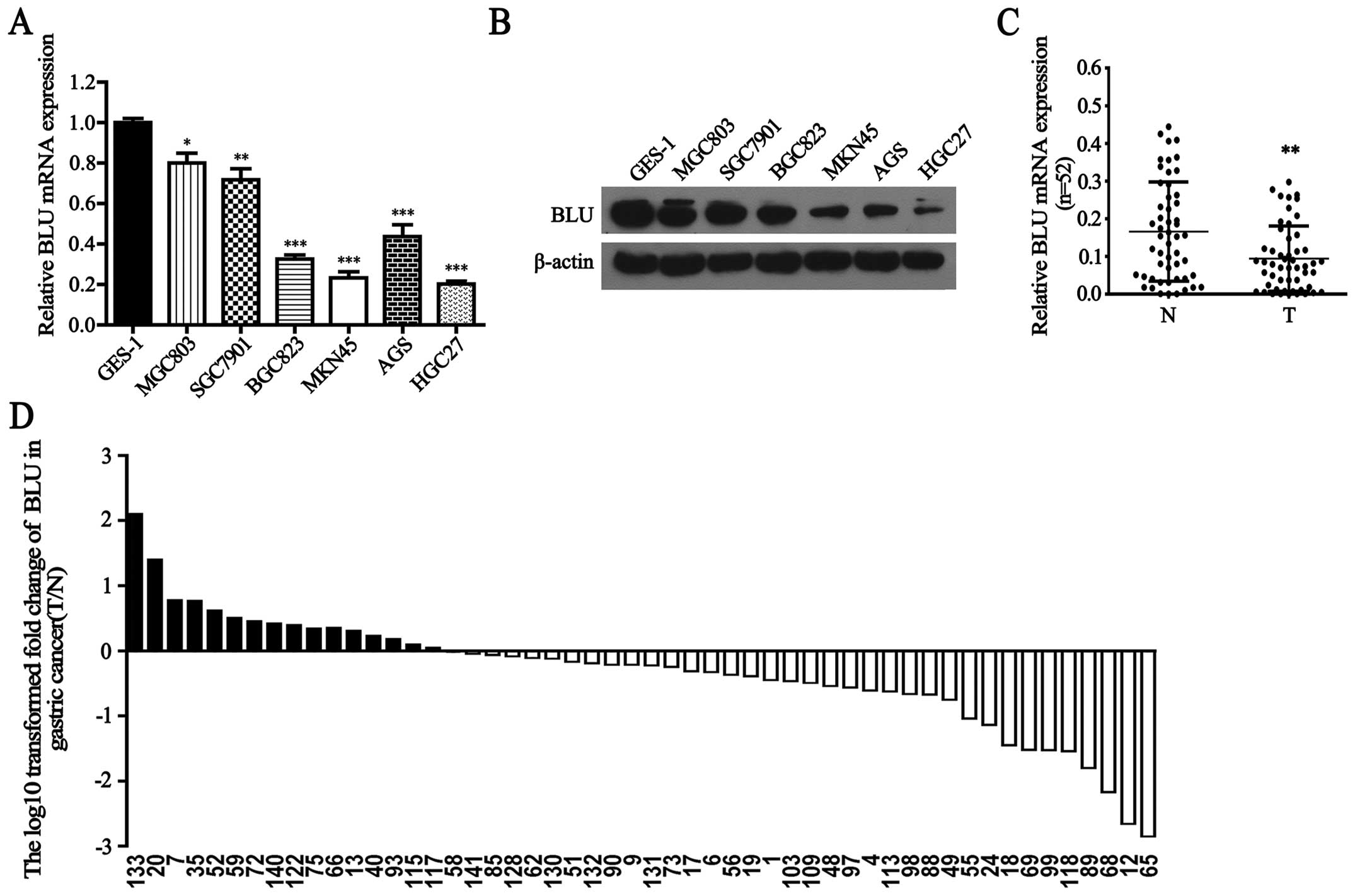
gastric carcinogenesis, we first detected BLU mRNA expression in
human gastric cancer cells and GES-1 cells. As shown in Fig. 1A, BLU mRNA was significantly lower
in MGC803, SGC7901, BGC823, MKN45, AGS and HGC27 cells than that in
GES-1 cells (P<0.05, P<0.01, P<0.001, P<0.001,
P<0.001 and P<0.001, respectively). Consistently, BLU protein
levels were also downregulated in gastric cancer cells (Fig. 1B). In addition, BLU mRNA expression
was notably reduced in 69% (36/52) gastric cancer tissues when
compared with their paired adjacent non-cancerous tissues
(P<0.01, Fig. 1C and D). No
significant difference in BLU mRNA level was observed between
gastric cancer tissues when classified by various clinicopathologic
characteristics (Table II).
| Table IICorrelation between
clinicopathological features and expression levels of BLU mRNA in
gastric cancer. |
Table II
Correlation between
clinicopathological features and expression levels of BLU mRNA in
gastric cancer.
| Characteristic | Case (n=52) | BLU mRNA |
|---|
| Gender |
| Male | 35 | 0.095±0.015 |
| Female | 17 | 0.091±0.018 |
| Z scorea | | −0.146 |
| P-value | | 0.884 |
| Age (year) |
| ≥60 | 28 | 0.084±0.015 |
| <60 | 24 | 0.105±0.018 |
| Z scorea | | −0.661 |
| P-value | | 0.509 |
| Lymphatic
invasion |
| Absent | 34 | 0.099±0.015 |
| Present | 18 | 0.084±0.020 |
| Z scorea | | −0.587 |
| P-value | | 0.557 |
| WHO
classification |
| W/D | 20 | 0.092±0.019 |
| M/D | 17 | 0.094±0.020 |
| P/D | 15 | 0.081±0.020 |
| H scoreb | | 0.077 |
| P-value | | 0.962 |
| TNM stage |
| I | 19 | 0.090±0.019 |
| II | 14 | 0.093±0.025 |
| III | 11 | 0.076±0.029 |
| IV | 8 | 0.075±0.019 |
| H scoreb | | 0.341 |
| P-value | | 0.952 |
Identification and characterization of a
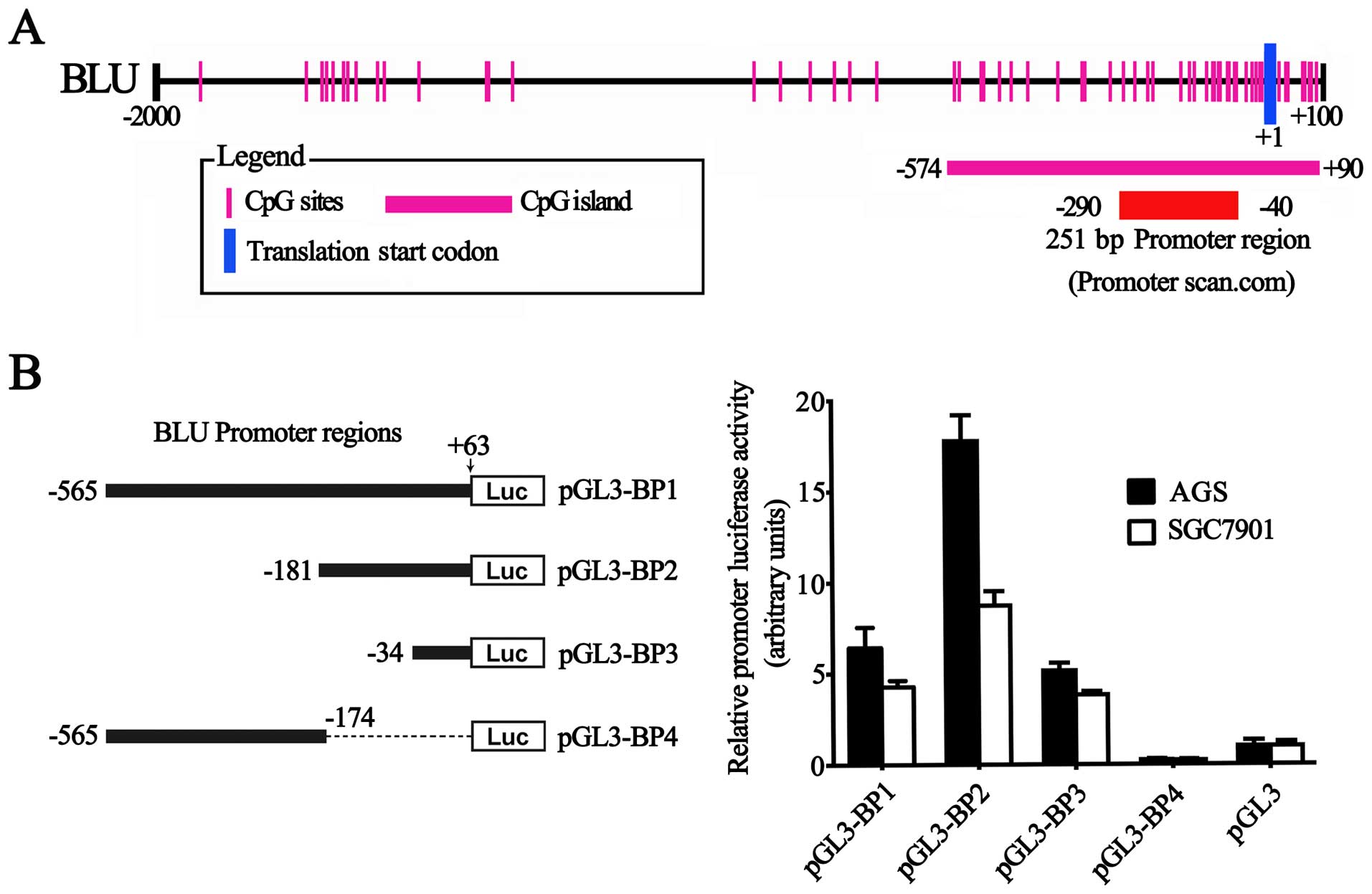
functional CpG-rich BLU promoter
To define the BLU promoter, a 2,100-bp sequence,
including BLU proximal promoter (−2,000–+1)) and 5′-UTR (+1–+100)
regions (RefSeq: NC_000003.12 and NM_015896), was available for
analysis. A 251-bp region spanning positions −290 to −40 relative
to the transcription start site was predicted to be the putative
promoter of the BLU gene using the Promoter Scan program (Fig. 2A). This region contains a GC box,
but lacks a TATA box. A potential CpG island spanning positions
−574 to +90 was identified by Methyl Primer Express®
software v1.0 (Applied Biosystems) (Fig. 2A).
To evaluate the effects of this sequence on
transcription, we generated the luciferase report constructs
containing various lengths of the BLU proximal promoter and 5′-UTR
region. As presented in Fig. 2B,
the fragment (−181 to +63) had the highest activity, while the
second largest fragment (−565 to −174) had less activity than the
largest and smallest fragments (−565 to +63; −34 to +63,
respectively) (Fig. 2B),
suggesting that an active cis-element was putatively at
position −181 to +63 and a repressive element resides was located
between positions −565 to −174 in the BLU promoter. Taken together,
the results indicated that the indentified BLU promoter is indeed
functional.
In silico prediction of functional CpG
sites in BLU promoter
Using Methyl Primer Express software (Applied
Biosystems), we identified that the 244-bp BLU promoter region
(−181 to +63) contains 22 CpG sites (Fig. 3A). Using the transcription factor
search program TFSEARCH (http://mbs.cbrc.jp/research/db/TFSEARCH.html), two
putative transcription factor Sp1-binding sites were predicted at
position −43/−35 and −89/−81 relative to the transcription start
site, each of them contains one CpG site (−39 CpG and −85 CpG,
respectively). Binding sites for several other potential
transcription factor, such as IK-2 (one site), CdxA (two sites),
GATA-1 (one site), E2F (one site), MZF1 (one site) were also noted
(Fig. 3A). Since the BLU promoter
lacks TATA box and the binding sites of transcription factor Sp1
was reported to be crucial for the basal transcription of TATA-less
promoters, we then investigated the putative functions of −39 CpG
and −85 CpG in the regulation of BLU expression.
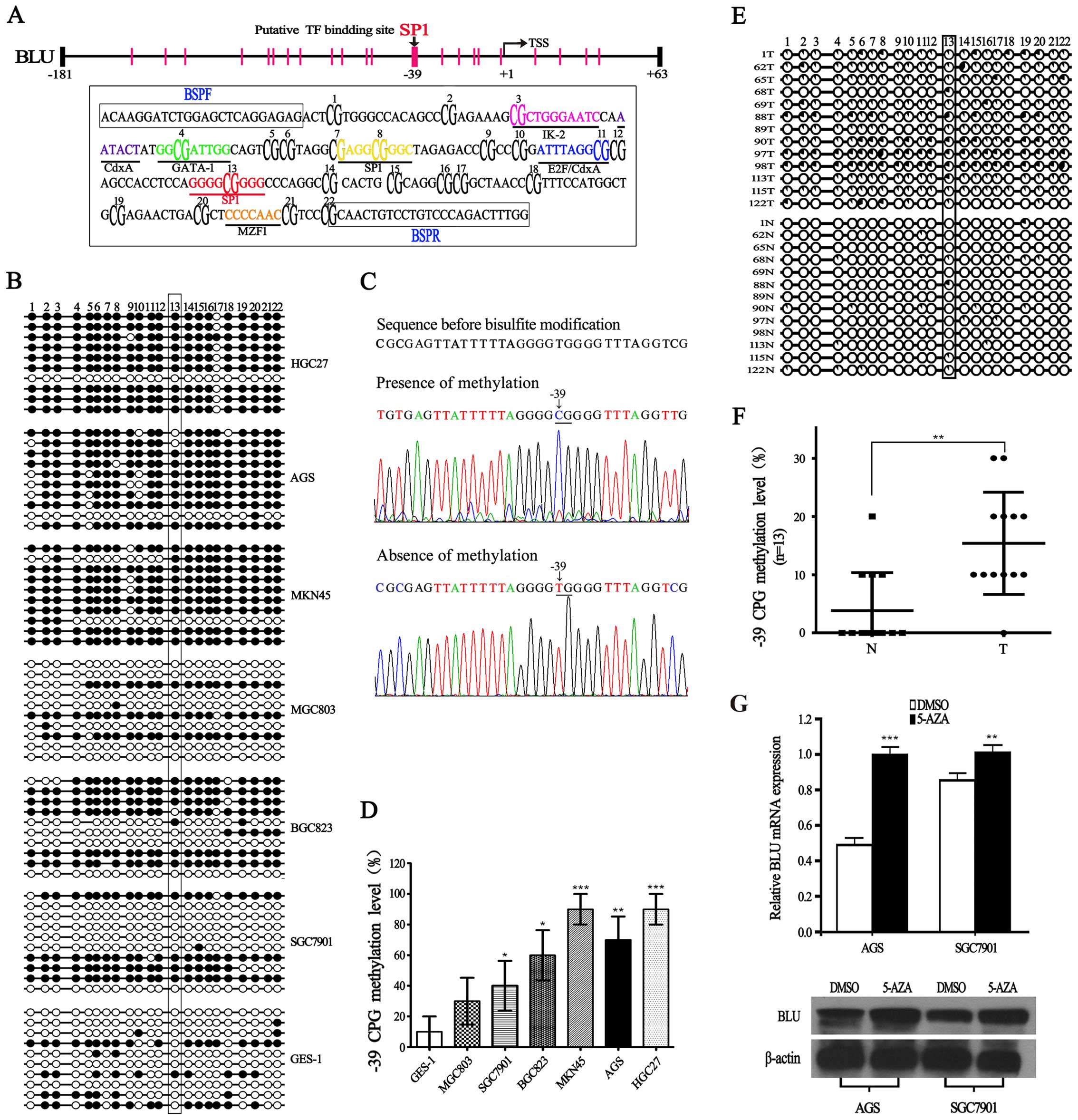 | Figure 3Characterization of BLU promoter, and
methylation level of BLU promoter in gastric cancer. (A) Locations
of 22 CpG sites analyzed in the BLU promoter, and each vertical bar
represents one CpG site. Sequence of the BLU promoter (−181 to +63)
is boxed below. The putative binding sites for IK-2, CdxA, GATA-1,
E2F, Sp1, and MZF1 are labeled and shown in different colors, CpG
sites are bold and numbered according to the order used in
bisulfite genomic sequencing, BSP primer sequences (BSPF and BSPR)
are boxed. (B) BSP analysis for methylation of cytosine residues in
22 CpG sites at the BLU promoter in gastric cancer cell lines and
gastric epithelial cell line GES-1. Methylation analysis was
performed in 10 clones. Open or filled circles represent
unmethylated or methylated cytosine residues, respectively. (C)
Representative sequences showing the presence or absence of
methylation at −39 CpG site. Unmethylated nucleotide C is converted
to T after bisulfite treatment, but methylated C remains unchanged.
(D) Summary of −39 CpG methylation level in all the cell lines. At
−39 CpG site, the methylation level was calculated as the ratio of
methylated clones over the total number of clones sequenced per
cell line. (E and F) Methylation status of BLU promoter in 13
randomly selected gastric tissues (N) and their paired
non-cancerous gastric tissues (T), methylation analysis was
performed in 10 clones, the percentage of methylation is indicated
as pie charts. (G) The expression of BLU mRNA and protein in AGS
and SGC7901 cells treated with or without 5-Aza (10 μM).
*P<0.05; **P<0.01;
***P<0.001. |
The BLU promoter is hypermethylated in
gastric cancer
We performed bisulfite sequencing (BSP) analysis to
investigate whether promoter CpG methylation is associated with the
loss of BLU expression. As shown in Fig. 3B, the hypermethylation of BLU was
detected in AGS, MKN45, HGC27, BGC823 and SGC7901 cells, while BLU
was hypomethylated in GES-1. Importantly, we observed that the
methylation level of −39 CpG is significantly higher in the five
gastric cancer cell lines with relatively low BLU mRNA expression
(Fig. 3B–D). In addition, we also
found a negative correlation between −39 CpG methylation level and
BLU expression in tumor samples (Fig.
3E and F). Furthermore, we treated AGS and SGC7901 cells with
demethylating agent 5-Aza, and found that BLU mRNA and protein
levels were restored in AGS and SGC7901 cells (Fig. 3G). These results implied that
hypermethylation of BLU promoter may play an important role in
transcription silencing of the BLU gene in human gastric
cancer.
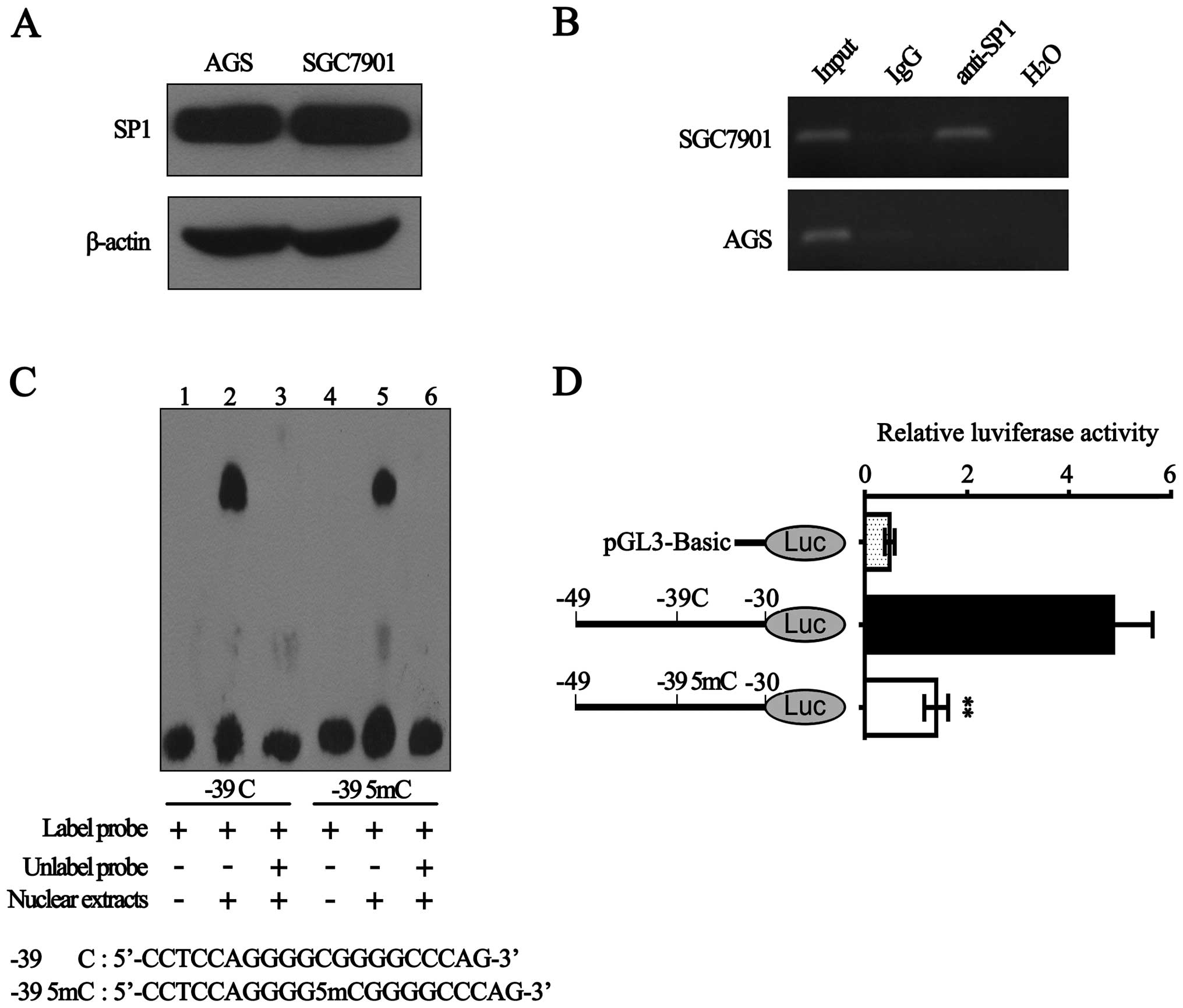
Sp1 specifically binds to the BLU
promoter and hypermethylation of −39 CpG decreases its binding
ability
It is well known that Sp1 is one of the first
identified eukaryotic transcription factors (30). Hypermethylation of CpG
dinucleotides within Sp1-binding sites could block Sp1 binding to
promoter (22,23). Being in silico predicted to
be within Sp1-binding sites in BLU promoter, the −39 CpG was
hypermethylated in 5 gastric cancer cell lines with low BLU mRNA
expression. Thus, we suggest the possibility that hypermethylated
−39 CpG may affect the recruitment of Sp1 to BLU promoter. To test
this, we performed the following experiments.
First, western blot analysis showed that Sp1 protein
expression in SGC7901 cells was nearly equaled with that in AGS
cells (Fig. 4A). Subsequently, we
performed ChIP assay in AGS and SGC7901 cells. As shown in Fig. 4B, with the DNA samples
immunoprecipitated by anti-Sp1 as templates, a 188 bp BLU promoter
sequence (−80 to +108 bp) containing −39 CpG was amplified in
SGC7901 cells, but not in AGS cells. Considering that the
methylation level of −39 CpG in AGS cells was higher than SGC7901
cells (Fig. 3B and C), the results
suggested that hypermethylation of −39 CpG significantly inhibited
the interaction between Sp1 and BLU promoter.
Next, EMSA assay showed that although both the probe
containing unmethylated −39 CpG and the probe containing methylated
−39 CpG could form DNA-protein complexes with nuclear extracts of
SGC7901 cells, binding ability of unmethylated probe is stronger
than the methylated probe (Fig.
4C). This indicated that methylation of −39 CpG within Sp1
binding-sites may reduce the complex formation.
Furthermore, we constructed the luciferase reporter
vectors containing the corresponding unmethylated and methylated
fragments (Sp1-binding sites) and then transiently transfected them
into SGC7901 cells. As shown in Fig.
4D, methylated −39 CpG significantly diminished the luciferase
activity by ~70%. Taken together, our results demonstrated that Sp1
could specifically bind with the BLU promoter and hypermethylation
of −39 CpG significantly reduced the recruitment of Sp1 to BLU
promoter.
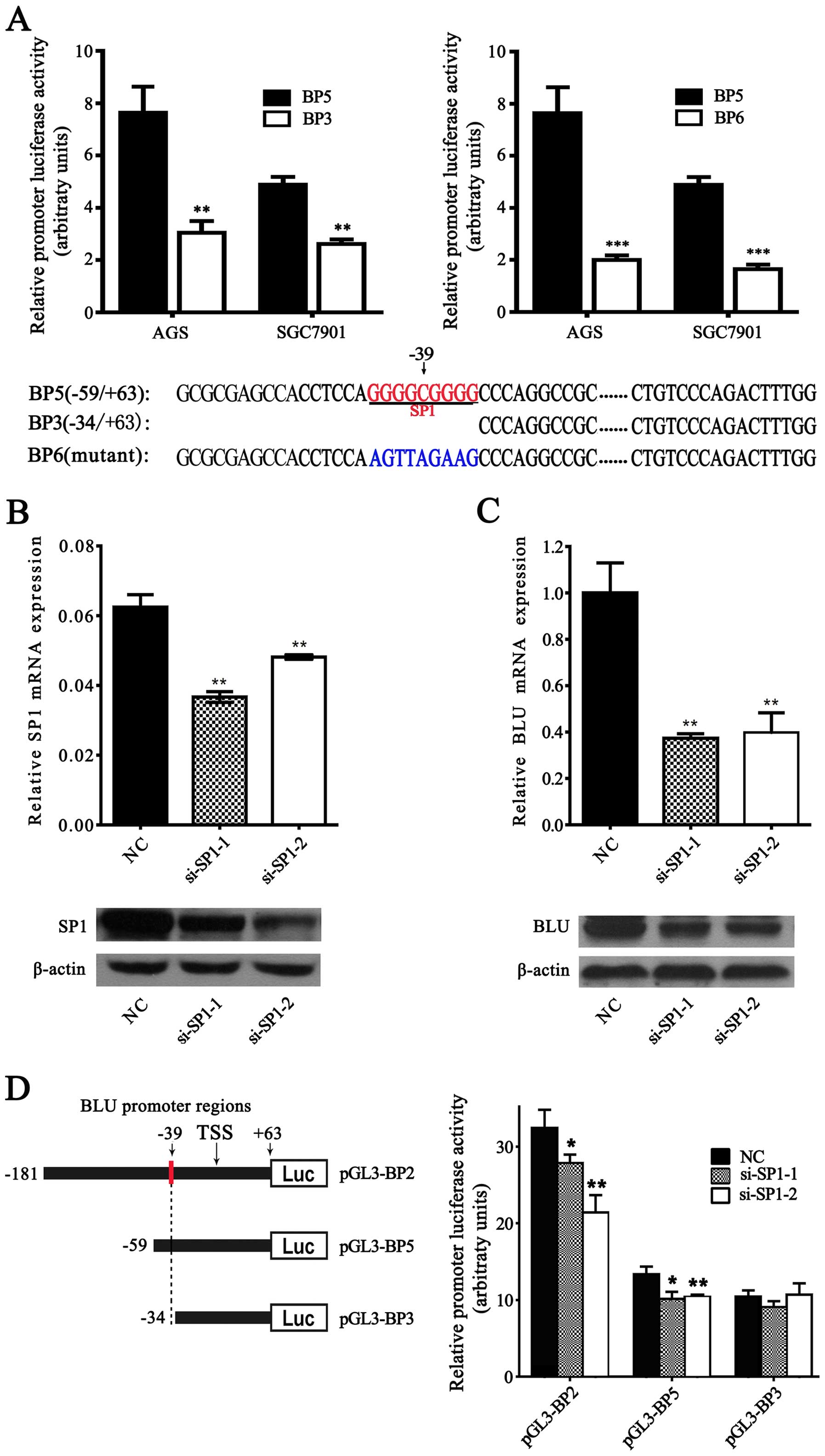
Sp1 activates the BLU promoter
To explore whether Sp1 could regulate
transcriptional activity of the BLU promoter, we performed the
luciferase report assay in SGC7901 cells. As a result, the
constructs containing the Sp1-binding site (BP5: −59/+63) showed
significant higher activity than the constructs with deletion of
Sp1-binding site (BP3: −34/+63) and the mutant constructs (BP6:
−59/+63) (Fig. 5A). Furthermore,
siRNA-mediated knockdown of endogenous Sp1 effectively decreased
the mRNA and protein expression level of BLU (Fig. 5B and C). After co-transfection of
the BLU promoter constructs (BP2: −181/+63 and BP5: −59/+63)
containing the Sp1-binding sites with si-Sp1, knockdown of Sp1
decreased the luciferase activities of these constructs when
compared with the negative control (NC) (Fig. 5D). By contrast, downregulation of
Sp1 presented no significant effects on the luciferase activity of
the construct (BP6: −34/+63) lacking the Sp1-binding sites. Our
results demonstrated that the sequence of the BLU promoter
(positions −43 to −35) is a functional Sp1-binding site and Sp1
could activate the BLU promoter.
Knockdown of BLU promotes the growth of
gastric cancer cells
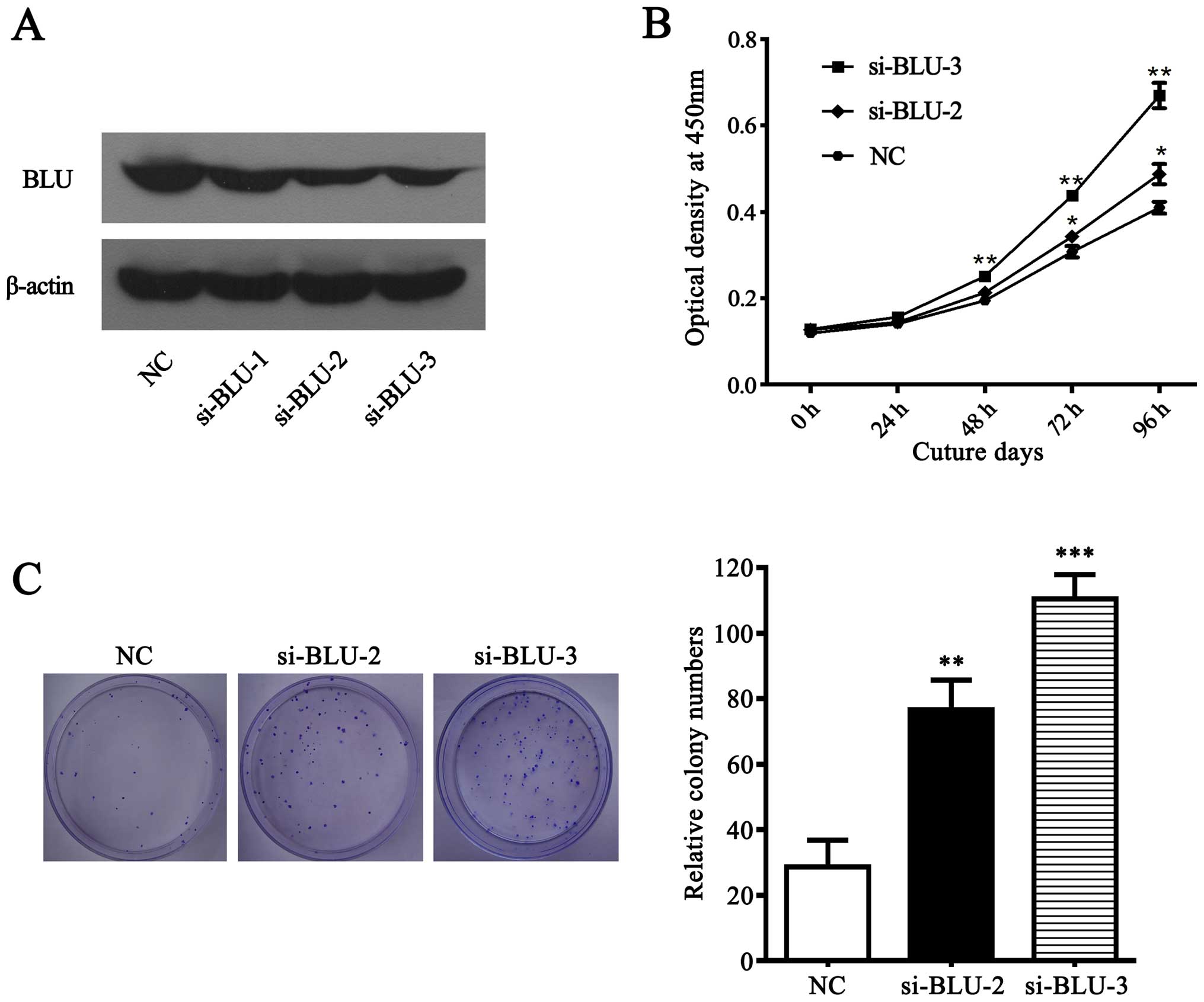
BLU was frequently downregulated in gastric cancer
cells and primary tumors, suggesting that BLU is likely a tumor
suppressor in gastric cancer. To investigate the affects of BLU on
the growth of gastric cancer cells, we performed CCK-8 and colony
formation assays in SGC7901 cells transfected with BLU siRNAs or
negative control. As expected (Fig.
6A), BLU was significantly silenced in SGC7901 cells
transfected with si-BLU-2 and si-BLU-3. Subsequently, CCK-8 and
colony formation experiment showed that BLU knockdown promoted cell
viability and clonogenicity (Fig. 6B
and C), suggesting that BLU acts as a tumor suppressor via
inhibiting cell growth in gastric carcinoma.
Discussion
Gastric carcinoma is the fifth most common cancer
and second leading causes of cancer related deaths worldwide
(1). During the past decades,
despite many improvements in the early detection and treatment of
gastric cancer, >50% of gastric cancers were diagnosed at
advanced stages and the overall prognosis is unsatisfactory
(31–34). Higher incidence rate of gastric
carcinoma leads to even more actual problems in China (35). Therefore, to further elucidate the
underlying mechanisms of gastric cancer has become an urgent
requirement.
In the present study, we demonstrated that the BLU
expression was commonly downregulated in gastric cancer. Moreover,
silencing BLU with siRNA significantly promoted the cellular
proliferation and clonogenicity, indicating that BLU served as a
tumor suppressor in gastric cancer. Our results were supported by a
panel of previous studies which detected the frequent loss of BLU
in nasopharyngeal carcinoma, ovarian carcinoma, non-small cell lung
carcinoma, glioma and esophageal cancer (18–21,36).
Promoter hypermethylation has been recognized as common mechanism
leading transcriptional inactivation of TSGs in many types of
cancer (37,38). In the present study, we found that
the expression of BLU in gastric cancer was notably repressed by
promoter CpG hypermethylation, especially by hypermethylated −39
CpG site which is located within Sp1-binding transcription element.
Moreover, the demethylating agent treatment significantly restored
the expression of BLU in gastric cancer cells. These results
provided evidence that loss of BLU in gastric cancer is caused at
least partly by the promoter hypermethylation. Similar phenomenon
was also reported in glioma, cervical neoplasia and other
malignancies (12,17,20).
Subsequently, we tried to clarify the mechanisms
underlying epigenetically silenced BLU expression in gastric
cancer. First, we identified a functional BLU promoter which lacks
a TATA-box and contains two GC boxes. As a transcription activator,
Sp1 binds to GC box and regulates the basal transcription of
TATA-less promoters (26,27,39).
Using ChIP, EMSA and luciferase reporter assays, we identified that
Sp1 could specifically bind to the BLU promoter (−43/−35) and
regulate its transcriptional activity. Moreover, −39 CpG was
located within the predicted Sp1-binding sites and frequently
hypermethylated in gastric cancer. In fact, DNA methylation was
considered to repress epigenetically gene expression via blocking
the binding of transcription factor such as E2F and Sp1 to
hypermethylated CpG dinucleotides within TF-binding sites (22–25).
Consistently, our data showed that −39 CpG methylation
significantly reduced transcription activity of BLU by ~70%.
Combined with ChIP and EMSA assay, the results indicate that
hypermethylation of −39 CpG may decrease BLU expression via
interfering with the binding of Sp1.
As previously reported, the hypermethylation
progress lacks alteration in the gene sequences and can be
reversed. The demethylated agent 5-Aza was able to restore the
function of the TSGs silenced by promoter hypermethylation
(29,40). In the present study, we also
observed that BLU expression was restored after treatment with
5-Aza in AGS and SGC7901 cells. In addition, we found that
knockdown of BLU promoted gastric cell proliferation and
clonogenicity. These results suggest that BLU may become a
potential target for epigenetic therapy of gastric cancer.
In conclusion, we identified a novel functional BLU
promoter and proved that BLU promoter activity was regulated by
Sp1. Furthermore, we found that hypermethylated −39 CpG in BLU
proximal promoter directly reduced its binding with Sp1, which may
be one of the mechanisms accounting for the inactivation of BLU in
gastric cancer.
Acknowledgements
The authors would like to thank the patients with
gastric cancer for their participation and cooperation. This study
was supported in part by the grants from Natural Science Foundation
of Jiangsu Province Colleges (14KJB320014), Suzhou Key Laboratory
for Molecular Cancer Genetics (SZS201209) and A Project funded by
the Priority Academic Program Development of Jiangsu Higher
Education Institutions (PAPD).
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Uemura N, Okamoto S, Yamamoto S, Matsumura
N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N and Schlemper RJ:
Helicobacter pylori infection and the development of gastric
cancer. N Engl J Med. 345:784–789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nobili S, Bruno L, Landini I, Napoli C,
Bechi P, Tonelli F, Rubio CA, Mini E and Nesi G: Genomic and
genetic alterations influence the progression of gastric cancer.
World J Gastroenterol. 17:290–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qu Y, Dang S and Hou P: Gene methylation
in gastric cancer. Clin Chim Acta. 424:53–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oue N, Mitani Y, Motoshita J, Matsumura S,
Yoshida K, Kuniyasu H, Nakayama H and Yasui W: Accumulation of DNA
methylation is associated with tumor stage in gastric cancer.
Cancer. 106:1250–1259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
8
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis: Epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lerman MI and Minna JD: The 630-kb lung
cancer homozygous deletion region on human chromosome 3p21.3:
Identification and evaluation of the resident candidate tumor
suppressor genes. The International Lung Cancer Chromosome 3p213
Tumor Suppressor Gene Consortium. Cancer Res. 60:6116–6133.
2000.PubMed/NCBI
|
|
10
|
Masselink H and Bernards R: The adenovirus
E1A binding protein BS69 is a corepressor of transcription through
recruitment of N-CoR. Oncogene. 19:1538–1546. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wolford JK and Prochazka M: Structure and
expression of the human MTG8/ETO gene. Gene. 212:103–109. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Agathanggelou A, Dallol A,
Zöchbauer-Müller S, Morrissey C, Honorio S, Hesson L, Martinsson T,
Fong KM, Kuo MJ, Yuen PW, et al: Epigenetic inactivation of the
candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene.
22:1580–1588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cheng Y, Ho RL, Chan KC, Kan R, Tung E,
Lung HL, Yau WL, Cheung AK, Ko JM, Zhang ZF, et al: Anti-angiogenic
pathway associations of the 3p213 mapped BLU gene in nasopharyngeal
carcinoma. Oncogene. Oct 27;2014 (Epub ahead of print). View Article : Google Scholar : 2014.
|
|
14
|
Zhang X, Liu H, Li B, Huang P, Shao J and
He Z: Tumor suppressor BLU inhibits proliferation of nasopharyngeal
carcinoma cells by regulation of cell cycle, c-Jun N-terminal
kinase and the cyclin D1 promoter. BMC Cancer. 12:2672012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park ST, Byun HJ, Kim BR, Dong SM, Park
SH, Jang PR and Rho SB: Tumor suppressor BLU promotes paclitaxel
antitumor activity by inducing apoptosis through the downregulation
of Bcl-2 expression in tumorigenesis. Biochem Biophys Res Commun.
435:153–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoo HJ, Kim BR, Byun HJ, Park SY and Rho
SB: BLU enhances the effects of anti-angiogenic activity in
combination with gemcitabine-based chemotherapeutic agents. Int J
Biochem Cell Biol. 45:1236–1245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lai HC, Lin YW, Chang CC, Wang HC, Chu TW,
Yu MH and Chu TY: Hypermethylation of two consecutive tumor
suppressor genes, BLU and RASSF1A, located at 3p21.3 in cervical
neoplasias. Gynecol Oncol. 104:629–635. 2007. View Article : Google Scholar
|
|
18
|
Ito M, Ito G, Kondo M, Uchiyama M, Fukui
T, Mori S, Yoshioka H, Ueda Y, Shimokata K and Sekido Y: Frequent
inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter
hypermethylation and allele loss in non-small cell lung cancer.
Cancer Lett. 225:131–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yi Lo PH, Chung Leung AC, Xiong W, Law S,
Duh FM, Lerman MI, Stanbridge EJ and Lung ML: Expression of
candidate chromosome 3p21.3 tumor suppressor genes and
downregulation of BLU in some esophageal squamous cell carcinomas.
Cancer Lett. 234:184–192. 2006. View Article : Google Scholar
|
|
20
|
Hesson L, Bièche I, Krex D, Criniere E,
Hoang-Xuan K, Maher ER and Latif F: Frequent epigenetic
inactivation of RASSF1A and BLU genes located within the critical
3p21.3 region in gliomas. Oncogene. 23:2408–2419. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava
G, Tsai ST, Tsao SW and Tao Q: The candidate tumor suppressor gene
BLU, located at the commonly deleted region 3p21.3, is an
E2F-regulated, stress-responsive gene and inactivated by both
epigenetic and genetic mechanisms in nasopharyngeal carcinoma.
Oncogene. 23:4793–4806. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Yang R, Jia Y, Cai D, Zhou B, Qu
X, Han H, Xu L, Wang L, Yao Y, et al: Hypermethylation of Sp1
binding site suppresses hypothalamic POMC in neonates and may
contribute to metabolic disorders in adults: Impact of maternal
dietary CLAs. Diabetes. 63:1475–1487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo D, Wu B, Yan J, Li X, Sun H and Zhou
D: A possible gene silencing mechanism: Hypermethylation of the
Keap1 promoter abrogates binding of the transcription factor Sp1 in
lung cancer cells. Biochem Biophys Res Commun. 428:80–85. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ehrlich S, Weiss D, Burghardt R,
Infante-Duarte C, Brockhaus S, Muschler MA, Bleich S, Lehmkuhl U
and Frieling H: Promoter specific DNA methylation and gene
expression of POMC in acutely underweight and recovered patients
with anorexia nervosa. J Psychiatr Res. 44:827–833. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Campanero MR, Armstrong MI and Flemington
EK: CpG methylation as a mechanism for the regulation of E2F
activity. Proc Natl Acad Sci USA. 97:6481–6486. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lania L, Majello B and De Luca P:
Transcriptional regulation by the Sp family proteins. Int J Biochem
Cell Biol. 29:1313–1323. 1997. View Article : Google Scholar
|
|
27
|
Emili A, Greenblatt J and Ingles CJ:
Species-specific interaction of the glutamine-rich activation
domains of Sp1 with the TATA box-binding protein. Mol Cell Biol.
14:1582–1593. 1994.PubMed/NCBI
|
|
28
|
Yuan H, Gong A and Young CY: Involvement
of transcription factor Sp1 in quercetin-mediated inhibitory effect
on the androgen receptor in human prostate cancer cells.
Carcinogenesis. 26:793–801. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qian Q, Shi X, Lei Z, Zhan L, Liu RY, Zhao
J, Yang B, Liu Z and Zhang HT: Methylated +58CpG site decreases DCN
mRNA expression and enhances TGF-β/Smad signaling in NSCLC cells
with high metastatic potential. Int J Oncol. 44:874–882.
2014.PubMed/NCBI
|
|
30
|
Dynan WS and Tjian R: The
promoter-specific transcription factor Sp1 binds to upstream
sequences in the SV40 early promoter. Cell. 35:79–87. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hundahl SA, Phillips JL and Menck HR: The
National Cancer Data Base Report on poor survival of U.S. gastric
carcinoma patients treated with gastrectomy: Fifth Edition American
Joint Committee on Cancer staging, proximal disease, and the
‘different disease’ hypothesis. Cancer. 88:921–932. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dikken JL, van de Velde CJ, Coit DG, Shah
MA, Verheij M and Cats A: Treatment of resectable gastric cancer.
Therap Adv Gastroenterol. 5:49–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mickevicius A, Ignatavicius P, Markelis R,
Parseliunas A, Butkute D, Kiudelis M, Endzinas Z, Maleckas A and
Dambrauskas Z: Trends and results in treatment of gastric cancer
over last two decades at single East European centre: A cohort
study. BMC Surg. 14:982014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Forman D and Burley VJ: Gastric cancer:
Global pattern of the disease and an overview of environmental risk
factors. Best Pract Res Clin Gastroenterol. 20:633–649. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rahman R, Asombang AW and Ibdah JA:
Characteristics of gastric cancer in Asia. World J Gastroenterol.
20:4483–4490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chiang YC, Chang MC, Chen PJ, Wu MM, Hsieh
CY, Cheng WF and Chen CA: Epigenetic silencing of BLU through
interfering apoptosis results in chemoresistance and poor prognosis
of ovarian serous carcinoma patients. Endocr Relat Cancer.
20:213–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wanajo A, Sasaki A, Nagasaki H, Shimada S,
Otsubo T, Owaki S, Shimizu Y, Eishi Y, Kojima K, Nakajima Y, et al:
Methylation of the calcium channel-related gene, CACNA2D3, is
frequent and a poor prognostic factor in gastric cancer.
Gastroenterology. 135:580–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Batistuzzo de Medeiros SR, Krey G, Hihi AK
and Wahli W: Functional interactions between the estrogen receptor
and the transcription activator Sp1 regulate the estrogen-dependent
transcriptional activity of the vitellogenin A1 io promoter. J Biol
Chem. 272:18250–18260. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Christman JK: 5-Azacytidine and
5-aza-2′-deoxycytidine as inhibitors of DNA methylation:
Mechanistic studies and their implications for cancer therapy.
Oncogene. 21:5483–5495. 2002. View Article : Google Scholar : PubMed/NCBI
|