Introduction
Ionizing radiation (IR) is the primary therapy for
nasopharyngeal carcinoma (NPC). Though most NPC cells are sensitive
to IR, there are still some tumor cells which are resistant to
irradiation. These radioresistant cancer cells often result in
local recurrence and metastasis after radiotherapy (1). Thus, finding drugs that can enhance
NPC therapy is of great significance.
In recent years, new drugs that target tyrosine
kinase have made a breakthrough in tumor therapy (2). Because targeted therapy is highly
specific and causes fewer side effects than chemotherapy, targeted
therapy has become increasingly widely accepted by patients and
doctors (3,4). To date a number of different kinds of
tyrosine kinase inhibitors (TKIs) have been approved by the Food
and Drug Administration (FDA), such as Imatinib (Gleevec),
Gefitinib (Iressa), Erlotinib (Tarceva), and Sorafenib (Nexavar).
In addition, there are still many other TKIs undergoing different
stages of preclinical and clinical trials (5).
CK2 is an important Ser/Thr kinase that is expressed
in most eukaryotes. The CK2 complex is a tetramer composed of two
catalytic subunits (αα or αα′) and two regulatory subunits (ββ)
(6). Using ATP and GTP as
phosphate group donors, CK2 phosphorylates the ser/thr residues of
its substrates. Substrates of CK2 such as nuclear factor-κB
(NF-κB), Dsh/Dvl, the product of the adenomatous polyposis coli
gene, transcription elements Lef/Tcf, engrailed, and β-catenin are
involved in signal transduction, DNA replication, transcription,
translation and many other important events (7). Therefore, CK2 plays a vital role in
cell growth, proliferation, differentiation, apoptosis and is
considered to be a potential target for regulation of some cell
processes such as cell cycle distribution, apoptosis and DNA damage
repair (8–10). Some studies have found that CK2
inhibitors can inhibit cancer cell growth and promote apoptosis
(10–12). Further research indicated that CK2
took part in DNA damage repair by affecting the binding of DNA-PKcs
and DNA (13). Lin et al
found that inhibiting CK2 activity increased radiosensitivity of
non-small cell lung cancer (NSCLC) cells through decreasing stat3
activation (14). These studies
suggest that CK2 may be a potential target to enhance
radiosensitivity of tumor cells, including NPC cells.
Traditional CK2 inhibitors include TBB, TBI/TBBz,
and DMAT. However, the selectivity of these inhibitors was not
found to be as narrow as it was originally believed (15). Therefore, new CK2 inhibitors have
been investigated. Quinalizarin is an ATP-site competitive CK2
inhibitor. It is highly selective and cell-permeable and the
selectivity of quinalizarin toward CK2 is superior to any other CK2
inhibitor known so far (16,17).
Thus, we used quinalizarin as a CK2 selective inhibitor in this
study and assessed its influence on NPC radiosensitivity.
SHP-1, also named PTPN6, PTP1C, HCP, HCPH, HPTP1C,
SH-PTP1 (18), is an SH2
domain-containing protein tyrosine phosphatase (PTP) consisting of
17 exons and 16 introns that spans ~17 kb of DNA (19,20).
SHP-1 shows high expression in normal hematopoietic cells and low
expression in many hematological malignancies (21), including Burkitt lymphomas
(22), natural killer cell
lymphoma (23), diffuse large cell
lymphoma, follicular lymphoma, Hodgkin's disease, mantle cell
lymphoma, peripheral T-cell lymphoma, adult T-cell lymphoma and
leukemia, plasmacytoma (24), and
chronic myeloid leukemia (25).
Thus, SHP-1 is traditionally regarded as a tumor suppressor.
However, some studies have found that SHP-1 is highly expressed in
some epithelial carcinoma cells, such as certain types of ovarian
cell lines and breast cell lines (26). Despite considerable research into
SHP-1 in hematological tumors, the functions of SHP-1 in solid
tumors, especially in NPC, are poorly understood. Our previous
investigation found that SHP-1 was highly expressed in NPC tissues
in contrast to normal nasopharyngeal mucosa and was associated with
local recurrence and metastasis after radiotherapy in NPC patients
(27). Knocking down SHP-1 by
siRNA in the NPC cell line CNE-2 and the non-small cell lung
carcinoma (NSCLC) cell line A549 enhanced radiosensitivity, which
was an outcome of cell cycle redistribution (28,29).
In this study, we used quinalizarin as a CK2
specific inhibitor and explored its impact on NPC cell
radiosensitivity and we also investigated its influence on NPC cell
proliferation, apoptosis, cell cycle distribution and DNA damage
repair. In addition, we explored how quinalizarin affects the
expression of SHP-1.
Materials and methods
Cell culture and irradiation
procedure
Human nasopharyngeal carcinoma cell lines CNE-1 and
CNE-2 were obtained from the Cell Bank of Sun Yatsen University
(Guangzhou, China). The nasopharyngeal carcinoma cell line 5–8F was
obtained from the Cell Bank of The Second Xiangya Hospital of
Central South University (Hunan, China). Cells were routinely
cultured in RPMI-1640 (Hyclone, USA) medium supplemented with 12%
fetal bovine serum (Hyclone), and 1% penicillin/streptomycin
(Hylcone). The cells were maintained at 37°C in a humidified
incubator with 5% CO2 and 95% air.
Irradiation was performed at room temperature with a
single dose of X-rays ranging from 2 to 8 Gy using a linear
accelerator (Primus K, Siemens, Munich, Bayern, Germany) with 6 MV
photons/100 cm focus-surface distance with a dose rate of 2.0
Gy/min.
Establishment of radioresistant
nasopharyngeal carcinoma cell sublines
Radioresistant NPC cells were established according
to a previously published method (27). Exponentially growing CNE-1 and
CNE-2 cells were irradiated with a dose of 6 Gy × 5. There was a
7–9-day break between the the doses. The surviving sublines (CNE-1R
and CNE-2R clones) were then passaged for three months and their
radiosensitivity was determined (Table
I).
 | Table IParameters of radiosensitivity in the
three cell lines treated with different concentrations of
quinalizarin. |
Table I
Parameters of radiosensitivity in the
three cell lines treated with different concentrations of
quinalizarin.
| Quinalizarin
concentration | CNE-1 | CNE-2 | 5–8F |
|---|
| 0 μM |
| N | 1.33 | 1.63 | 1.66 |
| SF2 | 0.47 | 0.53 | 0.58 |
| D0 | 2.08 | 2.02 | 2.25 |
| Dq | 1.02 | 1.19 | 1.32 |
| 12.5 μM |
| N | 1.24 | 2.07 | 1.65 |
| SF2 | 0.44 | 0.56 | 0.55 |
| D0 | 2.01 | 1.79 | 2.09 |
| Dq | 0.92 | 1.31 | 1.24 |
| 25 μM |
| N | 1.41 | 1.19 | 1.23 |
| SF2 | 0.34 | 0.31 | 0.33 |
| D0 | 1.48 | 1.53 | 1.55 |
| Dq | 0.74 | 0.60 | 0.64 |
| 50 μM |
| N2.12 | 1.65 | 1.14 | |
| SF2 | 0.22 | 0.17 | 0.17 |
| D0 | 0.91 | 0.88 | 1.06 |
| Dq | 0.66 | 0.38 | 0.18 |
| 100 μM |
| N | 0.46 | 1.09 | 0.88 |
| SF2 | 0.09 | 0.09 | 0.04 |
| D0 | 1.19 | 0.79 | 0.67 |
| Dq | −0.59 | −0.15 | −0.53 |
MTT assay
Cells (3,000–4,000 cells/well) were seeded into
96-well culture dishes, the next day they were treated with
quinalizarin (Merck, Germany) dissolved in DMSO at 50 mM then
diluted in RPMI-1640 medium at 0 (0.05% DMSO), 25, 50 and 100 μM.
The plates were then incubated for 24, 48 and 72 h, respectively.
MTT (20 μl) (Sigma, St. Louis, MO, USA) (5 mg/ml) was added to the
wells and incubated in the dark for 4 h. The culture media was
removed, and 150 μl DMSO added then the plates were slowly shaken
for 15 min. The OD value at 490 nm test wavelength and 630 nm
reference wavelength was measured with a microplate reader system
(Bio-Tek, USA). Cell viability rate = OD experimental
group/OD control group.
Colony formation assay
Cells were seeded into 6-well culture dishes, at
cell densities of 200, 300, 600, 1,500, and 4,000 cells/well, for
the 0, 2, 4, 6 and 8 Gy experiments, respectively, but with the
same cell density between 25 μM quinalizarin and 0.05% DMSO groups.
The cells were treated with 25 μM quinalizarin or 0.05% DMSO the
next day for 24 h and then irradiated at the different doses (0, 2,
4, 6, and 8 Gy) the radiotherapy time was based on the dose at 2
Gy/min. Plates were incubated for ~14 days, fixed with methanol,
stained with Giemsa, and colonies containing ≥50 cells were counted
as a clone. The multi-target click model was used to describe the
survival fraction. SF = 1 − (1-e−D/D0)N (SF,
cell survival fraction; D, radiation dose; e, the bottom of the
natural logarithm; D0, the mean death dose; N,
extrapolate number) was used to fit cell survival curves. The
sensitization enhancing ratio (SER) was calculated as a ratio of
D0. N reflects the ability of cells to repair damage
caused by radiation, if N increases, the dose required to kill the
cells increases. Dq represents the quasi-threshold amount required
for cell damage, as Dq increases the cell survival curve has a
widened shoulder area and enhanced radiation resistance.
Immunofluorescent assay (IFA)
Cells (5,000 cells/well) were treated with 25 μM
quinalizarin or 0.05% DMSO for 24 h then irradiated with a dose of
2 Gy for 0.5, 3, 6 and 24 h. At specific times after IR, cells were
harvested, and fixed in 4% paraformaldehyde for 15 min. After being
washed with PBS three times, the cells were permeabilized with
Triton X-100 for 15 min on ice. After being washed with PBS again
three times, the cells were blocked with 5% BSA for 30 min at room
temperature (RT), and then immunostained with anti-γ-H2AX (Abcam,
Cambridge, UK) at 4°C overnight. After further washes with PBS
three times, the cells were incubated with Dylight 488 labeled
secondary antibody (EarthOx Life Sciences, Millbrae, CA, USA) for 1
h, then washed with PBS three times, the nucleus was stained with
Hoechst 33258 (Wuhan Google Biological Technology Co., Ltd., China)
for 15 min at RT. Photographs were captured by Olympus Laser
scanning confocal microscopy (Olympus Optical Co., Tokyo, Honshu,
Japan). For each treatment condition, γ-H2AX foci number per cell
were counted in ≥50 cells under confocal microscopy with high
magnification (x800) by two independent reviewers who were blinded
to the grouping.
Flow cytometric analysis of the cell
cycle
Cells treated with 25 μM quinalizarin or 0.05% DMSO
for 24 h were then irradiated with 6 Gy X-rays. The cells were
harvested 24 h after IR, fixed overnight with 70% ethanol, then
resuspended in PBS containing 1 mg/ml RNase A (Sigma) and 50 μg/ml
propidium iodide (Sigma). The cellular DNA content was determined
on a flow cytometer FACScan (Becton-Dickinson, San Jose, CA, USA).
Quantifications of cells in the G0/G1, S, G2/M phases were
performed using CellQuest software (BD).
TUNEL analysis of apoptosis
Cells (5×105 cells/well) were seeded into
6-well plates, treated with 0.05% DMSO or 25 μM quinalizarin for 24
h and then irradiated with X-rays (6 Gy), and 24 h after IR, cells
were fixed with 4% paraformaldehyde, treated with Triton X-100. The
assay was performed according to the manufacturer's instructions of
the TUNEL kit (Roche Applied Science, Shanghai, China). Apoptosis
were observed by fluorescence microscopy (Olympus IX71, Olympus
Optical Co., Tokyo, Japan). Cells with green fluorescence were
regarded as apoptotic cells.
Overexpression of SHP-1
CNE-2 cells were planted at 24-well plates, and
incubated with 0.5 ml RPMI-1640 supplemented with 15%
heat-inactivated fetal bovine serum, 1% penicillin/streptomycin
(Hyclone). After 24-h incubation, 50 μl/well lentivirus-mediated
SHP-1 overexpression (8.6×109 copies/ml) (GeneCopoeia
Inc. Guangzhou, China) was added into the CNE-2 cells for 48 h. The
stably transfected cells were selected using medium containing 2
μg/ml puromycin (supplemented with 10% heat-inactivated fetal
bovine serum, penicillin/streptomycin) for 12 days. Positive clones
were obtained and screened by RT-PCR for transfection efficiency.
The medium containing puromycin was changed once every three
days.
Quantitative real-time RT-PCR
Total RNA was extracted by TRIzol (Invitrogen,
Carlsbad, NM, USA) and reverse transcription was used to obtain
cDNA, according to the Takara RT-PCR kit manufacturer's
instructions (Takara, Japan). Primer sequences were as follows:
SHP-1: forward, 5′-ACCAT CATCCACCTCAAGTACC-3′; reverse,
5′-CTGAGCACAGA AAGCACGAA-3′; β-actin: forward, 5′-GATGAGATTGGCA
TGGCTTT-3′; reverse, 5′-CACCTTCACCGTTCCAGTTT-3′. Then qPCR was
performed according to the SYBR Green manufacturer's instructions
(ABI, USA) in a PCR thermocycler (ABI PRISM 7000, USA) using 95°C
for 30 sec, then 95°C for 5 sec, 60°C for 35 sec, 95°C for 15 sec,
60°C for 1 min, and 95°C 15 sec, for 35 cycles. StepOne™ software
v2.1 was used to analyze the data. The Ct
value was defined as the number of PCR cycles in which the
fluorescence signal exceeded the detection threshold value. First,
ΔCt = Ct Gene − Ctβ-actin. Then, ΔΔCt = ΔCt treated − ΔCt control.
Lastly, 2−ΔΔCt was calculated to represent the relative
mRNA expression of target genes. β-actin was used as an internal
control.
Western blot analysis
Cells were harvested and lysed in RIPA buffer (Wuhan
Google Biological Technology Co., Ltd.). Protein concentrations of
the lysates were determined by the BCA protein assay system
(Google, Wuhan, China). Equal amounts of protein (40–80 μg) were
separated by 8–12% SDS-PAGE, and transferred to PVDF membrane
(Millipore, Billerica, MA, USA). The membranes were blocked with 5%
BSA, and then probed with either anti-SHP-1 (Epitomics, Burlingame,
CA, USA), anti-NF-κB p65 (Cell Signaling Technology, USA),
anti-phospho-NF-κB p65 (Cell Signaling Technology), or anti-GAPDH
(Santa Cruz, Dallas, TX, USA) primary antibodies. After washing,
the membrane was incubated with the appropriate horseradish
peroxidase secondary antibody (Invitrogen) and visualized by
chemiluminescence using a chemiluminescence kit (Invitrogen) and
the specific bands were recorded on X-ray film. GAPDH protein
levels were used as a control to verify equal protein loading.
EMSA
EMSA was performed using the gel-shift assay system
using the EMSA kit (Thermo Fisher Scientific, Waltham, MA, USA).
Probe sequences of SHP-1 promoter used in EMSA were as follows:
forward, 5′-GGCCACGCCTGGGCGCT TCC-3′; reverse,
5′-GGAAGCGCCCAGGCGTGGCC-3′. To prepare DNA probes, double-stranded
DNA probes were incubated at 95°C for 5 min, and slowly cooled down
to room temperature. Concentration of probe was 0.5 μM. The
following reagents were added to the tubes and incubated at 37°C
for 30 min: ultrapure water 25 μl, 5X TdT reaction buffer 10 μl,
unlabeled oligo (1 μM) 5 μl, biotin-11-UTP (5 μM) 5 μl, diluted TdT
(2 U/μl) Ul. Then 2.5 μl EDTA (0.2 M) was added to stop the
reaction. The labeled DNA probe was collected. The nuclear extracts
were prepared according to the manual of the EMSA kit. Then, the
nuclear extracts were incubated with the p65 antibodies (Cell
Signaling Technology) for 10 min at room temperature. The
protein/DNA complexes were resolved on 4–6% polyacrylamide gels and
transferred to the membrane (Millipore, Billerica, MA, USA).
Subsequently, the complex was visualized by chemiluminescence using
a chemiluminescence kit (Invitrogen).
Dual luciferase assay
p-GL3 basic was from Transgene (Bejing, China).
Linear vector was constructed using KpnI (10 U/μl) and
XholI (10 U/μl) DNase. The SHP-1 promoter sequence was
connected to the linear vectors. DNA plasmids were transiently
transfected into NPC cells with 10 μl Lipo2000 (Invitrogen). Cells
were harvested 60 h after transfection and lysed in 100 μl 1X cell
culture by 513 reagent. Upon adjusting protein concentration, 20 μl
of each lysate was assayed in replicates for luciferase activity
according to the manufacturer's instructions (Promega, Madison, WI,
USA).
SHP-1 activity assay
Cells were treated with DMSO or quinalizarin for 0,
12 and 24 h then total protein were extracted by RIPA lysis buffer
containing 1% PMSF. Total protein was incubated with anti-SHP-1
primary antibody overnight at 4°C. Protein A-Sepharose (Merck) were
added to each sample and then incubated for 3 h at 4°C with
rotation. After immuoprecipitation reaction, the protein-antibody
complex was separated from the protein A-sepharose by
centrifugation (4°C, 3,000 rpm, 3 min). Then the complex was
resuspended with PBS. SHP-1 activity was determined by RediPlate 96
EnzChek Tyrosine Phosphatase Assay kit (R-22067, Molecular Probes,
Invitrogen) according to the manufacturer's instructions.
Statistical analysis
SPSS 17.0 for Windows (SPSS Inc., Chicago, IL, USA)
was used for statistical analysis. Experimental data were expressed
as mean ± standard deviation (SD) from at least three independent
experiments. Differences in measured variables between experimental
and control groups were assessed using t-test and ANOVA with least
significant difference (LSD). The criterion for statistical
significance was P<0.05.
Results
Quinalizarin (25 μM) enhances
radiosensitivity but has little effect on NPC cell
proliferation
To investigate the effect of quinalizarin on
radiosensitivity of NPC cells, we performed colony formation
assays. Fig. 1A indicates that
irradiation killed the tumor cells exponentially in CNE-1, CNE-2
and 5–8F. All three NPC of the cell lines showed a lower survival
fraction after being treated with quinalizarin at different IR
doses. These effects were quinalizarin dose related. D0,
Dq, N, SF2 were also generally lower in the
quinalizarin groups than that in the dimethyl sulfoxide (DMSO)
groups, which represents higher sensitivity to IR. Although at some
doses N was increased suggesting that the capacity of the cells for
repair was increased. At 100 μM quinalizarin, Dq was
negative, possibly because the drug alone was enough to kill the
cells, and the role of radiotherapy was secondary. At 25 μM overall
the enhancement ratio of quinalizarin groups were 1.41, 1.18 and
1.52 for CNE-1, CNE-2 and 5–8F, respectively.
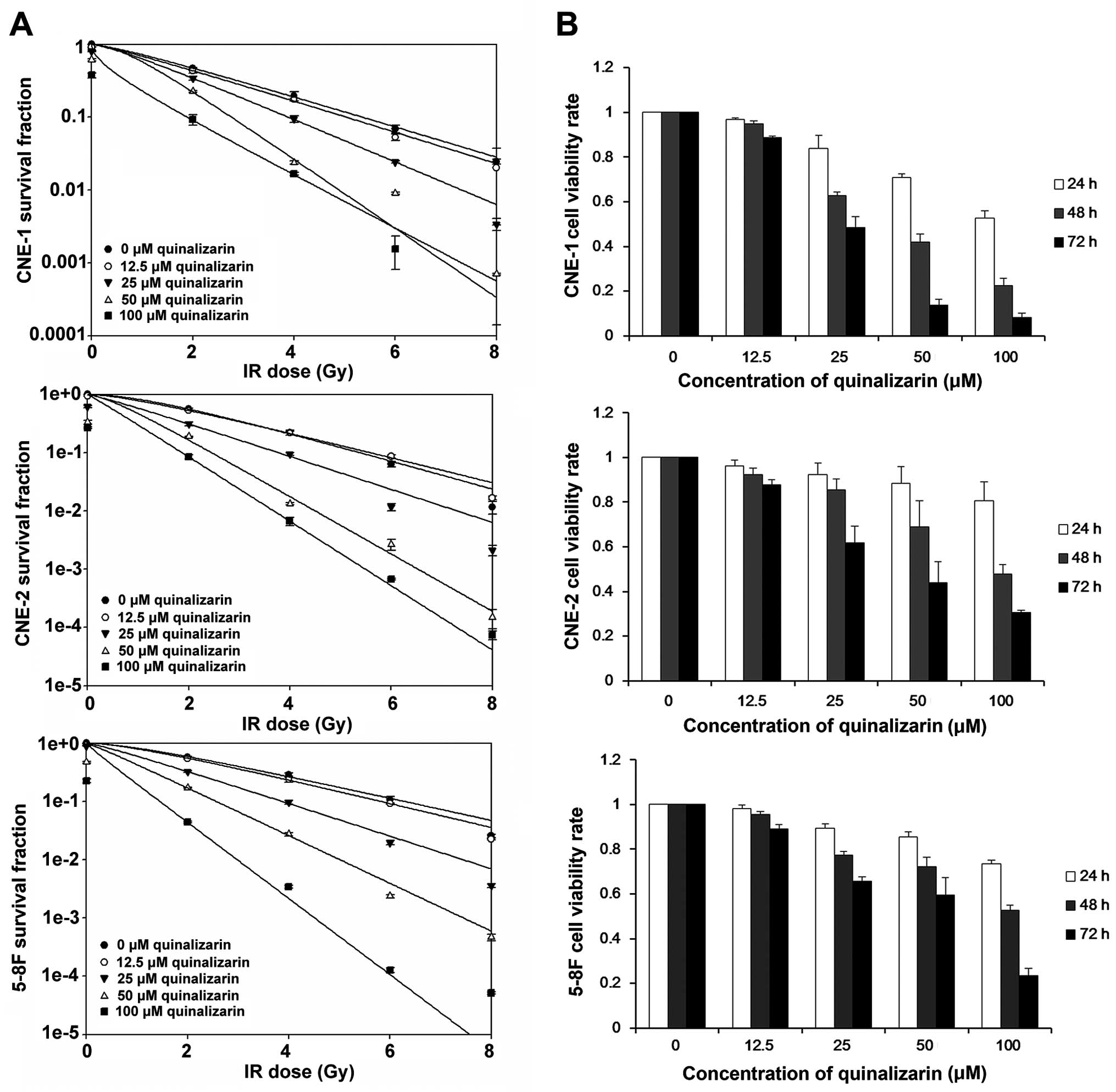 | Figure 1Effects of quinalizarin on
nasopharyngeal carcinoma (NPC) cell radiosensitivity and viability.
(A) CNE-1, CNE-2 and 5–8F cells were treated with 0, 12.5, 25, 50
and 100 μM quinalizarin for 24 h and then irradiated at the
different doses (0, 2, 4, 6 and 8 Gy) (radiation absorption rate is
2 Gy/min). After further incubation for ~14 days, clone formation
assays were used to examine radiosensitivity of the NPC cells.
Survival curves were fitted according to the multi-target
single-hit model. IR, ionizing radiation; (B) CNE-1, CNE-2 and 5–8F
cells were treated with 0, 12.5, 25, 50 and 100 μM quinalizarin for
24, 48 and 72 h, respectively. MTT assays were performed to examine
cell viability rates relative to the control group (0 μM
quinalizarin, 0.05% DMSO) (control group was normalized to 1). The
data are shown as mean ± standard deviation (SD). |
We used different concentrations of quinalizarin to
treat NPC cells for 24, 48 and 72 h, respectively. MTT assays were
performed to test cell viability. As shown in Fig. 1B, quinalizarin inhibited cell
viability in a concentration- and time-dependent manner. At 25 μM,
the inhibitory rates of all the three cell lines were <20%, at
24 h (0.839±0.0587 for CNE-1, 0.924±0.0527 for CNE-2 and
0.894±0.0209 for 5–8F compared to the normalized value of 1 for
controls). At 48 and 72 h, cell viability decreased sharply. When
concentration was increased to 50 μM, the inhibitory efficiency of
CNE-2 and 5–8F were ~20% while in CNE-1 was 50%. Consequently, we
used 25 μM quinalizarin treatment for 24 h in the later experiments
as this value enhanced radiosensitivity, but had little effect on
cell proliferation.
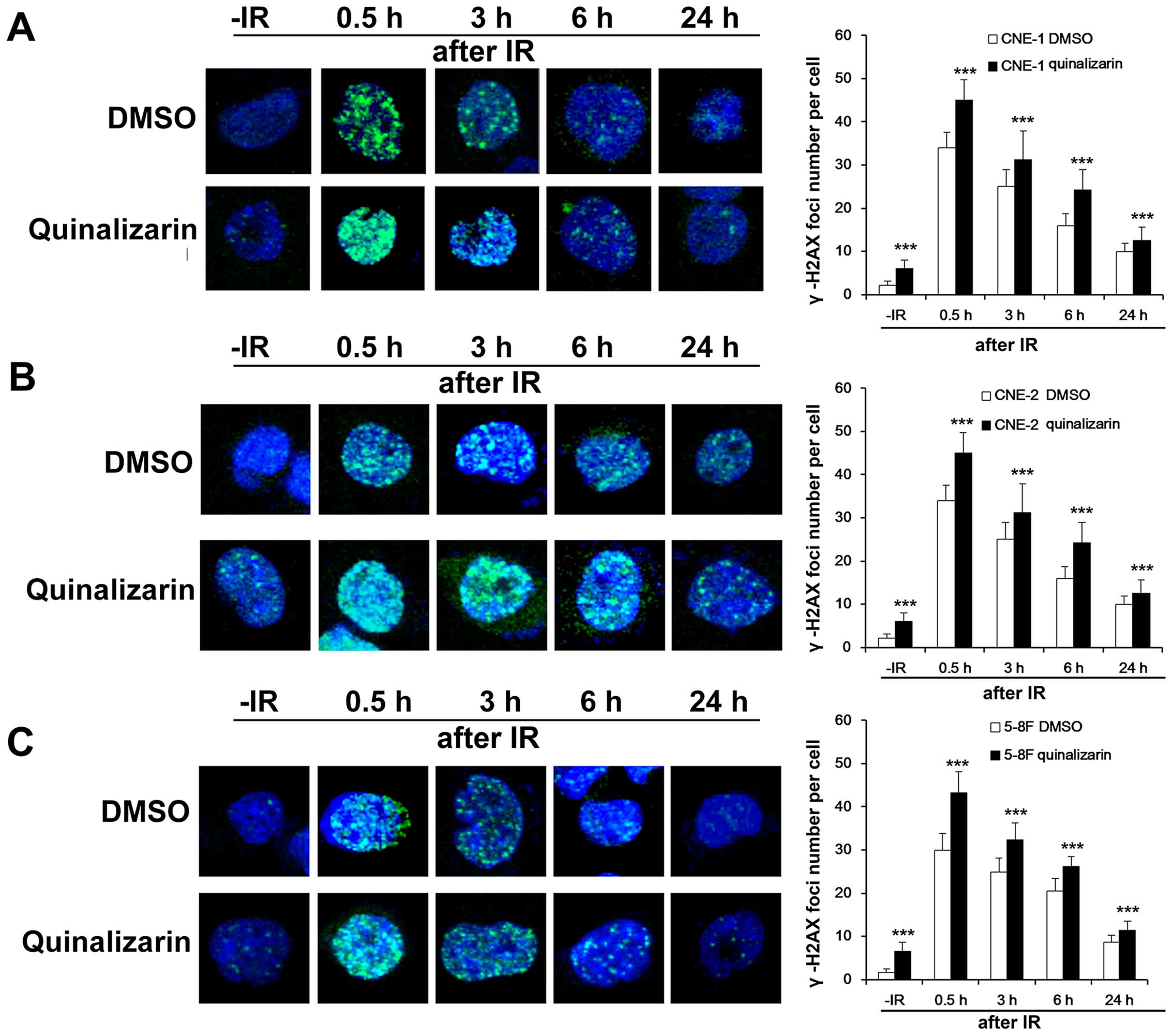
Quinalizarin slows down DSB repair
DNA double-strands break (DSB) is one of the most
important of DNA damage caused by irradiation and the ability to
repair DSB is one of the important influencing factors for
radiosensitivity. γ-H2AX foci are vital markers of DSB (1). To detect how quinalizarin influence
DSB repair in NPC cells, we used immunofluorescent assays (IFA) to
examine the formation of γ-H2AX foci in the DMSO and quinalizarin
groups before and at different times after IR. Before IR,
quinalizarin treated cells had more γ-H2AX foci than DMSO treated
cells. Not surprisingly, 0.5, 3, 6 and 24 h after IR, quinalizarin
treated cells still had more γ-H2AX foci than DMSO treated cells.
The three cell lines CNE-1, CNE-2 and 5–8F all showed similar
results and they were significantly different from the DMSO groups
at all time-points (P<0.001, Fig.
2). The results suggested that quinalizarin slowed down the
efficiency of DSB repair in NPC cells.
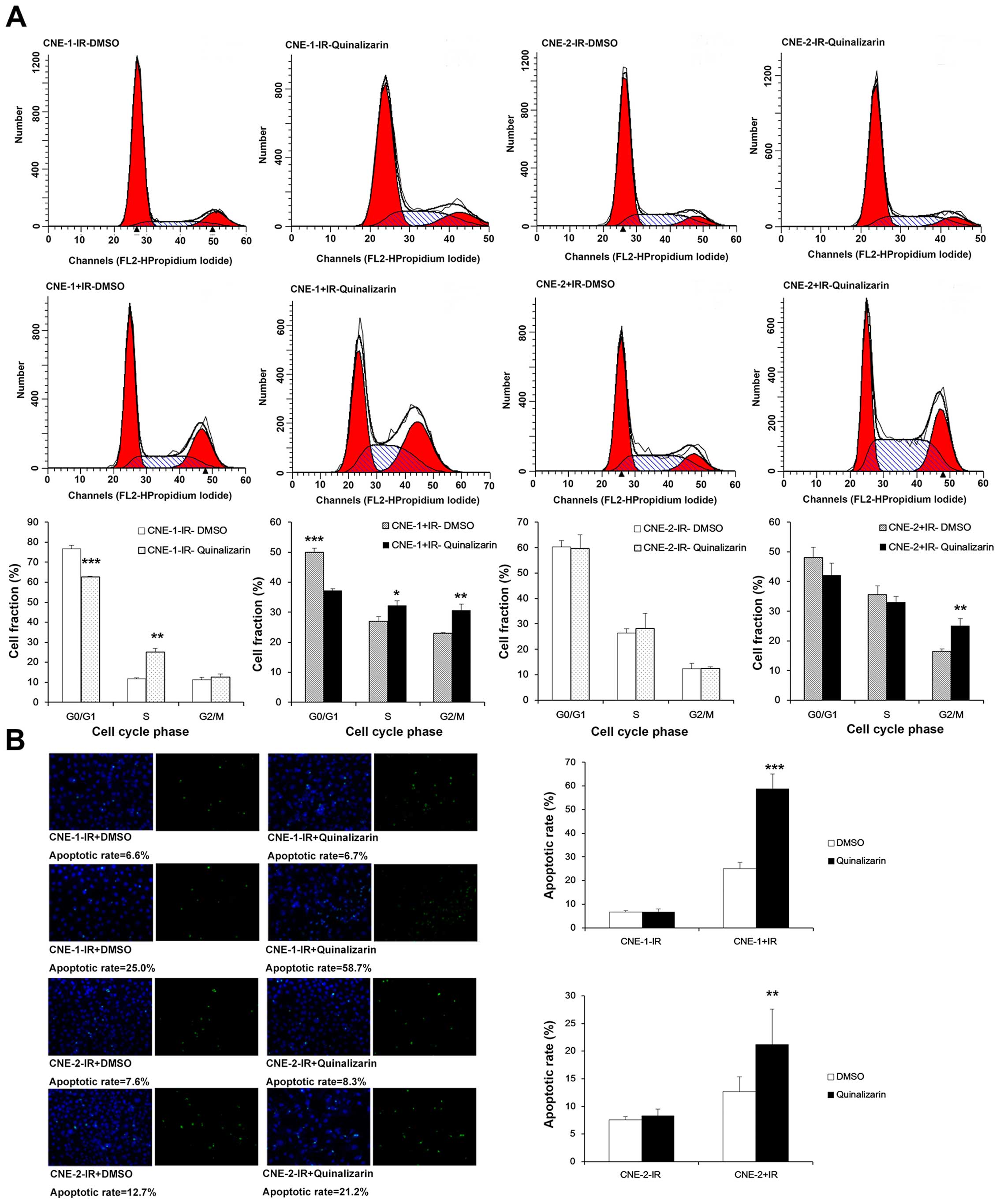
Quializarin increases the fraction of NPC
cells in G2/M phase and apoptosis
One of the mechanisms that influences
radiosensitivity is cell cycle distribution. It is well-known that
cells are most sensitive to irradiation in G2/M phase. Thus, we
used flow cytometry to detect the cell cycle distribution in the
two groups before or after IR. In CNE-1, the cell fractions in G2/M
phase were not significantly different between DMSO and
quinalizarin group before IR (11.38±1.21 vs. 12.40±1.76%,
respectively). After IR, however, cell proportion in G2/M phase was
significantly higher in quinalizarin group (23.02±0.16 vs.
30.55±2.22%, P=0.04), We also found that the change of cell cycle
distribution in CNE-2 was the same as with CNE-1. Cell fractions in
G2/M phase were not significantly different between DMSO and
quinalizarin group before IR (12.28±2.22 vs. 12.46±0.65%,
respectively). After IR, however, cells proportion in G2/M phase
was significantly higher in quinalizarin group (16.50±0.70 vs.
25.06±2.41%, P=0.004) (Fig.
3A).
To examine the effect of quinalizarin on apoptosis
before and after IR, we used TUNEL assays to determine the
apoptotic rate of NPC cells. Without irradiation by X-ray, the
apoptosis rates of the DMSO group was almost equal to the
quinalizarin group in both cell lines with rates of 6.6 and 6.7%,
respectively, in CNE-1, 7.6 and 8.3%, respectively, in CNE-2. After
irradiation by X-ray, apoptosis rates between the two groups showed
significant differences. The apoptosis rates of the quinalizarin
group were 34% higher than the DMSO group in CNE-1 and 9% higher in
CNE-2 (Fig. 3B). Based on these
results, we concluded that quinalizarin promoted apoptosis in NPC
cells after IR.
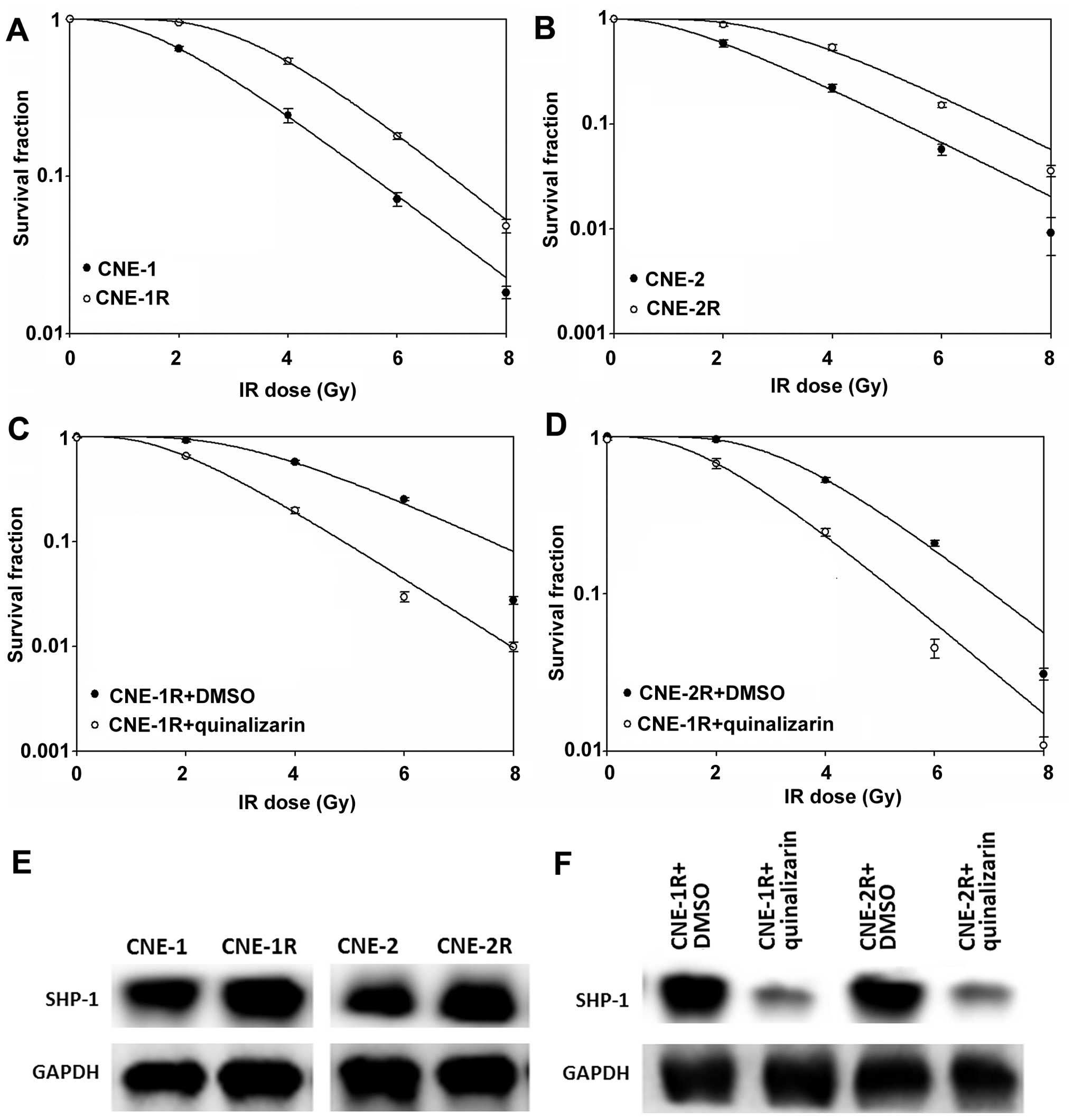
Quinalizarin suppresses SHP-1 expression
in radioresistant NPC cells
Radioresistant populations of CNE-1 and CNE-2 cells
(Fig. 4A and B) were treated with
quinalizarin and this successfully increased their radiosensitivity
(Fig. 4C and D). The levels of
SHP-1 were then evaluated in the cells by western blot analyses,
while the levels in the radioresitant populations were higher than
the parent cells (Fig. 4E), the
levels in the radioresistant cells that were then treated with
quinalizarin were obviously decreased (Fig. 4F).
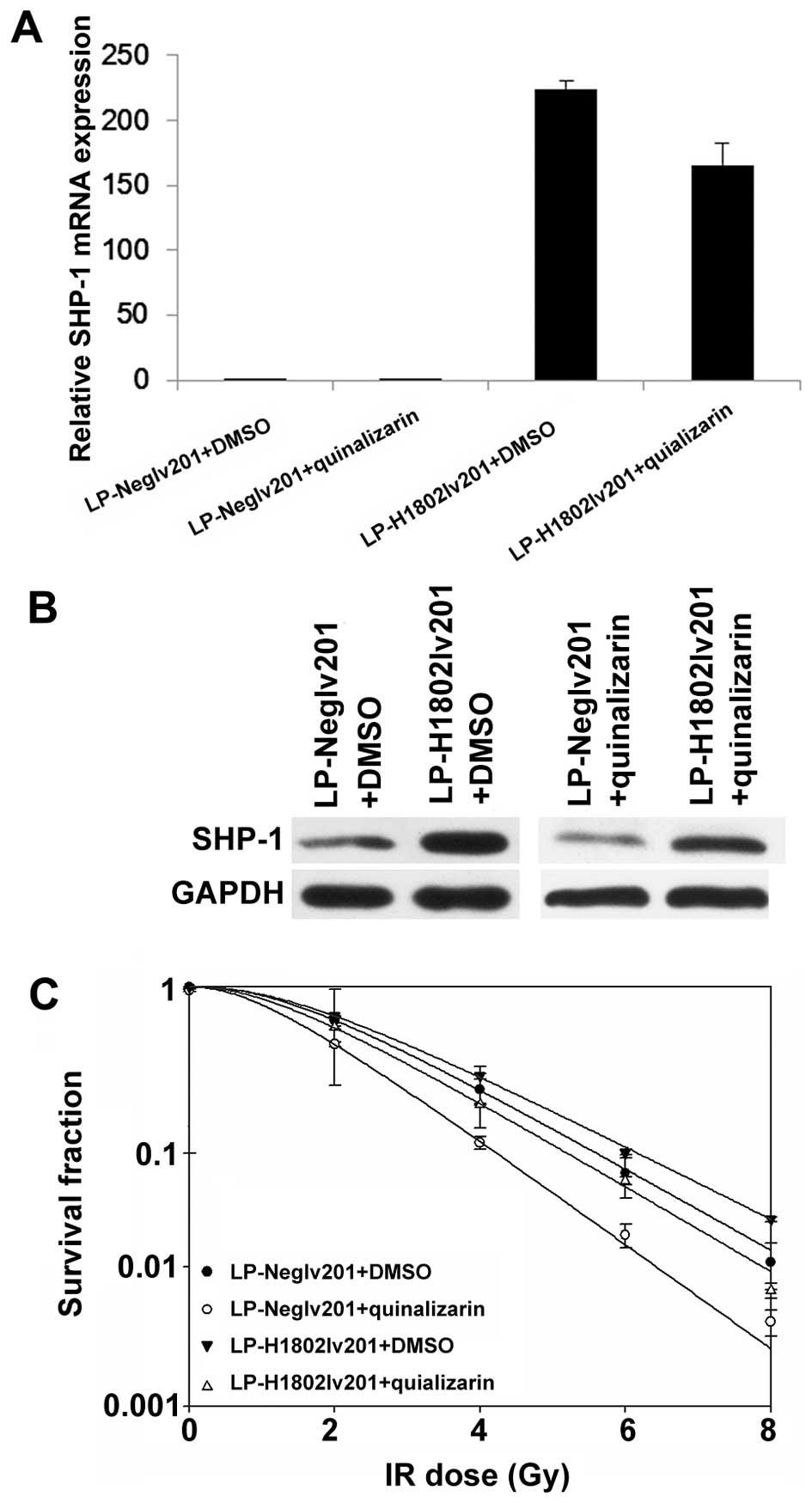
Overexpression of SHP-1 abolished the
radiosensitive effect of quinalizarin
Therefore, we thought that the enhanced
radiosensitivity acquired from quinalizarin may depend on its
downregulation of SHP-1. To verify our hypothesis, we used
lentivirus to transfect CNE-2 and produced a cell line with high
levels of SHP-1 expression (referred to as LP-H802Lv201) and a
negative control which only contained the vector (referred to as
LP-NegLv201) (Fig. 5A and B). We
then performed colony formation assays to test the effect of
quinalizarin on radiosensitivity of LP-Neglv201 and LP-H1802Lv201.
Survival curves showed that quinalizarin treated LP-NegLv201 had a
smaller shoulder area than DMSO treated LP-NegLv201, representing
higher radiosensitivity. In LP-H1802Lv201, however, being treated
with quinalizarin did not display significant enhancement of
radiosensitivity, compared with quinalizarin treated LP-NegLv201
(Fig. 5C). Results showed that
overexpressing SHP-1 reversed the radiosensitive effect of
quinalizarin. Thus, we can conclude, as least partially, that
quinalizarin radiosensitized NPC cells by suppressing the
expression of SHP-1.
Quinalizarin inhibits binding of p65 and
the promoter of SHP-1 and decreases the activity of SHP-1
promoter
Western blot analysis showed that quinalizarin
decreased SHP-1 expression, but not p65, a subunit of NF-κB;
however, the level of phosphorylated p65 was decreased with
quinlizarin (Fig. 6A). To verify
whether quinalizarin affected the binding of p65 and SHP-1
promoter, the SHP-1 promoter probe was used for EMSA. The CNE-1 and
CNE-2 nuclear proteins were extracted and an anti-p65 Ab was used
to precipitate p65. Then the complex of p65 Ab and p65 was used for
EMSA to detect its binding to SHP-1 promoter. We noted that
treatment with quinalizarin obviously reduced the binding of p65
and SHP-1 promoter, both in CNE-1 and CNE-2 cells. However, the
effect was not time-dependent (Fig.
6B).
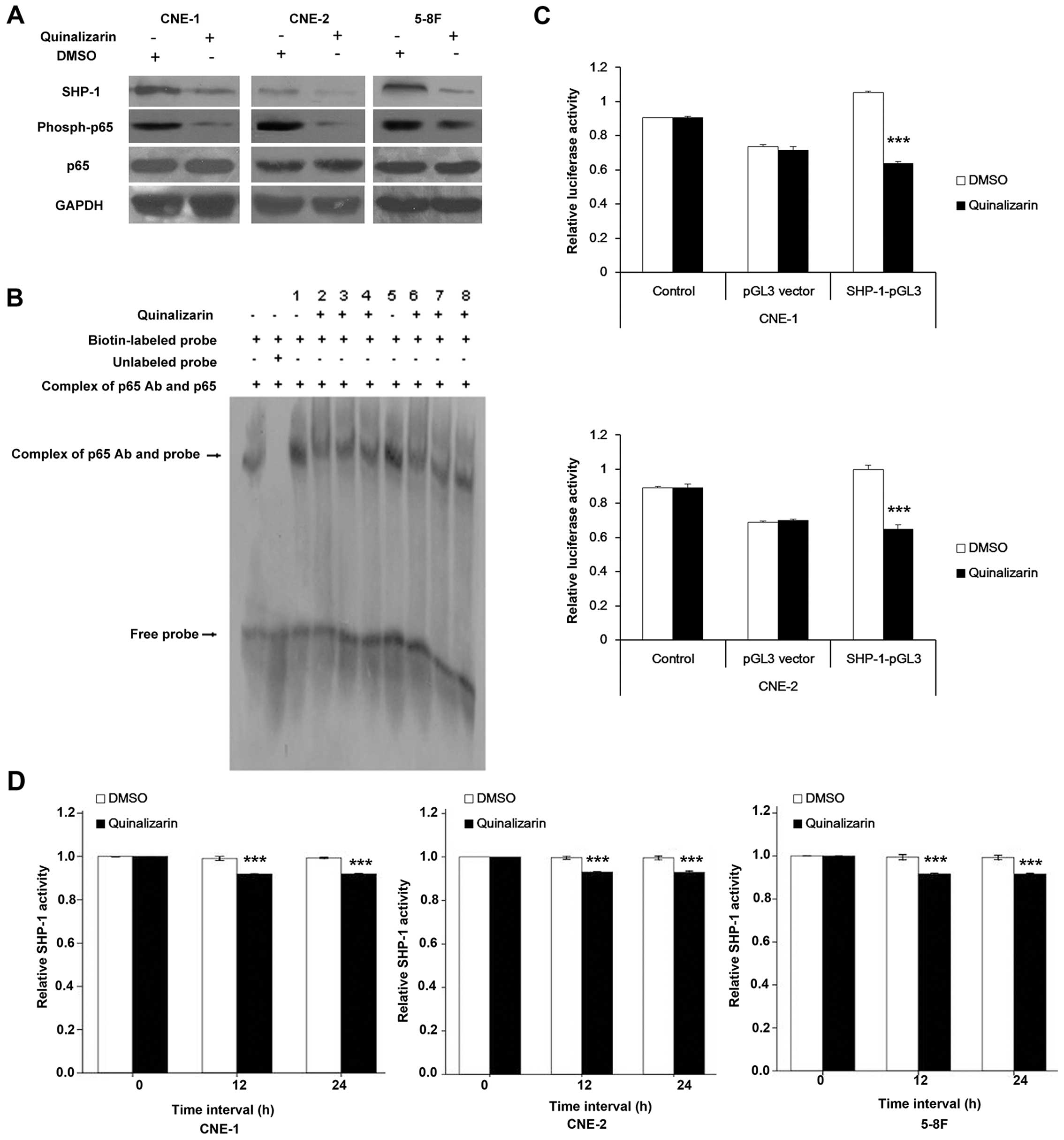 | Figure 6Quinalizarin inhibits binding of p65
and the promoter of SHP-1, and decreases the activities of SHP-1
promoter and SHP-1. (A) NPC cells were treated with 0.5% DMSO or 25
μM quinalizarin for 24 h. SHP-1, p65 and p-p65 protein expression
was determined by western blot analysis. (B) Binding of p65 and
SHP-1 promoter detected by EMSA. NPC cells CNE-1 and CNE-2 were
treated with 25 μM quinalizarin for 12, 24 and 48 h. Then nuclear
extract was incubated with p65 Ab to precipitate p65 and the
complex was used in EMSA. Binding of p65 and SHP-1 promoter was
inhibited by quinalizarin, but was not time-dependent. 1, CNE-1 not
treated with quinalizarin; 2, CNE-1 treated with quinalizarin for
12 h; 3, CNE-1 treated with quinalizarin for 24 h; 4, CNE-1 treated
with quinalizarin for 48 h; 5, CNE-2 not treated with quinalizarin;
6, CNE-2 treated with quinalizarin for 12 h; 7, CNE-2 treated with
quinalizarin for 24 h; 8, CNE-2 treated with quinalizarin for 48 h.
(C) Dual luciferase assay of SHP-1 promoter activity. NPC cells
CNE-1 and CNE-2 were transfected with SHP-1-pGL3 or pGL3-vector
(negative control). Then cells were treated with 0.5% DMSO or 25 μM
quinalizarin for 24 h and dual luciferase assay was used to
determine SHP-1 promoter activity. (D) SHP-1 activity of NPC cells.
NPC cells were treated with 0.5% DMSO or 25 μM quinalizarin for 12
or 24 h. Total protein was extracted, incubated with anti-SHP-1
antibodies and precipitated by protein A agarose beads to purify
SHP-1 protein. Then SHP-1 activity was assayed. The data are shown
as means ± SD from three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 vs. 0.05% DMSO group. |
Apart from the binding of p65 and SHP-1 promoter, we
also used dual luciferase assay to examine how quinalizarin
affected the activity of the SHP-1 promoter. As indicated in
Fig. 6C, quinalizarin treated
cells showed a significant decrease in SHP-1 promoter activity by
36 and 34% for CNE-1 and CNE-2, respectively.
Quinalizarin suppresses the activity of
SHP-1 in NPC cells
Besides regulating the expression of SHP-1, we were
also interested in the influence of quinalizarin on the phosphatase
activity of SHP-1. We incubated the protein extract with anti-SHP-1
antibodies and use immunoprecipitation to gain purified SHP-1
protein. Then a RediPlate 96 EnzChek Tyrosine Phosphatase Assay kit
(R-22067) was used to determine SHP-1 activity. As shown in
Fig. 6D, basic activity was
normalized to 1.0 (treated with quinalizarin or DMSO for 0 h).
After treatment with quinalizarin for 12 h, the activity of SHP-1
showed approximately a 10% decrease compared to the DMSO groups.
However, prolonging the treatment to 24 h did not cause further
decrease in SHP-1 activity, thus, the inhibition was not
time-dependent.
Discussion
The purpose of this study was to investigate whether
using quinalizarin would increase the radiosensitivity of NPC
cells. If this was the case then it may have potential as a method
of improving radiation therapy of NPC. The results show that
quinalizarin inhibited cell viability, and radiosensitized NPC
cells. The mechanism of this induced radiosensitivity involved
decreasing the efficiency of DSB repair, increasing the fraction of
cells in G2/M phase and promoting apoptosis. Quinalizarin decreased
SHP-1 expression and when SHP-1 was overexpressed the ability of
quinalizarin to induce radiosensitivity was partially suppressed
suggesting that reducing expression of SHP-1 is involved in the
mechanism of quinlizarin induced radiosensitivity. Firstly, we used
MTT assays to determine an appropriate drug concentration. At 25
μM, the drug itself did not show much cytotoxicity so that we could
observe its radiosensitive effect. Then using colony formation
assays, we found that quinalizarin enhanced NPC cells
radiosensitivity. The enhanced radiosensitivity probably results
from delayed DSB repair, increasing cell apoptosis, and larger
fraction of cells being in G2/M phase. S phase has been shown to
provide some degree of radioresistance and G0/G1 are relatively
radiosensitive, but G2/M phase shows the most sensitivity to
radiation (30,31). Similar results have been seen in
other cancers. Kroonen et al concluded that inhibiting CK2
delayed DNA damage repair, but did not radiosensitize malignant
glioma cells (32). Liu et
al used an RNA interference technique to silence the CK2α gene
in NPC cells and that resulted in higher radiosensitivity (33). Another study also found that CK2
inhibitors enhanced the radiosensitivity of human NSCLC cells by
inhibiting stat3 activation (14).
Our previous investigation found that SHP-1 was
highly expressed in NPC tissues in contrast to normal
nasopharyngeal mucosa and was associated with local recurrence and
metastasis after radiotherapy in NPC patients (27). Further research showed that SHP-1
overexpression increased the radioresistance of NPC cells by
enhancing DSB repair, increasing S phase arrest and decreasing cell
apoptosis (34). To assess whether
quinalizarin has an influence on SHP-1 expression, we treated NPC
cells with quinalizarin and detected the expression of SHP-1.
Expression of SHP-1 was obviously inhibited by quinalizarin. The
level of SHP-1 was apparently related to the radiosensitivity of
the NPC cells (28,29). This encouraged us to try
overexpression of SHP-1 and when this was done using lentivirus
expression the radiosensitivity induced by quinalizarin was
partially suppressed. Thus, it seems likely that quinalizarin
radiosensitized NPC cells via a CK2-SHP-1 pathway, at least
partially.
It is well known that NF-κB is a downstream target
of CK2 and is an important transcriptional factor in many cells.
The SHP-1 promoter contains a binding site of NF-κB (35), suggesting that expression of SHP-1
may be regulated by NF-κB. We treated NPC cells with quinalizarin
and detected the phosphorylation level of p65, and found that
quinalizarin inhibited p65 phosphorylation. Furthermore, EMSA and
dual luciferase assay suggested that quinalizarin suppressed the
activity of SHP-1 promoter by inhibiting p65 binding to SHP-1
promoter. In addition, we also discovered that quinalizarin
directly decreased the phosphatase activity of SHP-1. These results
further support the theory that CK2p65-SHP-1 pathway may have a
role in regulating radiosensitivity and cell cycle distribution in
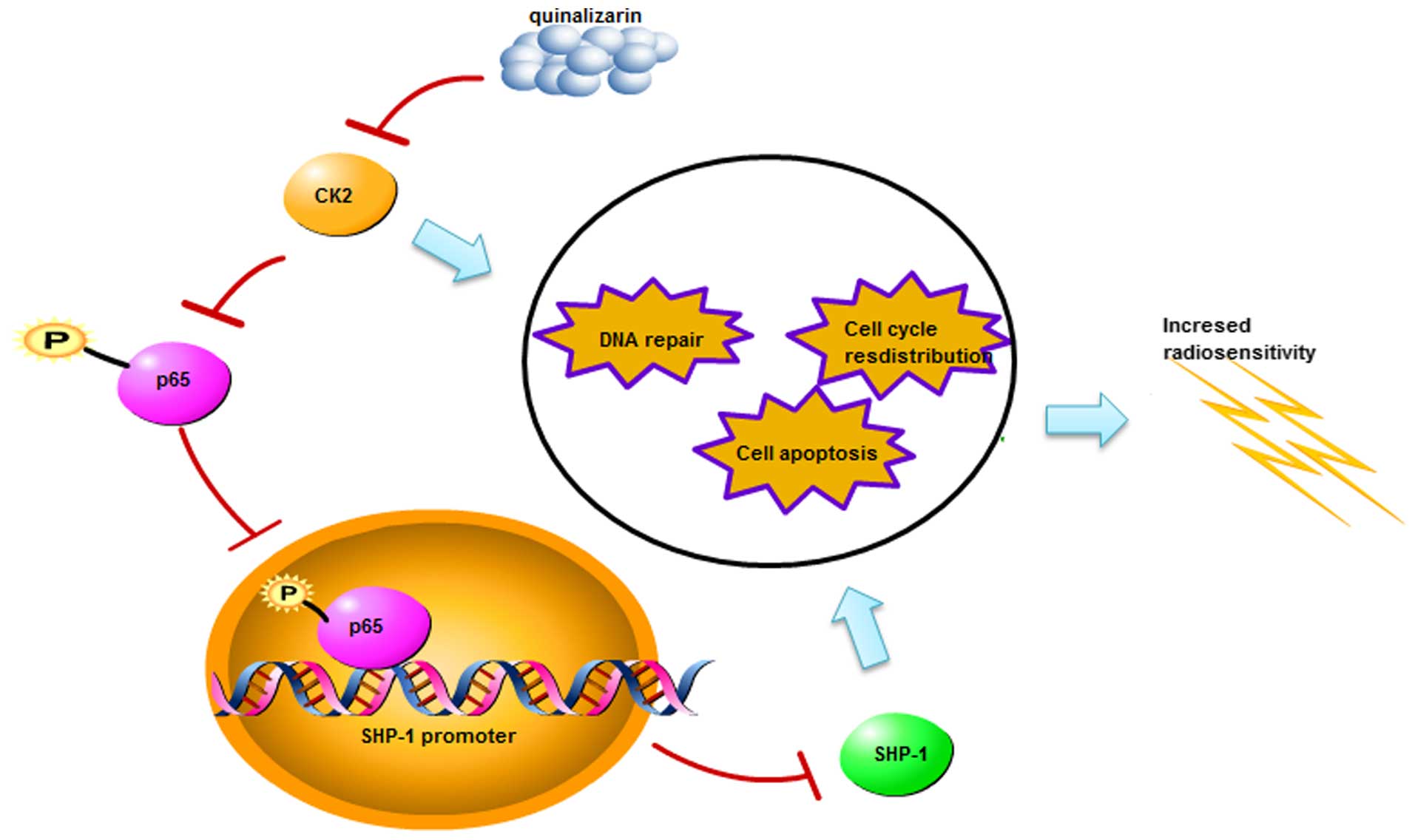
NPC cells (Fig. 7).
In this study, we firstly determined the
radiosensitive effect of quinalizarin on NPC cells. This
demonstrated that quinalizarin induced radiosensitivity on NPC
cells, an important finding that may well prove to have therapeutic
use in increasing the effectiveness of radiotherapy for NPC. We
also discovered the relationship between CK2 and SHP-1 for the
first time in this field. However, the details of the interaction
between CK2 and SHP-1 need further study to fully elucidate the
pathways involved. In conclusion, quinalizarin radiosensitized NPC
cells through inhibiting SHP-1 expression.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China in 2012 (no. 81101690), the National
Natural Science Foundation of China in 2013 (no. 81301976), and Wu
Jieping Medical Foundation in 2013.
References
|
1
|
Feng XP, Yi H, Li MY, Li XH, Yi B, Zhang
PF, Li C, Peng F, Tang CE, Li JL, et al: Identification of
biomarkers for predicting nasopharyngeal carcinoma response to
radiotherapy by proteomics. Cancer Res. 70:3450–3462. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iqbal N and Iqbal N: Imatinib: A
breakthrough of targeted therapy in cancer. Chemother Res Pract.
2014:3570272014.PubMed/NCBI
|
|
3
|
Sawyers C: Targeted cancer therapy.
Nature. 432:294–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giamas G, Man YL, Hirner H, Bischof J,
Kramer K, Khan K, Ahmed SS, Stebbing J and Knippschild U: Kinases
as targets in the treatment of solid tumors. Cell Signal.
22:984–1002. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Widmer N, Bardin C, Chatelut E, Paci A,
Beijnen J, Levêque D, Veal G and Astier A: Review of therapeutic
drug monitoring of anticancer drugs part two - targeted therapies.
Eur J Cancer. 50:2020–2036. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trembley JH, Chen Z, Unger G, Slaton J,
Kren BT, Van Waes C and Ahmed K: Emergence of protein kinase CK2 as
a key target in cancer therapy. Biofactors. 36:187–195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao Y and Wang HY: Casein kinase 2 is
activated and essential for Wnt/beta-catenin signaling. J Biol
Chem. 281:18394–18400. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Landesman-Bollag E, Song DH, Romieu-Mourez
R, Sussman DJ, Cardiff RD, Sonenshein GE and Seldin DC: Protein
kinase CK2: Signaling and tumorigenesis in the mammary gland. Mol
Cell Biochem. 227:153–165. 2001. View Article : Google Scholar
|
|
9
|
Trembley JH, Wang G, Unger G, Slaton J and
Ahmed K: Protein kinase CK2 in health and disease: CK2: a key
player in cancer biology. Cell Mol Life Sci. 66:1858–1867. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hagan CR, Regan TM, Dressing GE and Lange
CA: ck2-dependent phosphorylation of progesterone receptors (PR) on
Ser81 regulates PR-B isoform-specific target gene expression in
breast cancer cells. Mol Cell Biol. 31:2439–2452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu D, Hensel J, Hilgraf R, Abbasian M,
Pornillos O, Deyanat-Yazdi G, Hua XH and Cox S: Inhibition of
protein kinase CK2 expression and activity blocks tumor cell
growth. Mol Cell Biochem. 333:159–167. 2010. View Article : Google Scholar
|
|
12
|
Yefi R, Ponce DP, Niechi I, Silva E,
Cabello P, Rodriguez DA, Marcelain K, Armisen R, Quest AF and Tapia
JC: Protein kinase CK2 promotes cancer cell viability via
up-regulation of cyclooxygenase-2 expression and enhanced
prostaglandin E2 production. J Cell Biochem. 112:3167–3175. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Olsen BB, Wang SY, Svenstrup TH, Chen BP
and Guerra B: Protein kinase CK2 localizes to sites of DNA
double-strand break regulating the cellular response to DNA damage.
BMC Mol Biol. 13:72012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin YC, Hung MS, Lin CK, Li JM, Lee KD, Li
YC, Chen MF, Chen JK and Yang CT: CK2 inhibitors enhance the
radiosensitivity of human non-small cell lung cancer cells through
inhibition of stat3 activation. Cancer Biother Radiopharm.
26:381–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pagano MA, Bain J, Kazimierczuk Z, Sarno
S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F,
et al: The selectivity of inhibitors of protein kinase CK2: An
update. Biochem J. 415:353–365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cozza G, Mazzorana M, Papinutto E, Bain J,
Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene
M, et al: Quinalizarin as a potent, selective and cell-permeable
inhibitor of protein kinase CK2. Biochem J. 421:387–395. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sarno S, de Moliner E, Ruzzene M, Pagano
MA, Battistutta R, Bain J, Fabbro D, Schoepfer J, Elliott M, Furet
P, et al: Biochemical and three-dimensional-structural study of the
specific inhibition of protein kinase CK2 by
[5-oxo-5,6-dihy-droindolo-(1,2-a)quinazolin-7-yl]acetic acid (IQA).
Biochem J. 374:639–646. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lorenz U: SHP-1 and SHP-2 in T cells: Two
phosphatases functioning at many levels. Immunol Rev. 228:342–359.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Banville D, Stocco R and Shen SH: Human
protein tyrosine phosphatase 1C (PTPN6) gene structure: Alternate
promoter usage and exon skipping generate multiple transcripts.
Genomics. 27:165–173. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Evren S, Wan S, Ma XZ, Fahim S, Mody N,
Sakac D, Jin T and Branch DR: Characterization of SHP-1 protein
tyrosine phosphatase transcripts, protein isoforms and phosphatase
activity in epithelial cancer cells. Genomics. 102:491–499. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakase K, Cheng J, Zhu Q and Marasco WA:
Mechanisms of SHP-1 P2 promoter regulation in hematopoietic cells
and its silencing in HTLV-1-transformed T cells. J Leukoc Biol.
85:165–174. 2009. View Article : Google Scholar :
|
|
22
|
Delibrias CC, Floettmann JE, Rowe M and
Fearon DT: Downregulated expression of SHP-1 in Burkitt lymphomas
and germinal center B lymphocytes. J Exp Med. 186:1575–1583. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oka T, Yoshino T, Hayashi K, Ohara N,
Nakanishi T, Yamaai Y, Hiraki A, Sogawa CA, Kondo E, Teramoto N, et
al: Reduction of hematopoietic cell-specific tyrosine phosphatase
SHP-1 gene expression in natural killer cell lymphoma and various
types of lymphomas/leukemias: Combination analysis with cDNA
expression array and tissue microarray. Am J Pathol. 159:1495–1505.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato K, Horiuchi M, Yo R and Nakarai I: A
long survival case of small cell lung cancer synchronized with
renal cancer]. Kyobu Geka. 44:251–253. 1991.In Japanese. PubMed/NCBI
|
|
25
|
Amin HM, Hoshino K, Yang H, Lin Q, Lai R
and Garcia-Manero G: Decreased expression level of SH2
domain-containing protein tyrosine phosphatase-1 (Shp1) is
associated with progression of chronic myeloid leukemia. J Pathol.
212:402–410. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
López-Ruiz P, Rodriguez-Ubreva J, Cariaga
AE, Cortes MA and Colás B: SHP-1 in cell-cycle regulation.
Anticancer Agents Med Chem. 11:89–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peng G, Cao R, Xue J, Li P, Zou Z, Huang J
and Ding Q: Increased expression of SHP-1 is associated with local
recurrence after radiotherapy in patients with nasopharyngeal
carcinoma. Radiol Oncol. 48:40–49. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peng G, Cao RB, Li YH, Zou ZW, Huang J and
Ding Q: Alterations of cell cycle control proteins SHP-1/2, p16,
CDK4 and cyclin D1 in radioresistant nasopharyngeal carcinoma
cells. Mol Med Rep. 10:1709–1716. 2014.PubMed/NCBI
|
|
29
|
Cao R, Ding Q, Li P, Xue J, Zou Z, Huang J
and Peng G: SHP1-mediated cell cycle redistribution inhibits
radiosensitivity of non-small cell lung cancer. Radiat Oncol.
8:1782013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hall EJ and Giaccia A: Cell survival
curves. Radiobiology for the Radiologist. Lippincott Williams &
Wilkins; New York, NY: 2011
|
|
31
|
Peng G, Cao RB, Li YH, Zou ZW, Huang J and
Ding Q: Alterations of cell cycle control proteins SHP-1/2, p16,
CDK4 and cyclin D1 in radioresistant nasopharyngeal carcinoma
cells. Mol Med Rep. 10:1709–1716. 2014.PubMed/NCBI
|
|
32
|
Kroonen J, Artesi M, Capraro V,
Nguyen-Khac MT, Willems M, Chakravarti A, Bours V and Robe PA:
Casein kinase 2 inhibition modulates the DNA damage response but
fails to radiosensitize malignant glioma cells. Int J Oncol.
41:776–782. 2012.PubMed/NCBI
|
|
33
|
Liu L, Zou JJ, Luo HS and Wu DH: Effect of
protein kinase CK2 gene silencing on radiosensitization in human
nasopharyngeal carcinoma cells. Nan Fang Yi Ke Da Xue Xue Bao.
29:1551–1553. 2009.In Chinese. PubMed/NCBI
|
|
34
|
Pan X, Mou J, Liu S, Sun Z, Meng R, Zhou
Z, Wu G and Peng G: SHP-1 overexpression increases the
radioresistance of NPC cells by enhancing DSB repair, increasing S
phase arrest and decreasing cell apoptosis. Oncol Rep.
33:2999–3005. 2015.PubMed/NCBI
|
|
35
|
Wu C, Sun M, Liu L and Zhou GW: The
function of the protein tyrosine phosphatase SHP-1 in cancer. Gene.
306:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|