Introduction
The nuclear factor-κB (NF-κB) plays a key role in
several cellular functions, e.g., sustenance of proliferative
signaling, evasion of growth suppression, resistance to cell death,
ability of replicative immortality, and activation of invasion and
metastasis in hematological malignancies (1). The inflammatory process has emerged
as a useful marker of cancer progression (2,3).
NF-κB is also involved in the induction of inflammation (2,3).
Constitutive activation of NF-κB occurs in most malignant lymphomas
and plays a major role in lymphomagenesis and clinical
aggressiveness (1). Furthermore,
Epstein-Barr virus (EBV) latent membrane protein-1 (LMP-1) and CD30
overexpression have been shown to activate NF-κB and induce rapidly
progressing lymphomas (1,4).
EBV is associated with the development of lymphomas
including Burkitt's lymphoma (BL), Hodgkin's lymphoma (HL), diffuse
large B-cell lymphoma and natural killer/T-cell lymphoma (5). LMP-1, a transmembrane protein, is
essential for in vitro transformation of primary B cells
(6). The carboxyl-terminal
cytoplasmic domain of LMP-1 contains two carboxyl-terminal
activation regions (CTARs); CTAR-1 and CTAR-2. CTAR-1 binds to
tumor necrosis factor (TNF) receptor-associated factors (TRAFs)
(7), whereas CTAR-2 binds to the
TNF receptor-associated death domain (TRADD) (8). NF-κB activation by the CTAR-1 and
CTAR-2 domains of LMP-1 is probably mediated by the binding of
TRAFs directly or indirectly to both the CTAR-1 and CTAR-2 domains
(7–10).
CD30, a member of the TNF receptor superfamily, is
also a transmembrane protein and highly expressed in a variety of
lymphoma subsets including HL. Overexpression of CD30 was reported
to transduce signals independent of CD30 ligand in HL cells
(11). A region of ~100 amino
acids from the carboxyl-terminal region of CD30 is involved in
NF-κB activation (12). TRAFs
recognize the carboxyl-terminal D2 and D3 subdomains of CD30
(12).
Activation of NF-κB has often been linked to
recurrence, poor survival, tumor progression, aggressiveness and
chemoresistance (13). However,
there are also studies that found NF-κB or upstream activators
rather to act as tumor suppressors. Contributing to its anticancer
property, NF-κB has been shown to mediate apoptosis in a variety of
cell types (14). Overexpression
of RelA (p65) caused a cell cycle arrest followed by apoptosis
(15). Premature cellular
senescence is a terminal cell cycle arrest that can be induced by
oncogenic activation or chemotherapy (16,17).
NF-κB also participates in a senescence-associated cytokine
response (18). Therefore,
appropriate regulation of NF-κB is critical for the proper function
and survival of the cell.
IκB-ζ is an atypical nuclear member of the IκB
family (19). The activity of
NF-κB is modulated in a gene-specific manner by IκB-ζ. In contrast
to classical IκB proteins that are constitutively expressed and
controlled by inducible degradation, IκB-ζ expression is barely
detectable in resting cells but is rapidly induced by various
pro-inflammatory stimuli, such as lipopolysaccharides and
interleukin (IL)-1β (20). IκB-ζ
regulates NF-κB signaling, and reporter analyses suggested that
IκB-ζ may act as an inhibitor of NF-κB (19). In contrast, other studies have
reported that IκB-ζ can induce gene expression of individual NF-κB
target genes (21). A recent study
identified the nuclear IκB-ζ to be upregulated in activated
B-cell-like subtype of diffuse large B-cell lymphoma (22). We have also reported constitutive
expression of IκB-ζ in adult T-cell leukemia cells (23). The hypothesis tested in the present
study was that IκB-ζ is induced by LMP-1 and CD30 and that it is
also involved in regulation of NF-κB.
Materials and methods
Cell culture
Raji and Daudi are EBV-positive BL cell lines. In
contrast, BJAB and Ramos are EBV-negative BL cell lines. B95/Ramos
is Ramos infected with the B95-8 strain of EBV. L428, KM-H2, HDLM-2
and L540 are HL cell lines. These cell lines were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium supplemented
with 10 or 20% fetal bovine serum (FBS) and antibiotics. Human
embryonic kidney 293T cells were cultured in Dulbecco's modified
Eagle's medium supplemented with 10% FBS and antibiotics.
RNA detection
Total RNA was extracted from various cell cultures
by TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA)
according to the protocol provided by the manufacturer. The
first-strand cDNA was synthesized from 1 μg cellular RNA
using a PrimeScript RT-PCR kit (Takara Bio Inc., Otsu, Japan) with
random primers. The sequences of the primers used are summarized in
Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene name | Forward (5′) | Reverse (3′) |
|---|
| IκB-ζ |
GGAGCTTTTACTGAAGAATAAGA |
ATCTGTTCTCCCACAGGGCCATC |
| LMP-1 |
GTGACTGGACTGGAGGAGCC |
GAGGGAGTCATCGTGGTGGTG |
| CD30 |
CTGTGTCCCCTACCCAATCT |
CTTCTTTCCCTTCCTCTTCCA |
| IL-6 |
ATGAACTCCTTCTCCACAAGC |
CTACATTTGCCGAAGAGCCCTCAGGCTGGACTG |
| IL-8 |
ATGACTTCCAAGCTGGCCGTG |
TTATGAATTCTCAGCCCTCTTCAAAAACTTCTC |
| GAPDH |
GCCAAGGTCATCCATGACAACTTTGG |
GCCTGCTTCACCACCTTCTTGATGTC |
Plasmids, transfection and luciferase
assay
Cells (293T) were transfected by the calcium
phosphate DNA coprecipitation method. The expression plasmids
pSG5-LMP-1, pSG5-LMP-1Δ187-351, pSG5-LMP-1Δ349 and
pSG5-LMP-1Δ194–386 were previously described (24,25).
For CD30 expression, the plasmids wild-type human CD30 (pME-hCD30)
and its mutant [pCR-hCD30(Δ95)] were used (12). The wild-type and various mutants of
IκB-ζ, and pcDNA3-RelA were described previously (26,27).
The dominant-negative mutants of IκBα, IκBβ, IκB kinase (IKK) α,
IKKβ, IKKγ and NF-κB-inducing kinase (NIK) have been previously
described (28–31). The plasmid for truncated TRAF2
protein with retention of only the TRAF domain, ΔTRAF2, has been
described previously (32). The
human IκB-ζ promoter-luciferase gene constructs have already been
described (23,33). The single and combined internal
deletion mutants of NF-κB sites were constructed by deletion of the
NF-κB sites of the plasmid pGL3-hIκB-ζ(−853) (23). A reporter plasmid, expressing
luciferase through a minimal promoter linked to five copies of the
typical NF-κB responsive element from the IL-2 receptor α
chain (IL-2Rα) gene (κB-LUC), was used to measure the
NF-κB transcription competence (34). Two copies of the IL-8 activator
protein-1 (AP-1) binding site were inserted upstream of the IL-8
enhancer-less core promoter linked to luciferase gene (AP-1-LUC)
(35). Plasmids containing the
IL-8 promoter (−133 to +44 bp) and the IL-6 promoter (−225 to +14
bp) linked to luciferase expression vectors were constructed from
luciferase expression vectors (35,36).
Bcl-3 luciferase reporter construct was described previously
(37). In all cases, phRL-TK was
cotransfected to correct for transfection efficiency. After 24 h,
luciferase assays were conducted using the dual luciferase reporter
system (Promega Corp., Madison, WI, USA), in which the relative
luciferase activity was calculated by normalizing transfection
relative to the Renilla luciferase activities. Data were
expressed as mean ± SD of three experiments.
Preparation of nuclear extracts and
electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted and transcription
factors bound to specific DNA sequences were examined by EMSA, as
previously described (38). The
top strand sequences of the oligonucleotide probes or competitors
were as follows: for the NF-κB element (κB1) of the IκB-ζ
gene, 5′-GATCCGACGGGAATGTCCGGGACT-3′; for the mutated κB1 sequence,
5′-GATCCGACGtGtATGaCCGGG ACT-3′; for the
NF-κB element (κB2) of the IκB-ζ gene,
5′-GATCGGTCTGGGAATTTCCAGTG-3′; for the
mutated κB2 sequence, 5′-GATCGGTCTGtGtATaaCCAGTG; for the NF-κB
element of the IL-2Rα gene, 5′-GATCCGGCAGGGG AATCTCCCTCTC-3′; and for
the AP-1 element of the IL-8 gene, 5′-GATCGTGATGACTCAGGTT-3′. The above
underlined sequences are the NF-κB and AP-1 binding sites,
respectively. The sites of mutation are indicated in lowercase
letters. In competition experiments, the nuclear extract was
pre-incubated with 100-fold excess of unlabeled oligonucleotides
for 15 min. To identify NF-κB proteins in the DNA-protein complex
shown by EMSA, we used antibodies specific for various NF-κB family
proteins, including p50, RelA, c-Rel, p52 and RelB (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA). These antibodies were
incubated with the nuclear extracts for 45 min at room temperature
before incubation with radiolabeled probes.
Immunohistochemical analysis
Lymph node biopsy samples were obtained from
patients with BL and HL. IκB-ζ immunohistochemistry was performed
using an anti-IκB-ζ antibody (Cell Signaling Technology, Inc.,
Beverly, MA, USA) after pretreatment of deparafinized tissue
sections with ready-to-use proteinase K (Dako, Carpinteria, CA,
USA). The sections were counterstained with methyl green, hydrated
in ethanol, cleaned in xylene and mounted. Informed consent was
obtained from all tissue donors.
Results
Upregulated IκB-ζ expression in BL and
HL
To investigate the role of IκB-ζ in the pathogenesis
of BL and HL, we assessed IκB-ζ mRNA expression levels in
established BL and HL cell lines using reverse-transcription
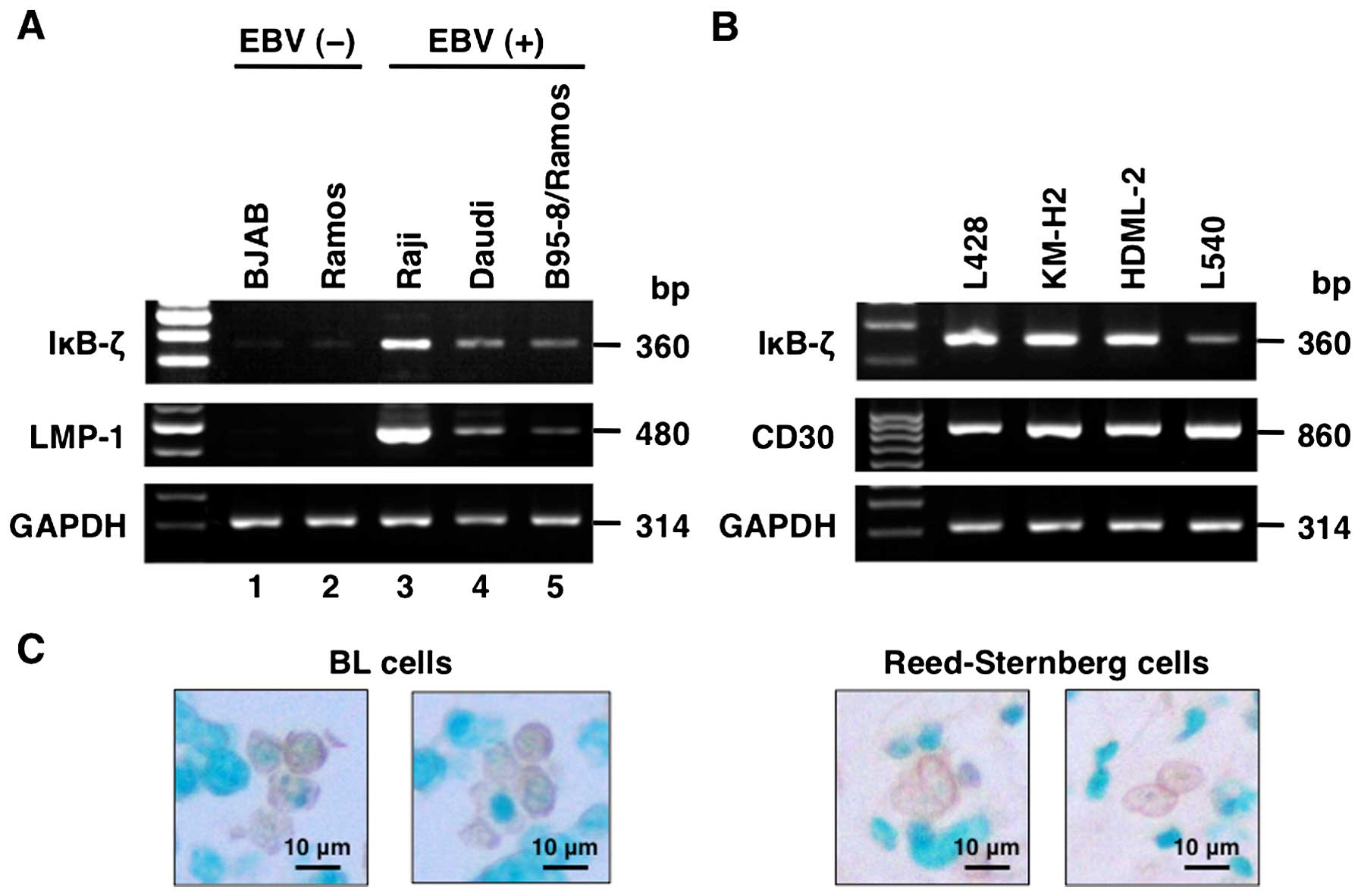
polymerase chain reaction (RT-PCR). We found that IκB-ζ mRNA
expression was limited to EBV-infected BL cell lines but not in
uninfected cells (Raji, Daudi and B95–8/Ramos) (Fig. 1A). On the other hand, all HL cell
lines showed IκB-ζ mRNA levels (Fig.
1B). All EBV-infected BL cell lines and HL cell lines
constitutively expressed LMP-1 and CD30, respectively (Fig. 1A and B). Immunohistochemical
staining of BL cells, and Hodgkin and Reed-Sternberg cells in lymph
nodes showed abundant IκB-ζ protein in the nuclei of these cells
(Fig. 1C).
LMP-1 and CD30 induce IκB-ζ mRNA
expression
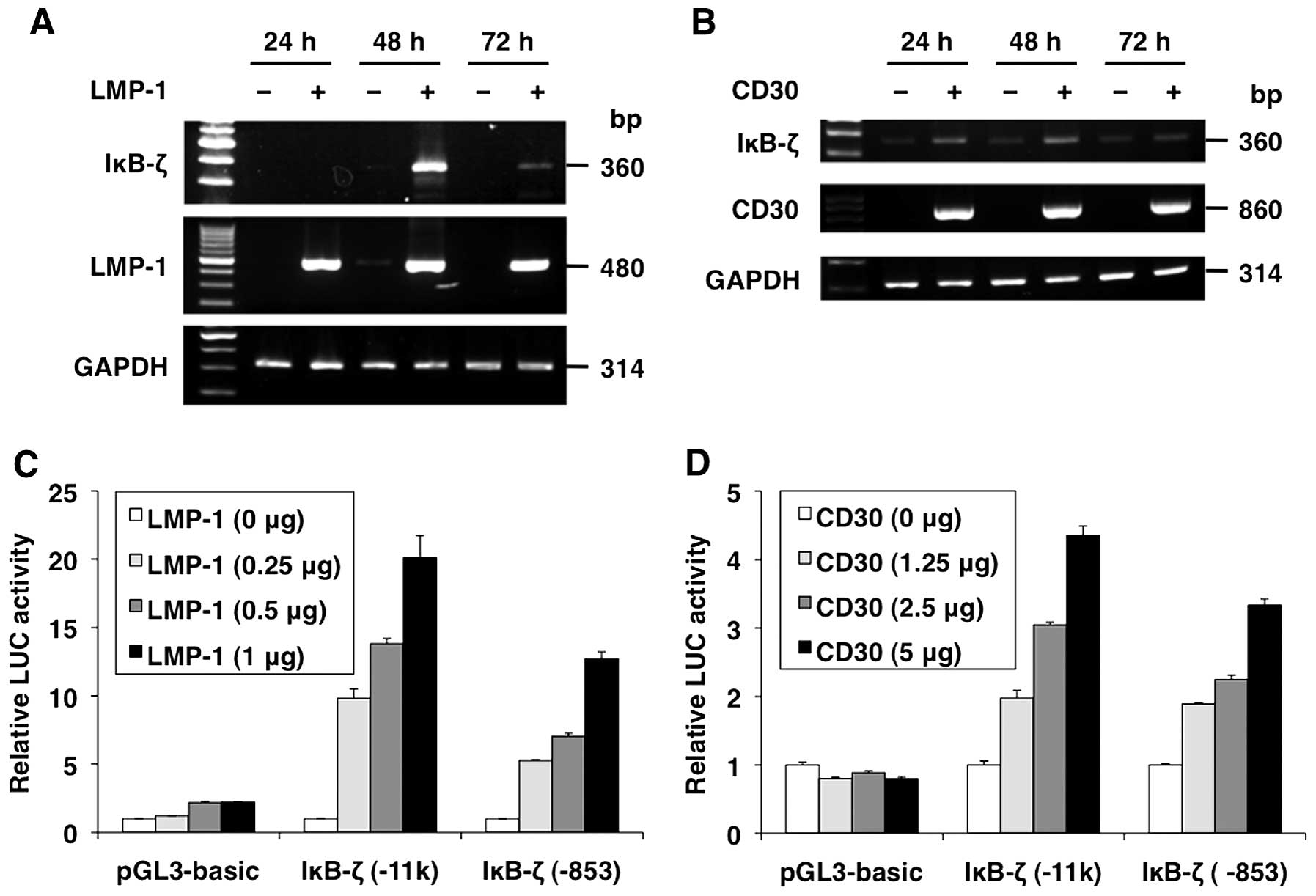
To investigate the induction of IκB-ζ in BL and HL,
we performed transient expression assays using mammalian expression
vectors for LMP-1 and CD30 in 293T cells. After the transfection,
RNA was extracted from the cells and the IκB-ζ mRNA levels were
analyzed by RT-PCR. IκB-ζ mRNA induction was observed at 48 and 24
h after LMP-1 and CD30 transfection, respectively. In contrast,
IκB-ζ mRNA was hardly detected in cells transfected with empty
vectors (pSG5 and pME18S) (Fig. 2A and
B).
LMP-1 and CD30 activate the IκB-ζ
promoter
To determine whether LMP-1 and CD30 regulate IκB-ζ
promoter activity, transient expression assays were performed using
the reporter plasmids, pGL3-hIκB-ζ(-11k) and pGL3-hIκB-ζ(−853), and
expression vectors for LMP-1 and CD30. LMP-1 and CD30
transactivated the −11 kb and −853 IκB-ζ promoter fragments, but
this effect was lost in pGL3-basic. LMP-1 and CD30 induced relative
levels of IκB-ζ promoter-directed luciferase expression in a
dose-dependent manner, suggesting that LMP-1 and CD30 functionally
activate minimum IκB-ζ promoter between −853 and −17 bp (Fig. 2C and D).
Importance of carboxyl-terminal regions
of LMP-1 and CD30 for IκB-ζ promoter activation
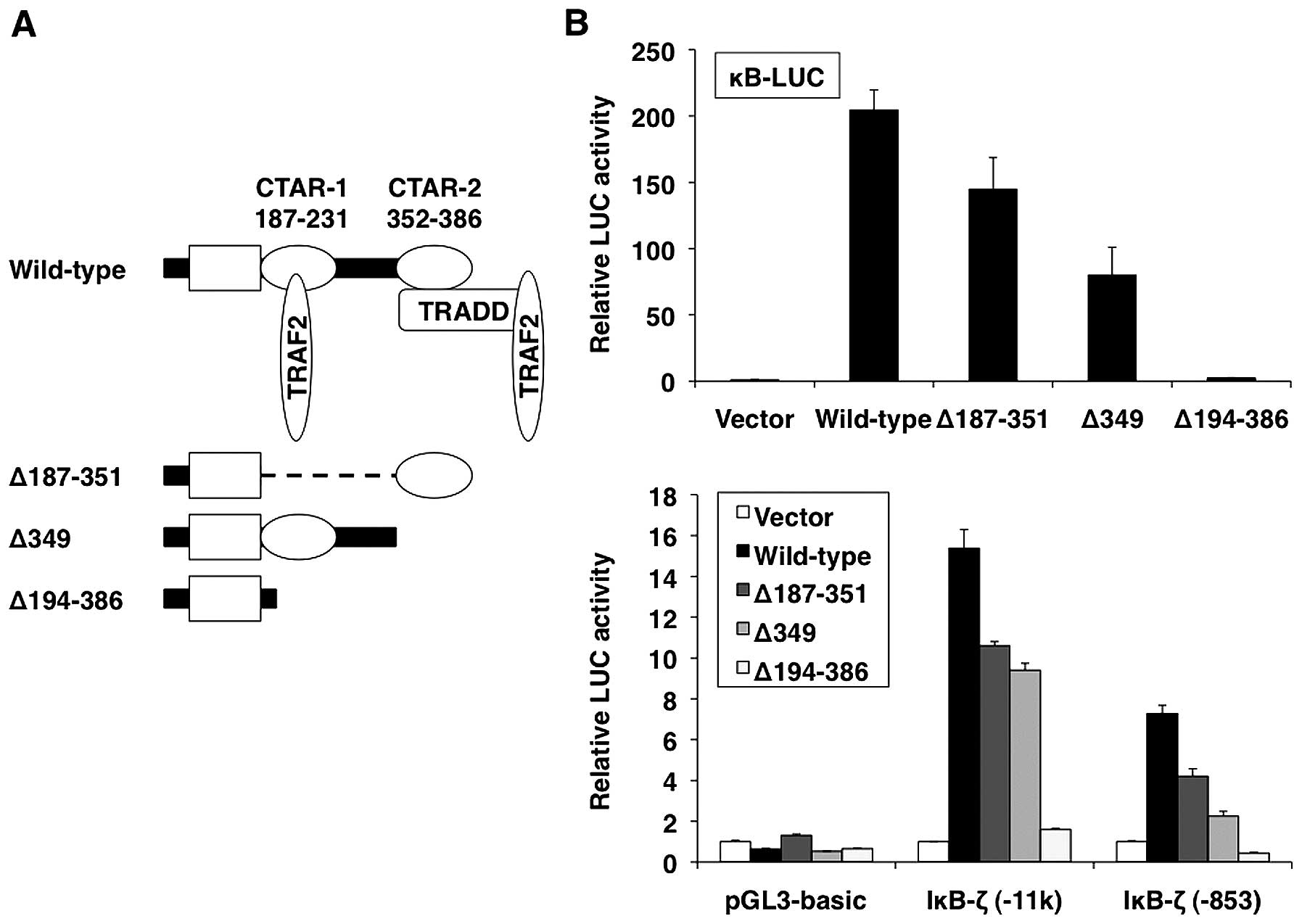
To map the regions in the LMP-1 and CD30 proteins
that mediate activation of IκB-ζ promoter, LMP-1 and CD30 mutants
were expressed and their effect on IκB-ζ promoter activity was
investigated. The LMP-1 mutants used included LMP-1Δ187–351 (which
contains only CTAR-2 in the carboxyl-terminus), LMP-1Δ349 (which
lacks CTAR-2) and LMP-1Δ194–386 (in which the entire
carboxyl-terminal cytoplasmic region is deleted) (Fig. 3A). In cells that expressed
CTARs-free LMP-1Δ194–386, IκB-ζ promoter activity was not
increased. In contrast, activation of pGL3-hIκB-ζ( −11k) and
pGL3-hIκB-ζ( −853) was observed by both CTAR-1-free LMP-1Δ187–351
and CTAR-2-free LMP-1Δ349, although to a lesser extent than by
wild-type LMP-1 (Fig. 3B, lower
panel). These results suggest that LMP-1 activates IκB-ζ expression
via the cooperative activity of CTAR-1 and CTAR-2 signaling motifs.
As measured in an NF-κB-dependent luciferase reporter gene assay,
LMP-1 increased NF-κB activation via CTAR-1 and CTAR-2 (Fig. 3B, upper panel).
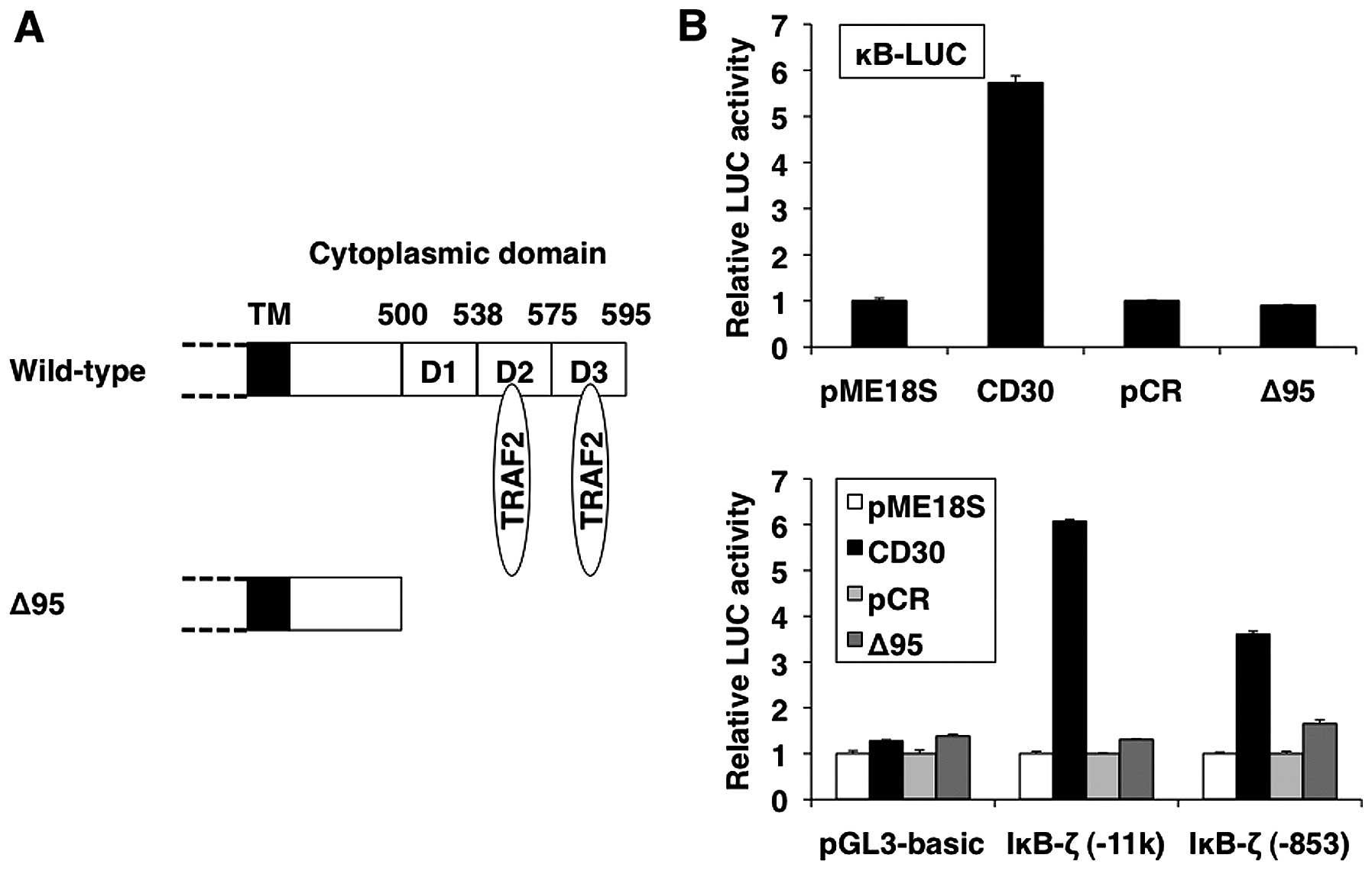
The carboxyl-terminal region of CD30 is also
essential for signal transduction (Fig. 4A) (12). Next, we investigated whether the
carboxyl-terminal region of CD30 plays a role in the induction of
IκB-ζ. As shown in Fig. 4B, lower
panel, IκB-ζ-driven reporter gene activity was not increased by
CD30Δ95, which lacks the carboxyl-terminal region of CD30. Notably,
the effect of the structural context of the carboxyl-terminal
region of LMP-1 and CD30 on IκB-ζ promoter activation correlated
with NF-κB activation as reporter analyses by mutant constructs
(Figs. 3B and 4B, upper panels). These results suggest
that LMP-1 and CD30 activate IκB-ζ promoter through the NF-κB
signaling pathway.
LMP-1 and CD30 activate IκB-ζ promoter
activity via the NF-κB signaling pathway
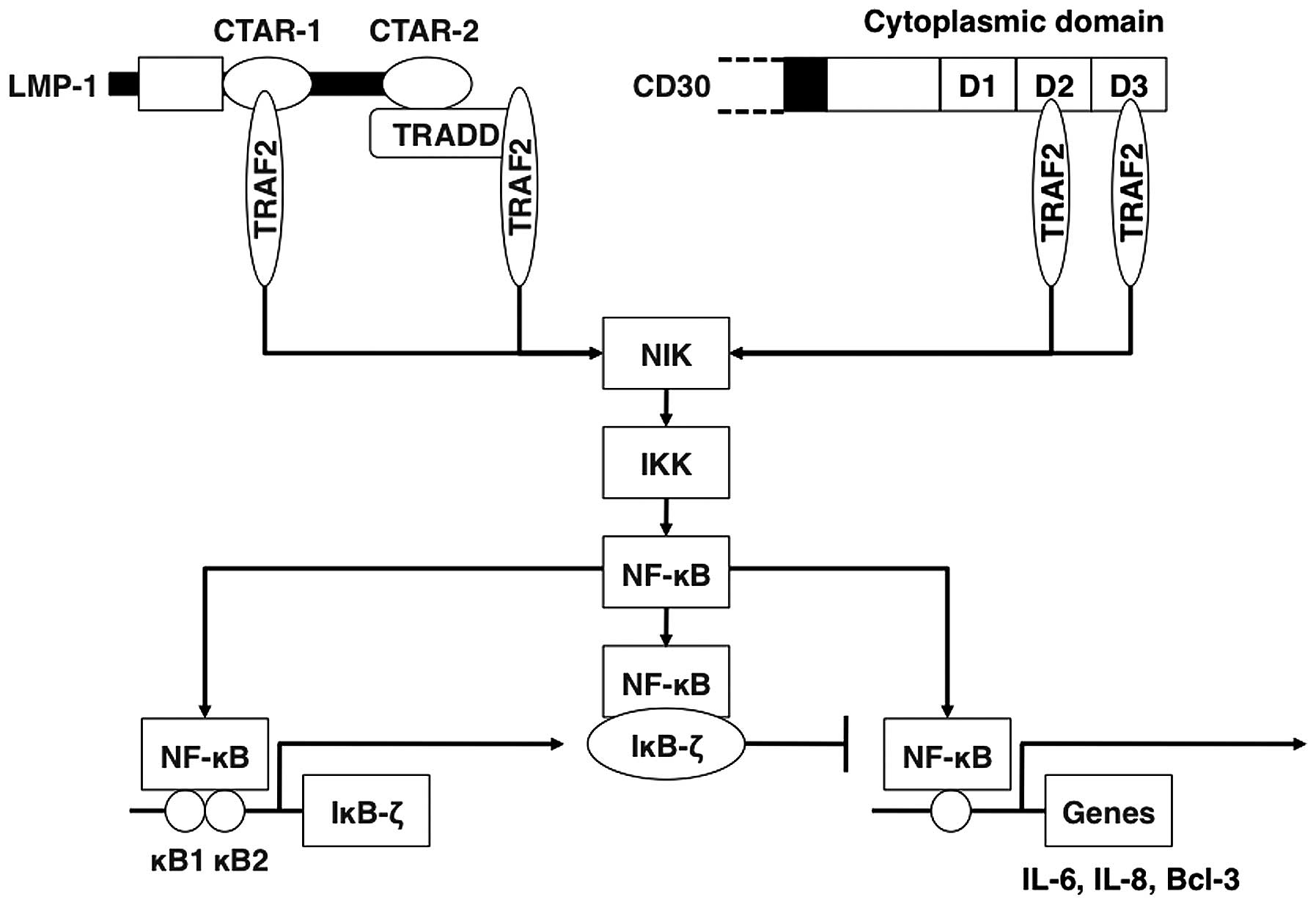
LMP-1 and CD30 are constitutively aggregated
pseudo-TNF receptors that activate NF-κB through their
carboxyl-terminal cytoplasmic domains associated with TRAF2
(7–12). Aggregated TRAF2 activates NIK and
its downstream target, the IKK complex, which is composed of two
catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ
(39,40). The IKK complex phosphorylates the
inhibitory IκB proteins, which are bound to NF-κB in the cytosol.
Their phosphorylation is followed by their degradation,
dissociation of NF-κB from the inhibitors, and NF-κB translocation
into the nucleus (41). In order
to determine the role of TRAF2/NIK/IKK/IκB proteins in mediating
IκB-ζ activation induced by LMP-1 and CD30, 293T cells were
cotransfected with LMP-1 or CD30 expression plasmid and plasmids
expressing dominant-negative forms of TRAF2, NIK, IKKα, IKKβ, IKKγ,
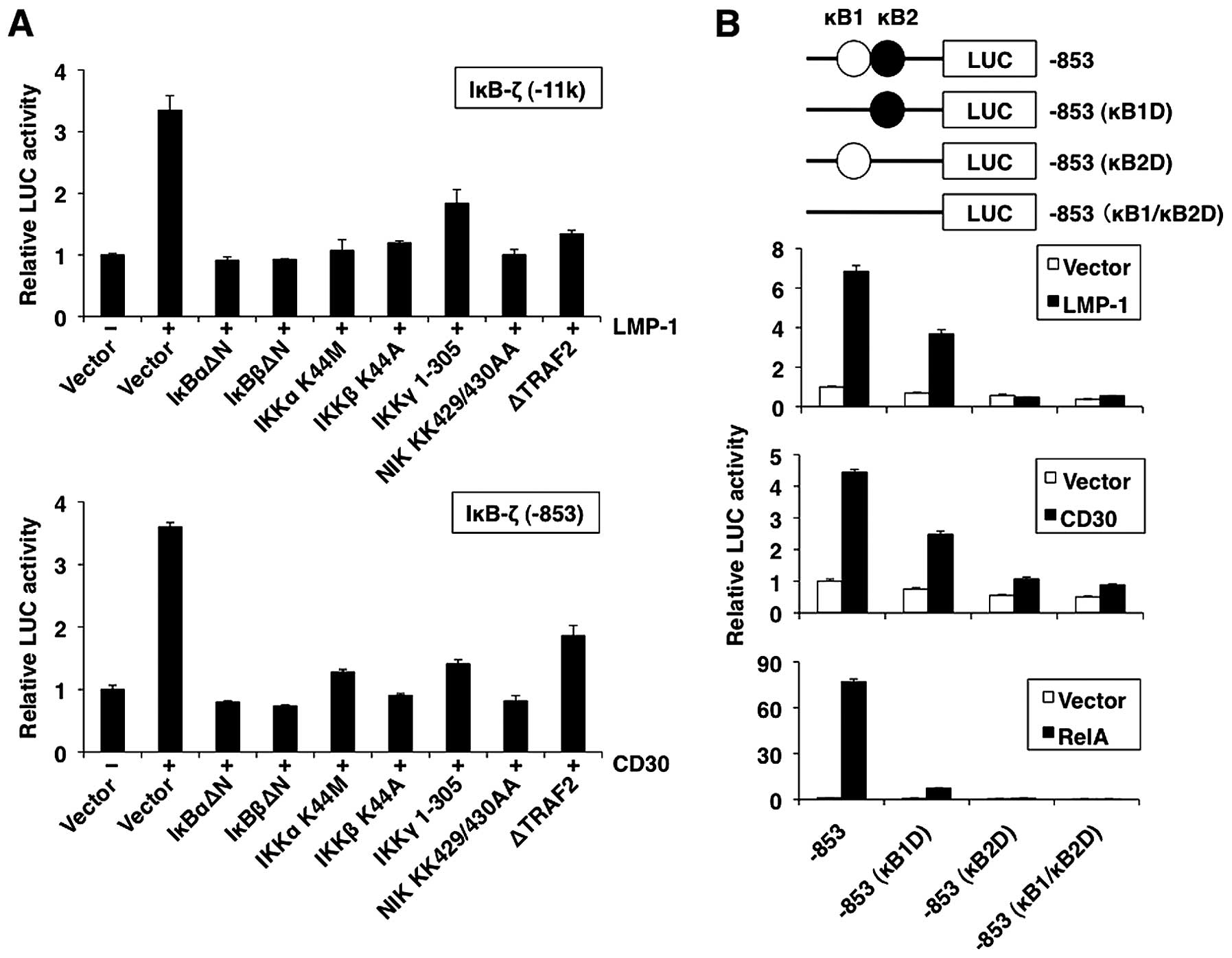
IκBα or IκBβ. All dominant-negative mutants reduced IκB-ζ promoter
activation by LMP-1 and CD30 (Fig.
5A). These data indicate that the activation of NF-κB through
TRAF2/NIK/IKK plays a role in the activation of IκB-ζ promoter by
LMP-1 and CD30.
NF-κB sites in the promoter are essential
for the transcriptional upregulation of the IκB-ζ gene
The human IκB-ζ promoter contains two NF-κB motifs
(κB1 and κB2) (Fig. 5B, top panel)
(33). To determine the
involvement of κB1 and κB2 in the induction of IκB-ζ gene
expression by LMP-1 and CD30, we investigated the activity of IκB-ζ
promoter with deletions in κB1 and κB2 sites. As shown in Fig. 5B, LMP-1 and CD30 activated the
wild-type promoter pGL3-hIκB-ζ( −853) activity. A single deletion
of the κB2 site from the IκB-ζ reporter plasmid (κB2D) markedly
inhibited LMP-1- or CD30-induced transactivation, whereas a single
deletion of the κB1 site (κB1D) resulted in moderate activation.
These data indicate that IκB-ζ κB2 site was necessary for
transcription of IκB-ζ. Furthermore, double deletions (κB1/κB2D)
completely abolished the LMP-1- or CD30-induced transactivation.
Expression of RelA is sufficient to induce IκB-ζ expression. The
predominant role of κB2 in the induction of IκB-ζ expression was
further supported by the finding that a promoter with a deleted κB2
site failed to respond to overexpressed RelA (Fig. 5B, bottom panel). In contrast, a
promoter with a deleted κB1 was slightly activated by RelA.
Considered together, these data suggest that upregulation of IκB-ζ
by LMP-1 and CD30 requires both the κB1 and κB2 sites of the IκB-ζ
promoter.
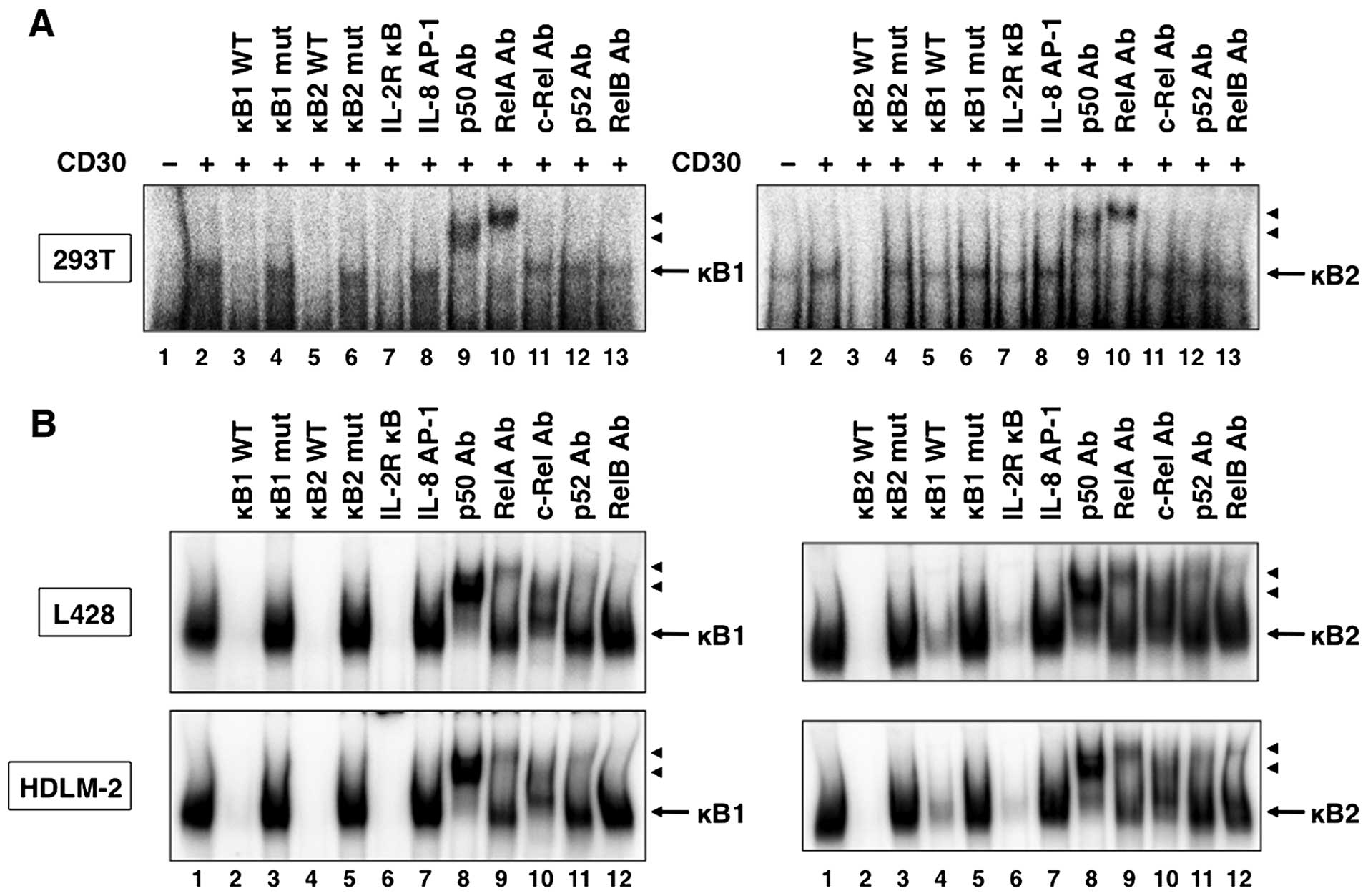
Since the deletional analysis of the IκB-ζ promoter
indicated that LMP-1 and CD30 activated transcription through both
the κB1 and κB2 sites, it was important to identify the nuclear
factors that bind to these sites. Using the κB1 and κB2 sequences
in the IκB-ζ promoter as the probes in EMSA, NF-κB binding was
detected in 293T cells transfected with LMP-1 (Fig. 6A, lane 2) and CD30 (Fig. 7A, lane 2). In addition, formation
of these complexes was competed with excess of unlabeled wild-type
κB1, κB2 and consensus IL-2R κB oligonucleotides (Figs. 6A and 7A, lanes 3, 5 and 7). In contrast,
mutated oligonucleotides, κB1 mut and κB2 mut, and irrelevant
consensus IL-8 AP-1 oligonucleotide, did not compete with the
labeled probes (Figs. 6A and
7A, lanes 4, 6 and 8). Supershift
analyses demonstrated that the κB1 and κB2 complexes contained both
p50 and RelA subunits of the NF-κB family (Figs. 6A and 7A, lanes 9 and 10). To determine the role
of LMP-1 and CD30 on endogenous NF-κB binding to DNA, we measured
NF-κB binding to respective NF-κB sites in the IκB-ζ promoter in
LMP-1-expressing Daudi cells and CD30-expressing L428 and HDLM-2
cells. As expected, protein complexes bound to both κB1 and κB2
sites were detected in nuclear extracts from Daudi, L428 and HDLM-2
cells (Figs. 6B and 7B, lane 1). The specificity of
DNA-protein complexes in these extracts was determined by
competition studies using unlabeled competitors. As observed in
nuclear extracts from 293T cells transfected with LMP-1 and CD30
expression plasmids, unlabeled κB1 and κB2 oligonucleotides, and
consensus NF-κB site from the IL-2Rα promoter, but not the mutated
κB1 and κB2 oligonucleotides, and consensus AP-1 element,
efficiently competed with the labeled probes (Figs. 6B and 7B, lanes 2–7). Antibodies against p50,
RelA, c-Rel and RelB induced a supershift of the DNA-protein
complexes in nuclear extracts of Daudi cells (Fig. 6B, lanes 8–10 and 12), whereas the
κB1 and κB2 complexes contained p50, RelA, c-Rel, p52 and RelB in
nuclear extracts of L428 and HDLM-2 cells (Fig. 7B, lanes 8–12). Taken together, the
results indicate that NF-κB proteins bind to both κB elements of
the IκB-ζ promoter in Daudi, L428 and HDLM-2 cells.
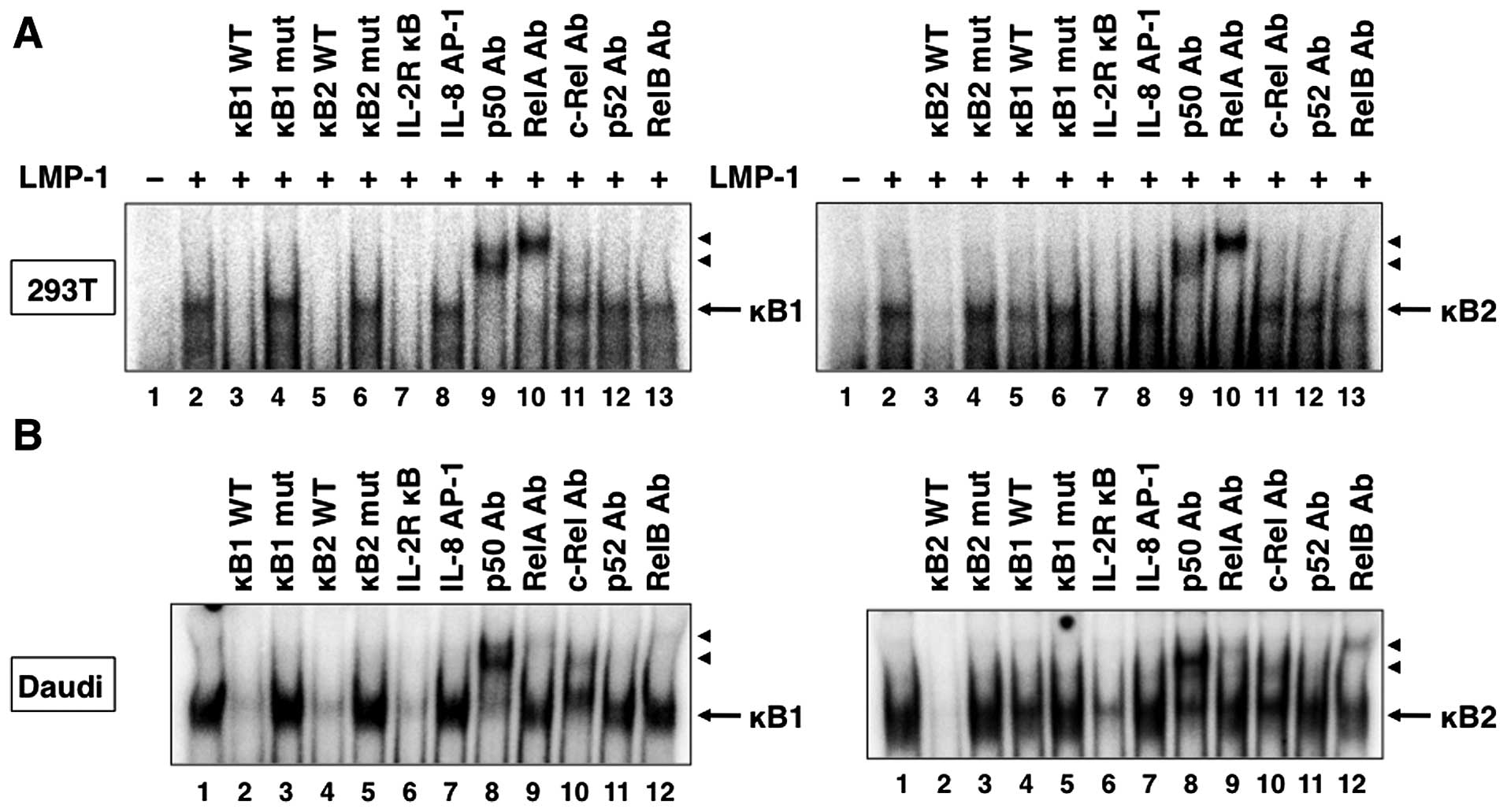 | Figure 6LMP-1 induces NF-κB binding to the
NF-κB sites in the IκB-ζ promoter. (A) Cells (293T) were
transfected with control or LMP-1 expression plasmid. Nuclear
proteins were extracted 48 h after transfection. Nuclear extracts
from transfected 293T cells (A) or Daudi cells (B) were incubated
with the labeled DNA probes representing the IκB-ζ κB1 and κB2
sites. Nuclear extracts were subjected to competition analysis with
an excess of unlabeled oligonucleotides representing the IκB-ζ κB1
and κB2 sites (lanes 3 and 5 in A, and lanes 2 and 4 in B,
respectively), IκB-ζ mutated κB1 and κB2 sites (lanes 4 and 6 in A,
and lanes 3 and 5 in B), a consensus NF-κB site from the IL-2Rα
promoter (lane 7 in A, and lane 6 in B) or an AP-1 site from the
IL-8 promoter (lane 8 in A, and lane 7 in B). Nuclear extracts were
also subjected to supershift assays with either no antibody (lane 2
in A, and lane 1 in B) or the indicated antibodies (Ab) (lanes 9–13
in A, and lanes 8–12 in B). Arrows, specific complexes; arrowheads,
DNA binding complex supershifted by the antibody. |
Role of IκB-ζ in the expression of NF-κB
target genes
Unlike other IκB family members, IκB-ζ has dual
opposite functions on the expression of different cellular genes
activated by NF-κB (19–21,27).
To identify the genes whose expression is regulated by IκB-ζ, we
examined the promoter activities of several NF-κB target genes in
293T cells transfected with LMP-1, CD30 or RelA, and IκB-ζ.
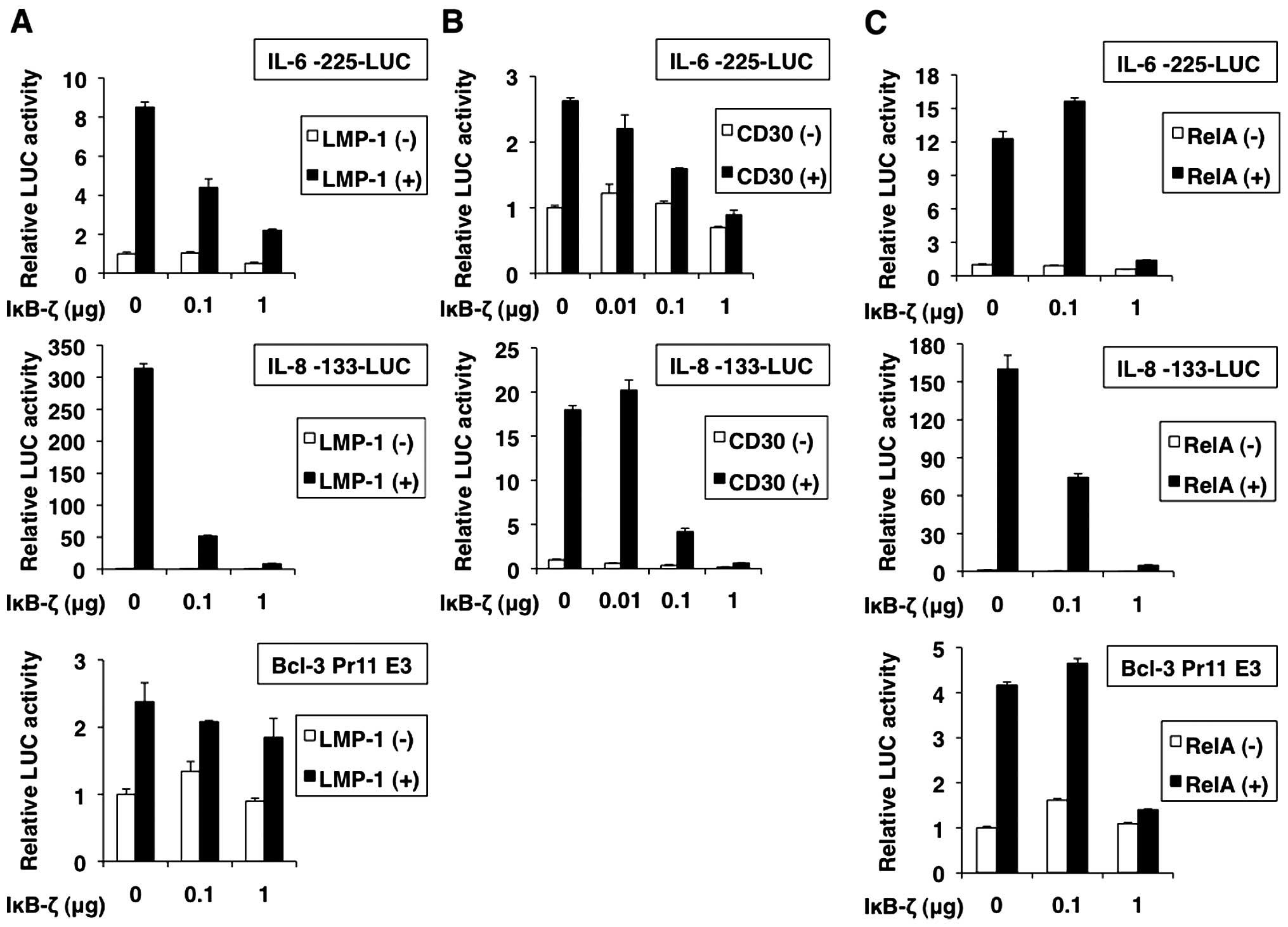
IL-6, IL-8 and Bcl-3 genes are known to be
activated by NF-κB (35–37). Luciferase reporter analyses
indicated that promoters of IL-6, IL-8 and Bcl-3 were activated by
transfection of LMP-1, CD30 and RelA as expected (Fig. 8A–C). Cotransfection of IκB-ζ
dose-dependently inhibited the LMP-1-, CD30- and RelA-induced
activation of promoters of IL-6, IL-8 and Bcl-3. In addition, we
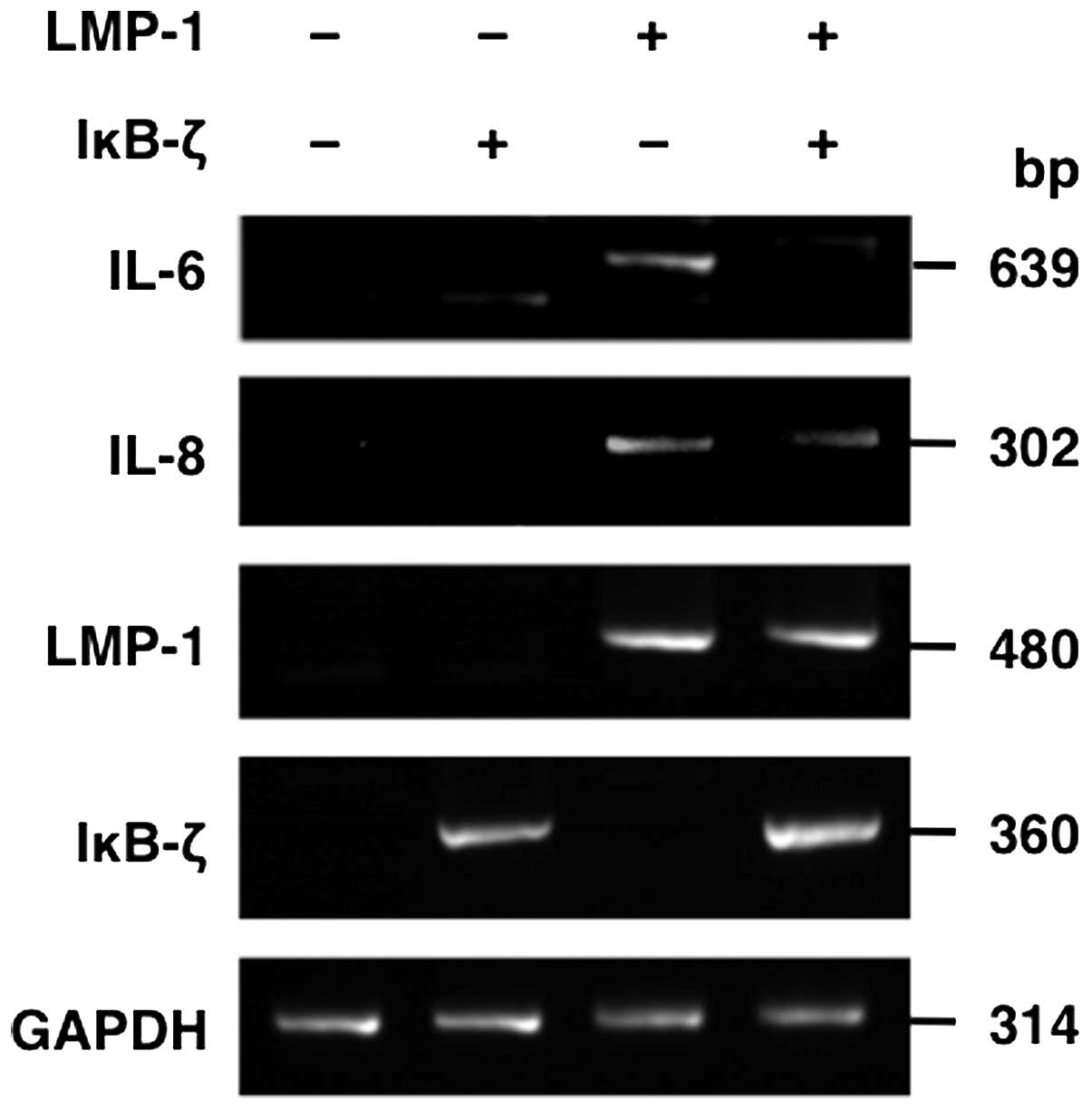
analyzed whether IκB-ζ overexpression leads to downregulation of
known NF-κB targets on mRNA level. To this end, we determined the
effect of IκB-ζ overexpression on IL-6 and IL-8 mRNA expression in
the presence of LMP-1 by RT-PCR. These analyses demonstrated that
IL-6 and IL-8 mRNA levels were downregulated after IκB-ζ
overexpression (Fig. 9),
suggesting that IκB-ζ plays a role in negatively regulating NF-κB
targets.
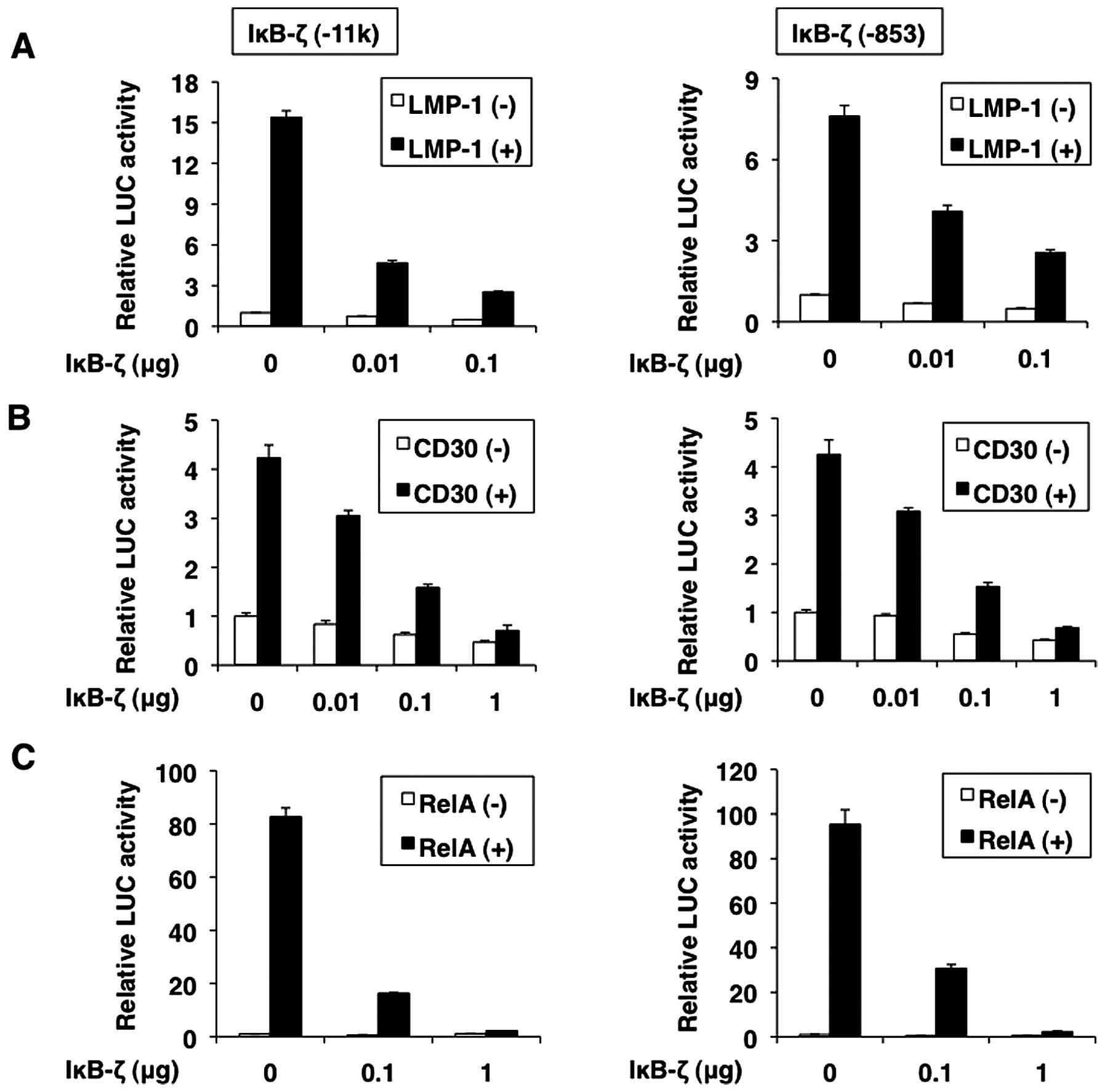
We also analyzed the effect of IκB-ζ overexpression
on the activation of its promoter induced by LMP-1, CD30 and RelA.
LMP-1-, CD30- and RelA-induced IκB-ζ promoter activation was
dose-dependently repressed by IκB-ζ over-expression (Fig. 10A–C). Thus, IκB-ζ can repress its
own transcription. IκB-ζ expression itself was regulated by NF-κB,
suggesting that its activity is controlled through a negative
feedback loop.
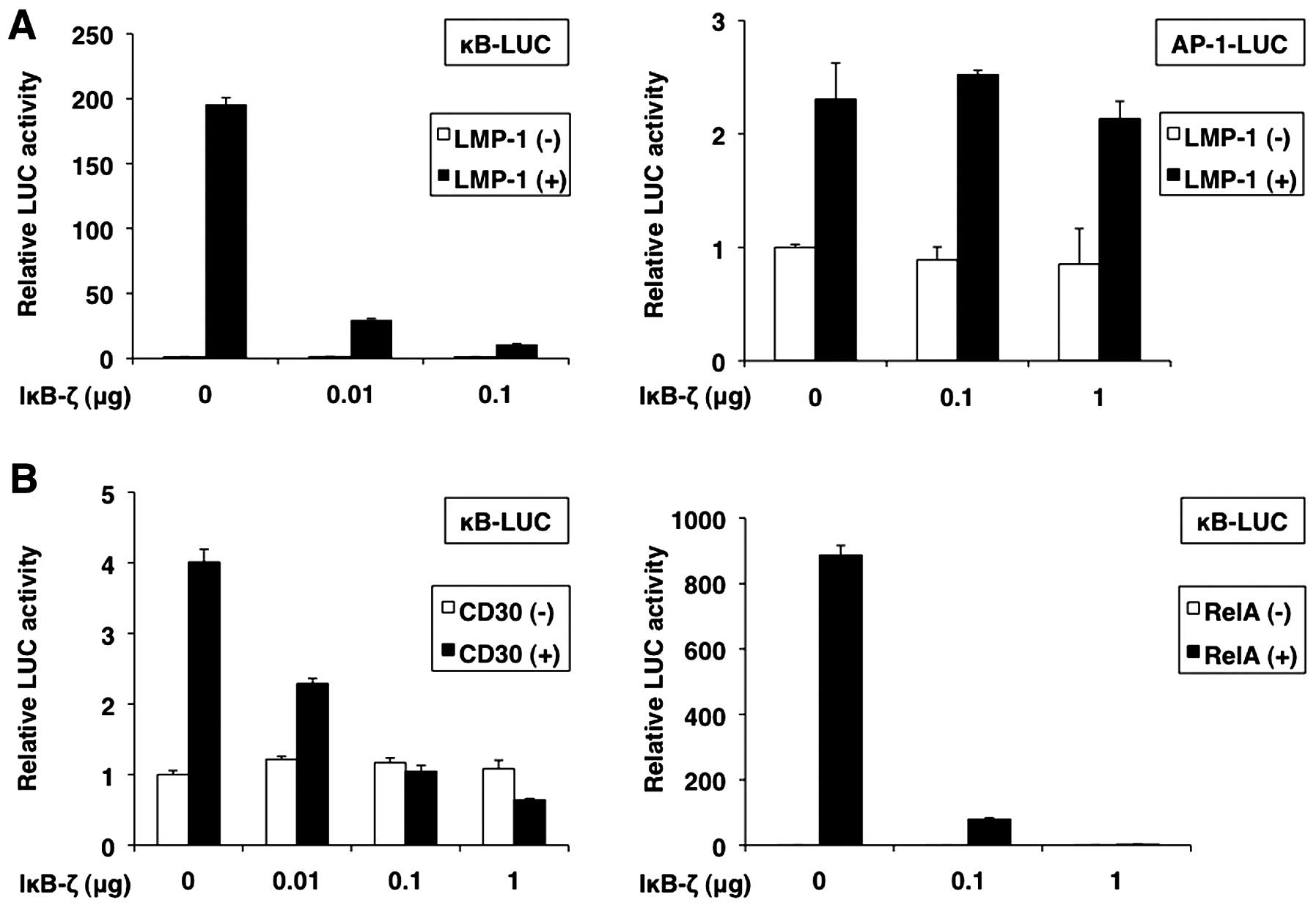
To confirm the role of IκB-ζ in NF-κB activity, we
transfected 293T cells with IκB-ζ, and LMP-1, CD30 or RelA, and
measured the activity of κB-LUC, an NF-κB reporter construct. As
expected, we found that NF-κB reporter activity induced by LMP-1,
CD30 and RelA was repressed by IκB-ζ overexpression in a
dose-dependent manner (Fig. 11A,
left panel, and 11B). However, the results showed that IκB-ζ did
not affect AP-1 reporter activity induced by LMP-1 (Fig. 11A, right panel).
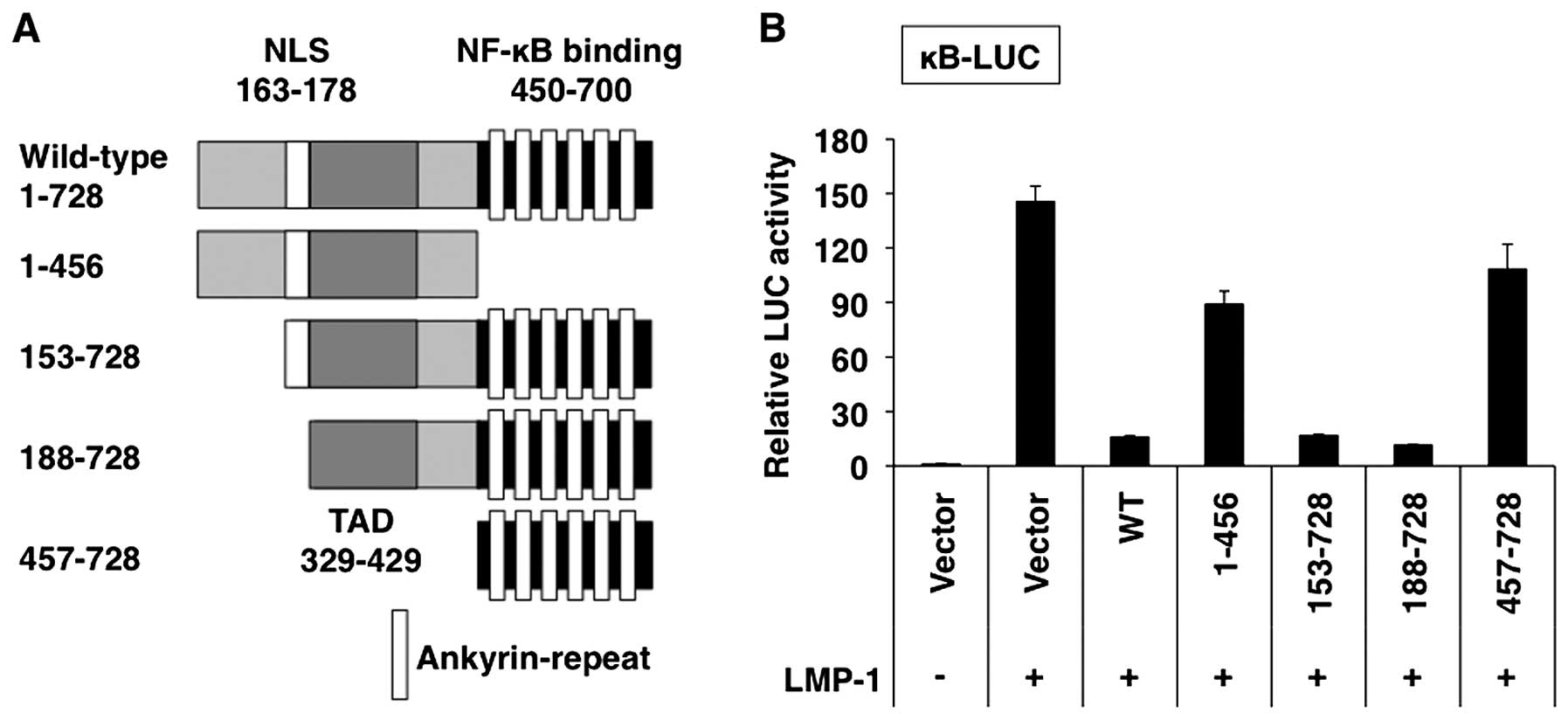
Mutants of IκB-ζ truncated from the amino- and
carboxyl-termini were expressed in the presence of LMP-1 in 293T
cells, and the NF-κB reporter activity was measured (Fig. 12A). The amino-terminal truncated
mutants (153–728 and 188–728) as well as the full-length IκB-ζ
(1–728), showed inhibitory activities against LMP-1-induced NF-κB
activation, whereas the mutants consisting of the amino-terminus to
amino acid 456 (1–456) and the amino-terminal truncated mutant
(457–728) exhibited less activity than the full-length IκB-ζ
(1–728) (Fig. 12B). These results
indicate that the region between amino acids 188–728 harbors a
domain with transcriptional inhibitory activity.
Discussion
Constitutive activation of the oncogenic NF-κB
pathway is a characteristic hallmark of several lymphoma subtypes
(1). EBV LMP-1 and CD30 have been
demonstrated to activate the NF-κB signaling pathways in lymphomas
(1). It has become increasingly
clear that activation of NF-κB is not only controlled in the
cytoplasm but, presumably even more importantly, also modulated in
the nucleus. The nuclear IκB family member IκB-ζ acts a
multifaceted modulator of NF-κB activity (20). We demonstrated high IκB-ζ
expression in LMP-1-expressing BL and CD30-expressing HL cell
lines. Nuclear IκB-ζ expression was also shown in lymph nodes from
patients with BL and HL by immunohistochemical staining. In
contrast, normal lymph nodes did not express IκB-ζ (23). Due to the potential significance of
these observations on the two lymphoma types, we investigated the
transcriptional basis for LMP-1- and CD30-induced IκB-ζ expression.
Our results demonstrated that LMP-1 and CD30 activate IκB-ζ
transcription primarily through two NF-κB sites in its promoter.
The TRAF/NIK/IKK pathway also contributed to the activation of the
IκB-ζ promoter as shown by the use of dominant-negative constructs.
These data provide the molecular basis for the observed LMP-1- and
CD30-induced overexpression of IκB-ζ (Fig. 13).
We next considered the consequence of IκB-ζ
overexpression in BL and HL cells. The results showed that IκB-ζ
potently repressed the LMP-1- and CD30-induced NF-κB activation in
a negative feedback loop, suggesting the presence of an NF-κB-IκB-ζ
autoregulatory loop (Fig. 13).
IκB-ζ associates with the NF-κB subunit p50 and IκB-ζ inhibits the
DNA binding of the RelA/p50 heterodimer and the p50/p50 homodimer
(19). Negative autoregulatory
loop provides an effective mechanism for the control of NF-κB
activation. The inhibitory roles of the negative autoregulatory
loop on NF-κB-mediated transcription may be critical in fine tuning
the balance between activators and suppressors of tumors to
maintain lymphoma in vivo. The relatively high frequency of
expression of another nuclear IκB family protein, Bcl-3, was
reported in some lymphoma types (42). Like IκB-ζ, Bcl-3 is a multifaceted
modulator of the NF-κB activity and has multiple functions
(43). Because the ankyrin-repeats
of IκB-ζ are homologous to that of Bcl-3 (20), Bcl-3 may act as a competitor for
IκB-ζ, or vice versa. Appropriate cellular responses are regulated
by the control of precise balance between accelerators, brakes and
steering wheels to maintain homeostasis following environmental
change (20). Elucidation of the
precise mechanism that determines the atypical nuclear IκB family
effects should be paramount to our understanding of the role of
NF-κB family in lymphomas.
Acknowledgements
We thank Dr Ryuichiro Kimura for excellent
assistance and discussion. We express our gratitude to Dr Tatsushi
Muta for providing the expression vectors for IκB-ζ and its
mutants, and reporter plasmids for IκB-ζ. We also thank Drs Martin
Rowe, Toshiki Watanabe, Lionel Larue, Dean W. Ballard, Romas
Geleziunas, Kuan-Teh Jeang, Jun-Ichi Fujisawa, Ken-Ichi Yamamoto,
Naofumi Mukaida, Timothy W. McKeithan for providing expression
vectors for LMP-1 and its mutants; expression vectors for CD30 and
its mutant, and TRAF2-dominant-negative mutant; expression vectors
for RelA; for IκBα- and IκBβ-dominant-negative mutants; for NIK-,
IKKα- and IKKβ-dominant-negative mutants; for IKKγ-dominant
negative mutant; reporter plasmids for NF-κB; for IL-6; for IL-8
and AP-1; and for Bcl-3. We acknowledge Dr Takeshi Sairenji for
providing B95–8/Ramos. The present study was supported in part by
JSPS KAKENHI grant nos. 90542358 and 25461428.
References
|
1
|
Gasparini C, Celeghini C, Monasta L and
Zauli G: NF-κB pathways in hematological malignancies. Cell Mol
Life Sci. 71:2083–2102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carbone A, Tripodo C, Carlo-Stella C,
Santoro A and Gloghini A: The role of inflammation in lymphoma. Adv
Exp Med Biol. 816:315–333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
4
|
Horie R and Watanabe T: The biological
basis of Hodgkin's lymphoma. Drug News Perspect. 16:649–656. 2003.
View Article : Google Scholar
|
|
5
|
Vockerodt M, Yap L-F, Shannon-Lowe C,
Curley H, Wei W, Vrzalikova K and Murray PG: The Epstein-Barr virus
and the pathogenesis of lymphoma. J Pathol. 235:312–322. 2015.
View Article : Google Scholar
|
|
6
|
Kaye KM, Izumi KM and Kieff E:
Epstein-Barr virus latent membrane protein 1 is essential for
B-lymphocyte growth transformation. Proc Natl Acad Sci USA.
90:9150–9154. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Devergne O, Cahir McFarland ED, Mosialos
G, Izumi KM, Ware CF and Kieff E: Role of the TRAF binding site and
NF-kappaB activation in Epstein-Barr virus latent membrane protein
1-induced cell gene expression. J Virol. 72:7900–7908.
1998.PubMed/NCBI
|
|
8
|
Izumi KM and Kieff ED: The Epstein-Barr
virus oncogene product latent membrane protein 1 engages the tumor
necrosis factor receptor-associated death domain protein to mediate
B lymphocyte growth transformation and activate NF-kappaB. Proc
Natl Acad Sci USA. 94:12592–12597. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schultheiss U, Püschner S, Kremmer E, Mak
TW, Engelmann H, Hammerschmidt W and Kieser A: TRAF6 is a critical
mediator of signal transduction by the viral oncogene latent
membrane protein 1. EMBO J. 20:5678–5691. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soni V, Cahir-McFarland E and Kieff E:
LMP1 TRAFficking activates growth and survival pathways. Adv Exp
Med Biol. 597:173–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Horie R, Watanabe T, Morishita Y, Ito K,
Ishida T, Kanegae Y, Saito I, Higashihara M, Mori S, Kadin ME, et
al: Ligand-independent signaling by overexpressed CD30 drives
NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene.
21:2493–2503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Horie R, Aizawa S, Nagai M, Ito K,
Higashihara M, Ishida T, Inoue J and Watanabe T: A novel domain in
the CD30 cytoplasmic tail mediates NFkappaB activation. Int
Immunol. 10:203–210. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arkan MC and Greten FR: IKK- and
NF-κB-mediated functions in carcinogenesis. Curr Top Microbiol
Immunol. 349:159–169. 2011.
|
|
14
|
Ryan KM, Ernst MK, Rice NR and Vousden KH:
Role of NF-kappaB in p53-mediated programmed cell death. Nature.
404:892–897. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sheehy AM and Schlissel MS: Overexpression
of RelA causes G1 arrest and apoptosis in a pro-B cell line. J Biol
Chem. 274:8708–8716. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmitt CA: Cellular senescence and cancer
treatment. Biochim Biophys Acta. 1775:5–20. 2007.
|
|
18
|
Jing H and Lee S: NF-κB in cellular
senescence and cancer treatment. Mol Cells. 37:189–195. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamazaki S, Muta T and Takeshige K: A
novel IkappaB protein, IkappaB-zeta, induced by proinflammatory
stimuli, negatively regulates nuclear factor-kappaB in the nuclei.
J Biol Chem. 276:27657–27662. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Muta T: IkappaB-zeta: An inducible
regulator of nuclear factor-kappaB. Vitam Horm. 74:301–316. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamamoto M, Yamazaki S, Uematsu S, Sato S,
Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, et
al: Regulation of Toll/IL-1-receptor-mediated gene expression by
the inducible nuclear protein IkappaBzeta. Nature. 430:218–222.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nogai H, Wenzel S-S, Hailfinger S, Grau M,
Kaergel E, Seitz V, Wollert-Wulf B, Pfeifer M, Wolf A, Frick M, et
al: IκB-ζ controls the constitutive NF-κB target gene network and
survival of ABC DLBCL. Blood. 122:2242–2250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimura R, Senba M, Cutler SJ, Ralph SJ,
Xiao G and Mori N: Human T cell leukemia virus type I tax-induced
IκB-ζ modulates tax-dependent and tax-independent gene expression
in T cells. Neoplasia. 15:1110–1124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Floettmann JE and Rowe M: Epstein-Barr
virus latent membrane protein-1 (LMP1) C-terminus activation region
2 (CTAR2) maps to the far C-terminus and requires oligomerisation
for NF-kappaB activation. Oncogene. 15:1851–1858. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huen DS, Henderson SA, Croom-Carter D and
Rowe M: The Epstein-Barr virus latent membrane protein-1 (LMP1)
mediates activation of NF-kappa B and cell surface phenotype via
two effector regions in its carboxy-terminal cytoplasmic domain.
Oncogene. 10:549–560. 1995.PubMed/NCBI
|
|
26
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-kappaB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Motoyama M, Yamazaki S, Eto-Kimura A,
Takeshige K and Muta T: Positive and negative regulation of nuclear
factor-kappaB-mediated transcription by IkappaB-zeta, an inducible
nuclear protein. J Biol Chem. 280:7444–7451. 2005. View Article : Google Scholar
|
|
28
|
Brockman JA, Scherer DC, McKinsey TA, Hall
SM, Qi X, Lee WY and Ballard DW: Coupling of a signal response
domain in I kappa B alpha to multiple pathways for NF-kappa B
activation. Mol Cell Biol. 15:2809–2818. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geleziunas R, Ferrell S, Lin X, Mu Y,
Cunningham ET Jr, Grant M, Connelly MA, Hambor JE, Marcu KB and
Greene WC: Human T-cell leukemia virus type 1 Tax induction of
NF-kappaB involves activation of the IkappaB kinase alpha
(IKKalpha) and IKKbeta cellular kinases. Mol Cell Biol.
18:5157–5165. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iha H, Kibler KV, Yedavalli VRK,
Peloponese JM, Haller K, Miyazato A, Kasai T and Jeang K-T:
Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax
and TNFalpha: Implications for stimulus-specific interruption of
oncogenic signaling. Oncogene. 22:8912–8923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McKinsey TA, Brockman JA, Scherer DC,
Al-Murrani SW, Green PL and Ballard DW: Inactivation of IkappaBbeta
by the tax protein of human T-cell leukemia virus type 1: A
potential mechanism for constitutive induction of NF-kappaB. Mol
Cell Biol. 16:2083–2090. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aizawa S, Nakano H, Ishida T, Horie R,
Nagai M, Ito K, Yagita H, Okumura K, Inoue J and Watanabe T: Tumor
necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are
involved in CD30-mediated NFkappaB activation. J Biol Chem.
272:2042–2045. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamazaki S, Muta T, Matsuo S and Takeshige
K: Stimulus-specific induction of a novel nuclear factor-kappaB
regulator, IkappaB-zeta, via Toll/Interleukin-1 receptor is
mediated by mRNA stabilization. J Biol Chem. 280:1678–1687. 2005.
View Article : Google Scholar
|
|
34
|
Suzuki T, Hirai H, Murakami T and Yoshida
M: Tax protein of HTLV-1 destabilizes the complexes of NF-kappa B
and I kappa B-alpha and induces nuclear translocation of NF-kappa B
for transcriptional activation. Oncogene. 10:1199–1207.
1995.PubMed/NCBI
|
|
35
|
Okamoto S, Mukaida N, Yasumoto K, Rice N,
Ishikawa Y, Horiguchi H, Murakami S and Matsushima K: The
interleukin-8 AP-1 and kappa B-like sites are genetic end targets
of FK506-sensitive pathway accompanied by calcium mobilization. J
Biol Chem. 269:8582–8589. 1994.PubMed/NCBI
|
|
36
|
Shimizu H, Mitomo K, Watanabe T, Okamoto S
and Yamamoto K: Involvement of a NF-kappa B-like transcription
factor in the activation of the interleukin-6 gene by inflammatory
lymphokines. Mol Cell Biol. 10:561–568. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ge B, Li O, Wilder P, Rizzino A and
McKeithan TW: NF-kappa B regulates BCL3 transcription in T
lymphocytes through an intronic enhancer. J Immunol. 171:4210–4218.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mori N and Prager D: Transactivation of
the interleukin-1alpha promoter by human T-cell leukemia virus type
I and type II Tax proteins. Blood. 87:3410–3417. 1996.PubMed/NCBI
|
|
39
|
Horie R and Watanabe T, Ito K, Morisita Y,
Watanabe M, Ishida T, Higashihara M, Kadin M and Watanabe T:
Cytoplasmic aggregation of TRAF2 and TRAF5 proteins in the
Hodgkin-Reed-Sternberg cells. Am J Pathol. 160:1647–1654. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sylla BS, Hung SC, Davidson DM,
Hatzivassiliou E, Malinin NL, Wallach D, Gilmore TD, Kieff E and
Mosialos G: Epstein-Barr virus-transforming protein latent
infection membrane protein 1 activates transcription factor
NF-kappaB through a pathway that includes the NF-kappaB-inducing
kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc Natl Acad
Sci USA. 95:10106–10111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Canoz O, Rassidakis GZ, Admirand JH and
Medeiros LJ: Immunohistochemical detection of BCL-3 in lymphoid
neoplasms: A survey of 353 cases. Mod Pathol. 17:911–917. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Palmer S and Chen YH: Bcl-3, a
multifaceted modulator of NF-kappaB-mediated gene transcription.
Immunol Res. 42:210–218. 2008. View Article : Google Scholar : PubMed/NCBI
|