Introduction
Hepatocellular carcinoma (HCC) represents the second
leading cause of cancer-related deaths worldwide (1), resulting in ~700,000 deaths per year.
Although therapeutic options such as surgical resection and liver
transplantation are used to improve the outcomes and decrease
mortality of HCC patients, the 5-year survival rate is still less
than 30% (1). In many cases, HCC
is detected at advanced stage with limited therapeutic options.
Thus, a better understanding of the molecular mechanism underlying
HCC carcinogenesis will help to indentify novel diagnostic
biomarkers and develop targeted therapies.
MicroRNAs (miRNAs) are small non-coding RNAs with a
length of 20–25 nucleotides that post-transcriptionally regulate
gene expression by complementary binding to the 3'untranslated
region (3'UTR) of target mRNAs, inducing degradation or
translational repression (2).
miRNAs have been well established to closely associate with
carcinogenesis and progression, where they function as oncogenes or
tumor suppressors depending on their targets. miR-26a, an early
discovered miRNA, comprises miR-26a-1 and miR-26a-2, which are
located in the intron of CTD small phosphatase like (CTDSPL) and
CTD small phosphatase 2 (CTDSP2), respectively. miR-26a-1 and
miR-26a-2 are expressed concomitantly with their host genes in
physiological and pathological conditions (3). It has been reported that miR-26a is
downregulated in several types of cancers including HCC (4,5).
miR-26a suppressed tumor growth and metastasis by targeting
multiple oncogenes and cell cycle-related genes such as IL-6
(5), Zcchc11 (6), ITGA5 (7), CCND2 and CCNE2 (8) in HCC. However, the underlying
mechanism of how miR-26a is repressed remains largely unknown.
Epigenetic regulation plays a central role in the
regulation of genes. Polycomb repressive complex 2 (PRC2) is an
important epigenetic regulator that represses tumor suppressor
genes such as KFL2, ID4 and E-cadherin by chromatin modification
(9–12). The enhancer of zest homologue 2
(EZH2) is the catalytically active subunit of PRC2. It suppresses
gene expression by inducing the trimethylation of lysine 27 on
histone 3 (H3K27) (13). EZH2 has
been shown overexpressed in a variety of cancers including HCC
(14,15). EZH2 plays an important role in
carcinogenensis, progression and metastasis, where it functions as
an oncogene (16–18). Recently, several studies reported
that miRNAs such as miR-214 (19),
miR-101 (20), miR-200s (21–23),
miR-218 (24), miR-143 (25), miR-622 (26) and miR-31 (27), could be epigenetically suppressed
by EZH2-mediated H3K27 trimethylation. It seems that overexpression
of EZH2 may partly explain the suppression of tumor suppressor
miRNAs. Thus, we presume that miR-26a could be silenced by
EZH2-mediated epigenetic mechanisms. Moreover, EZH2 has been
reported to be a direct target of miR-26a in nasopharyngeal
(28), lung (29) and prostate cancer (30). However, the regulatory mechanism of
EZH2 expression and the relationship between EZH2 and miR-26a in
HCC are still unclear.
In the present study, we demonstrate that miR-26a is
repressed by EZH2-mediated H3K27 trimethylation within the miR-26a
promoter and confirm that EZH2 is a direct target of miR-26a in HCC
cells. EZH2 and miR-26a form a double-negative feedback loop,
contributing to miR-26a deregulation and regulating HCC cell
growth. Our findings provide a better understanding of HCC
pathogenesis.
Materials and methods
Cell culture and transfection
LO2, HepG2 and SMMC-7721 cell lines were obtained
from the Cell Bank of Chinese Academy of Sciences (Shanghai,
China), and were maintained at 37°C (5% CO2) in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS; Gibco, Grand Island, NY, USA). All
experiments were performed when cells were at logarithmic growth
phase. For transfection, cells were plated in 6-well plates for 24
h to ensure 70–80% confluence. Then, the cells were transfected
with 4 μg pcDNA3.1, pcDNA3.1-miR-26a, pcDNA3.1-EZH2 and sh-EZH2, or
100 nM miR-26a mimics and control mimics using Lipofectamine 2000
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instruction. The miR-26a mimics and control mimics
were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou,
China).
Construction of expression plasmids
To construct miR-26a expression plasmid
(pcDNA3.1-miR-26a), a 254-bp DNA fragment containing pre-miR-26a
and the 5′- and 3′-flanking sequence was amplified from human
genomic DNA and cloned into the KpnI and EcoRI sites
of pcDNA3.1 vector. The primers were 5′-AGGGGTACCGGCTGGGGTCAGAAAT-3′
(forward) and 5′-GCAGAATTCGCTACATGCAAAGGGC AG-3′
(reverse). The underlined bases represent the restriction enzyme
cutting sites. EZH2 expression plasmid (pcDNA3.1-EZH2) was
generated by cloning the coding sequence of EZH2 without 3'UTR into
pcDNA3.1+ vector. For sh-EZH2 plasmid construction, two shRNAs
targeting human EZH2 mRNA (shRNA-1 5′-CCCAACATAGATGGACCAAAT-3′;
shRNA-2 5′-GAGTATGTTTAGTTCCAATAA-3′) were designed and cloned into
pGenesil-1 plasmid.
Quantitative real-time PCR
TRIzol reagent (Invitrogen) was used to extract
total RNA according to the manufacturer's instructions. The
expression of miR-26a was determined by a stem-loop qRT-PCR assay.
Reverse transcription of miR-26a and U6 was performed by using
PrimeScript™ RT reagent kit (Takara Bio, Shiga, Japan) with
specific RT primer. For mRNA analysis, 1 μg of total RNA was
reverse-transcribed using PrimeScript™ RT Master Mix (Takara Bio).
Subsequently, real-time PCR was performed using SYBR-Green PCR
Master Mix (Takara). All PCR reactions were performed under the
conditions of 5 min at 95°C, followed by 40 cycles of 5 sec at
95°C, 30 sec at 60°C and 30 sec at 72°C on a Stratagene Mx3000P
system. The relative expression levels of miR-26a and mRNAs were
calculated by 2−ΔΔCt method using GAPDH or U6 as
internal control. The primers used for qRT-PCR were as follows:
miR-26a, 5′-GTCGTATCCAGTGCAGGGTCC GAGGTATTCGCACTGGATACGACAGCCA-3′
(RT), 5′-GC CCGCTTCAAGTAATCCAGG-3′ (forward) and 5′-GTGCA
GGGTCCGAGGT-3′ (reverse); U6, 5′-CTCGCTTCGGCAG CACA-3′ (forward)
and 5′-AACGCTTCACGAATTTGC GT-3′ (reverse); EZH2,
5′-AGTTTGCTGCTGCTCTCAC-3′ (forward) and 5′-GTTCTCTCCCCCCGTTTC-3′
(reverse); CTDSPL, 5′-TGCTGAGGGAGGGGAGTGAG-3′ (forward) and
5′-GCAGCATGCCACAGGTTGTC-3′ (reverse); CTDSP2,
5′-TCACCAGGTGTATGTGCTCAA-3′ (forward) and 5′-AA
GACTCACGGAATAGGCG-3′ (reverse); E-cadherin, 5′-CC
CCATACCAGAACCTCG-3′ (forward) and 5′-TTCTTGGG TTGGGTCGTT-3′
(reverse); GAPDH, 5′-GAAGGTGAAG GTCGGAGTC-3′ (forward) and
5′-GAAGATGGTGATGG GATTTC-3′ (reverse).
Western blot analysis
Cells were lysed using RIPA reagent (Beyotime
Institute of Biotechnology, Jiangsu, China). Protein lysates were
sonicated and centrifuged at 12,000 rpm for 5 min. Then protein
extracts were subjected into 12% SDS-PAGE and transferred to NC
membrane (Milipore, Billerica, MA, USA). After blocked with 5%
non-fat milk, the membrane was incubated with rabbit anti-human
specific primary antibodies (anti-EZH2, 1:1,000 and anti-GAPDH,
1:2,000; Cell Signaling Technology, Danvers, MA, USA) at 4°C
overnight, followed by incubation with HRP-conjugated goat
anti-rabbit secondary antibody (1:5,000; Wuhan Boster Biological
Technology, Ltd., Wuhan, China) at room temperature for 1 h. The
membrane was detected with BeyoECL plus kit (Beyotime Institute of
Biotechnology) and the intensity of protein fragments was
quantified using Image Lab software (Bio-Rad Laboratories,
Hercules, CA, USA). The signals of the detected bands were
normalized to GAPDH band, and the values were presented as the
ratio between treated cells and control cells.
CCK-8 assay
Cells were seeded in 96-well plates
(2×103/well) at a final volume of 100 μl. After
transfection, the cells were cultured for another 24, 48, 72 and 96
h. Then, 10 μl of CCK-8 reagent (Beyotime Institute of
Biotechnology) was added into each plate at different time-points
and incubated at 37°C for 1 h. The absorbance was measured by a
microplate reader at 450 nm.
Colony formation assay
Forty-eight hours after transfection, cells were
seeded in 6-well plates (500 cells/well) and maintained in DMEM
medium containing 10% FBS for 2 weeks. The cells were then fixed
and stained with 1% crystal violet. The number of colonies was
counted under a microscope (Olympus IX83; Olympus, Tokyo, Japan).
All experiments were repeated three times and the values were
reported as the ratio between treated cells and control cells.
Luciferase reporter assay
To construct EZH2-3'UTR-WT plasmid, a 274-bp DNA
fragment of EZH2 3'UTR containing the predicted binding site of
miR-26a was amplified from human genomic DNA and cloned into a
reporter vector as previously described (31). The primers for EZH2-3'UTR-WT were
as follows: 5′-ATAGAATTCCATCTGCTACCTCC TCC-3′
(forward) and 5′-CGCAAGCTTGATTCAACAAGG AC-3′ (reverse). The
EZH2-3'UTR-MUT plasmid, which converted the miR-26a binding site
‘TACTTGAA’ to ‘CTGCAGAA’, was generated by site-specific
mutagenesis based on EZH2-3'UTR-WT plasmid. The primers for
EZH2-3'UTR-MUT plasmid were: 5′-AACTTTGAATAAAGAACT
GCAGAACTTGTCCTTGTTG-3′ (forward) and 5′-AACAA
GGACAAGTTCTGCAGTTCTTTATTCAAAGTTG-3′ (reverse).
To investigate the transcriptional regulation of
miR-26a, a 2147-bp fragment of CTDSPL/miR-26a-1 promoter
(−1521/+626) and a 1569-bp fragment of CTDSP2/miR-26a-2 promoter
(−1322/+247) were amplified by PCR and cloned into the pGL3-basic
plasmid, generating pGL3-26a-1 and pGL3-26a-2. The primers for
pGL3-26a-1 were as follows: 5′-ACGGGTACCCAGAGTGTGGCCTGCAAGTTGGAA-3′
(forward) and 5′-TCAAAGCTTCGCACACACATTCCACA
ACCCCTCA-3′ (reverse). The primers for pGL3-26a-2 were:
5′-TCACTCGAGATTGGGGTTGGATTTAGC-3′
(forward) and 5′-AACAAGCTTCGAAAACAAGAGGAGGAAAT-3′
(reverse).
For luciferase reporter assay, HepG2 cells were
seeded in 96-well plates until 70–80% confluence. Then, 50 ng of
the constructed EZH2 3'UTR reporter plasmids were cotransfected
with either 100 nM miR-26a mimics or control mimics, and 5 ng of
pRL-TK plasmid (Promega, Madison, WI, USA); 50 ng of the
constructed miR-26a promoter reporter plasmids were cotransfected
with 200 ng of either pcDNA3.1-EZH2 or pcDNA3.1 and 5 ng of pRL-TK.
The pRL-TK plasmid was used as internal control. Forty-eight hours
after transfection, the luciferase activity was measured using the
Dual-luciferase reporter assay system (Promega).
Chromatin immunoprecipitation
CHIP assay was performed using EpiQuik™ chromatin
immunopercipitation kit (Epigentek, Farmingdale, NY, USA) according
to the manufacturer's protocol. Briefly, the cells were transfected
with sh-EZH2 or sh-control for 48 h prior to formaldehyde fixation.
After cell lysis, the chromatin was fragmented to ~200–1,000 bp.
The DNA fragment was then enriched with anti-EZH2, anti-H3K27me3 or
normal IgG antibodies (Cell Signaling Technology). The purified DNA
was used as template and quantified by qRT-PCR method. The primers
for CHIP were as follows: 26a1s1-F 5′-AGGCTGAGGAGGCACTTTG-3′,
26a1s1-R 5′-AGTGGGCATTTTCGGGTG-3′, 26a1s2-F 5′-GC
CGACTGAGCCAGTGTGA-3′, 26a1s2-R 5′-AACTTCTCT GCGGGCGTG-3′, 26a2s-F
5′-GAGAGCCAATCGGAGC AC-3′, 26a2s-R 5′-TGGGACCCTCCATCTGTG-3′.
Statistical analysis
Data were analyzed using GraphPad Prism 5.0 software
(GraphPad Software, Inc., La Jolla, CA, USA). The significance of
differences between two groups was evaluated by the Student's
two-tail t-test. P<0.05 was considered to indicate a
statistically significant result.
Results
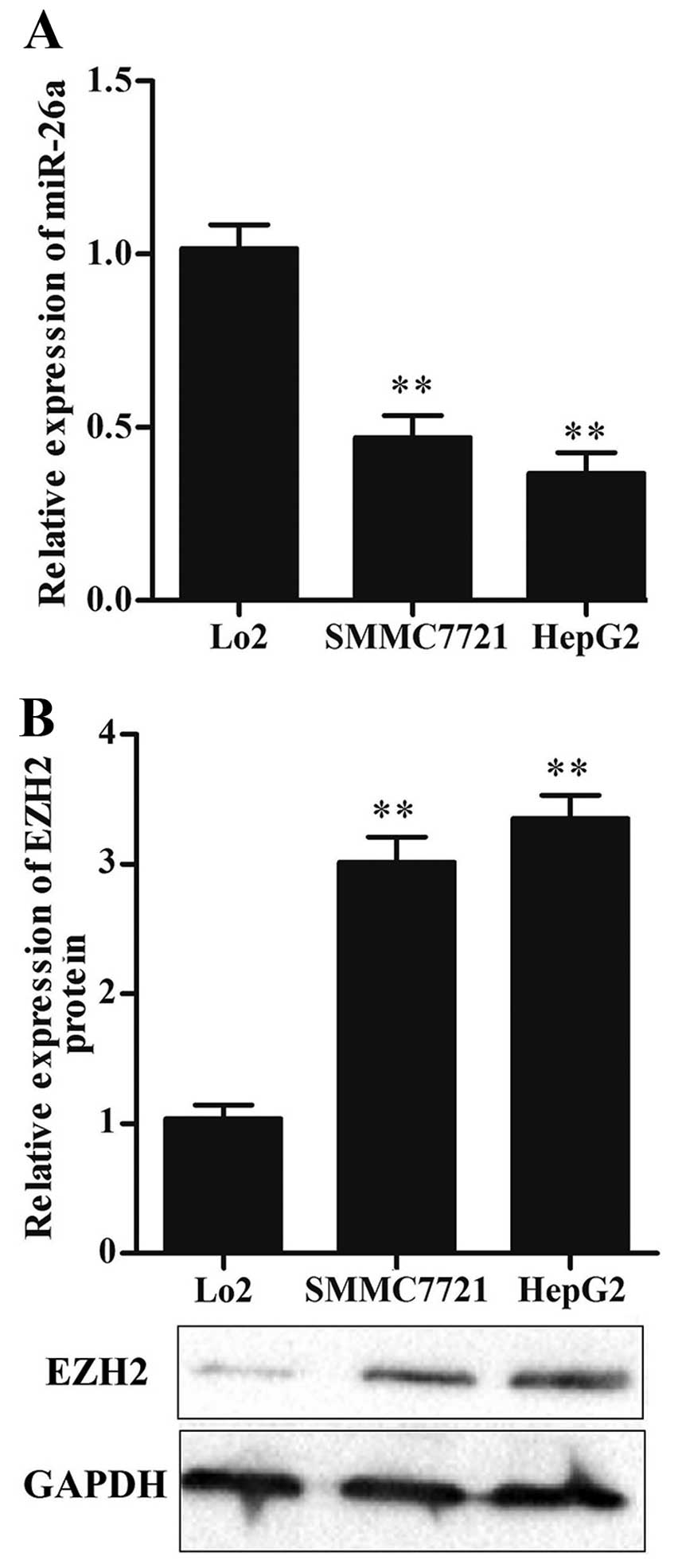
The expression of miR-26a is
downregulated while EZH2 is upregulated in HCC cell lines
To determine the expression level of miR-26a in HCC
cells, two HCC cell lines (HepG2 and SMMC7721) and a normal liver
cell line (Lo2) were used to detect miR-26a expression levels by
qRT-PCR. The result showed that the expression level of miR-26a was
significantly decreased in HCC cells compared with that in Lo2
cells (P<0.01) (Fig. 1A).
Moreover, we detected the level of EZH2 by western blot analysis,
the result showed that the expression of EZH2 was significantly
increased in HCC cells (P<0.01) (Fig. 1B).
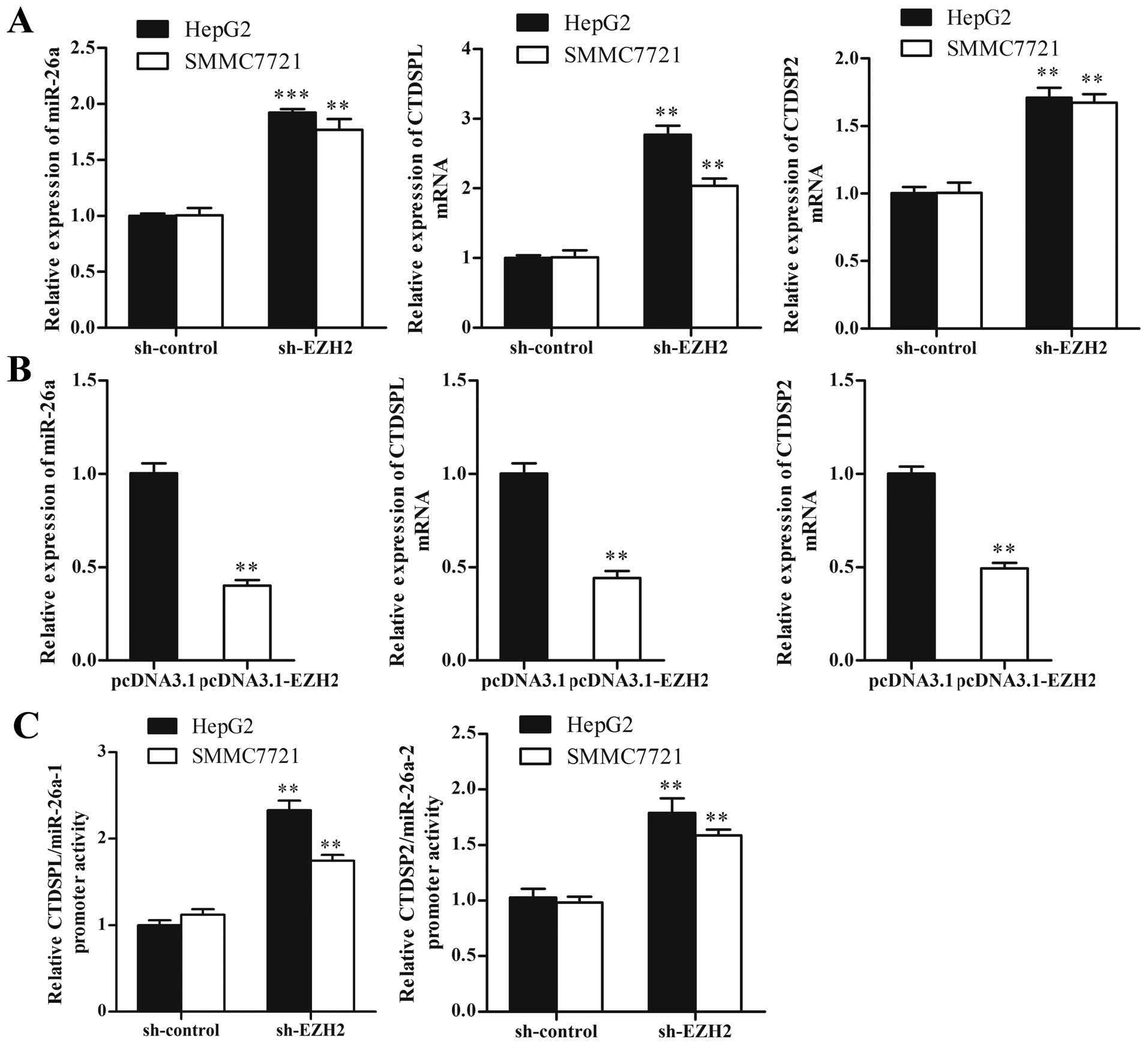
miR-26a is negatively regulated by
EZH2
EZH2 as an oncogene, has been reported to
epigenetically repress expressions of various tumor suppressor
miRNAs such as miR-101, miR-200s and miR-31. To investigate whether
miR-26a could be inhibited by EZH2 in HCC cells, we performed loss-
and gain-of-function studies. The result showed that knockdown of
EZH2 led to a 1.9- and a 1.8-fold increase of miR-26a in HepG2 and
SMMC7721 cells, respectively (P<0.01) (Fig. 2A). We also detected the expression
of miR-26a host genes, CTDSPL and CTDSP2, which were reported to be
concomitantly expressed with miR-26a. As shown in Fig. 2B, knockdown of EZH2 increased the
expressions of CTDSPL and CTDSP2 (P<0.01). Ectopic expression of
EZH2 significantly decreased the expression of miR-26a and its host
genes by ~60% in Lo2 cells (P<0.01) (Fig. 2B). Futhermore, luciferase reporter
assay revealed that transcriptional activities of CTDSPL/miR-26a-1
and CTDSP2/miR-26a-2 increased significantly in sh-EZH2 treated HCC
cells compared with sh-control (P<0.01) (Fig. 2C).
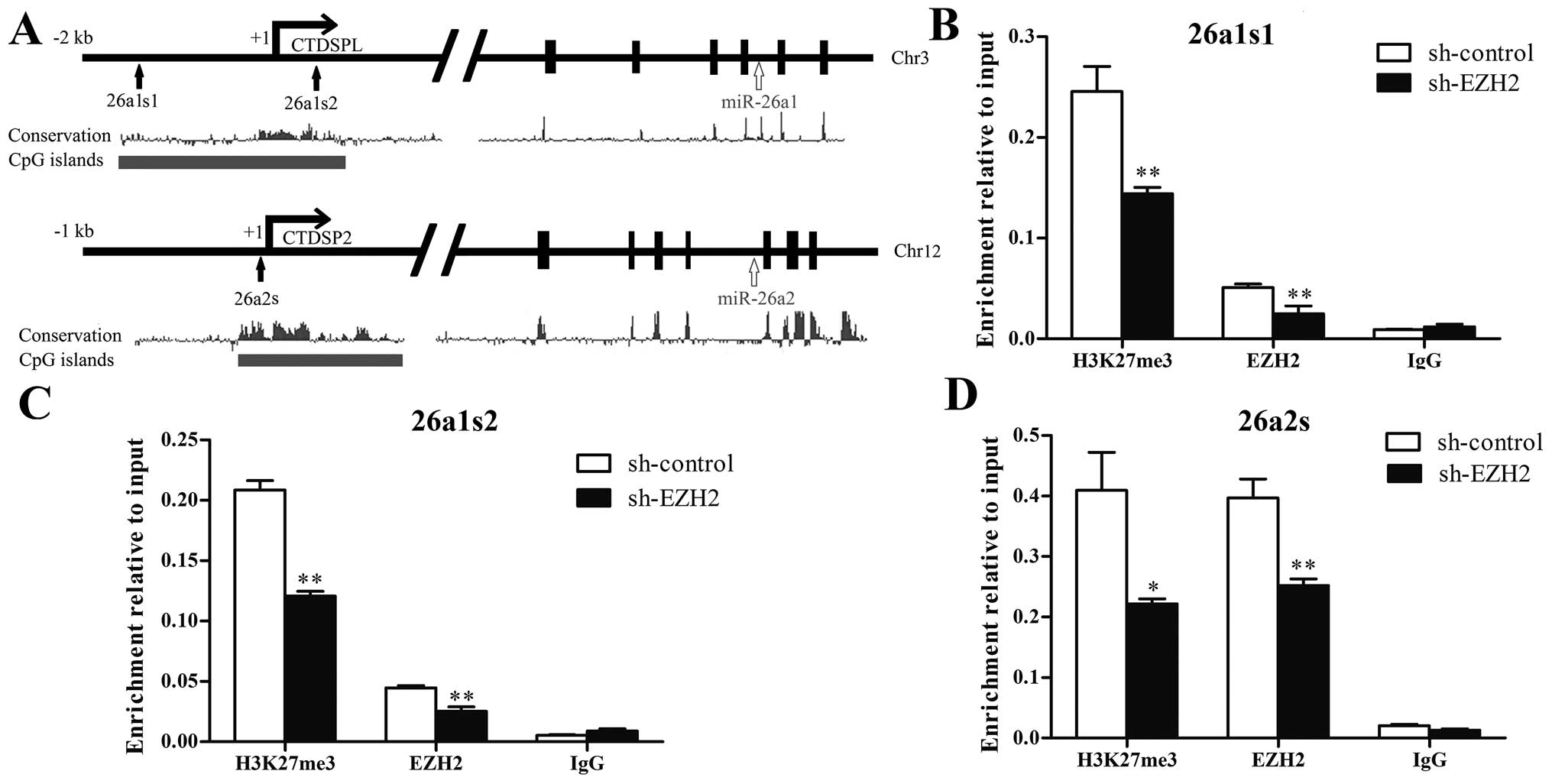
EZH2 regulates H3K27 methylation on the
miR-26a promoter
To determine whether EZH2 could regulate histone
H3K27 methylation on the miR-26a promoter, we performed CHIP assay
near the transcription start site on CTDSPL/miR-26a-1 promoter and
CTDSP2/miR-26a-2 promoter, which containing rich CpG islands and
were highly conserved according to UCSC Genome Browser (Fig. 3A). As shown in Fig. 3B–D, EZH2 and H3K27me3 were enriched
at both CTDSPL/miR-26a-1 and CTDSP2/miR-26a-2 promoters. Knockdown
of EZH2 decreased the binding of EZH2 and the level of H3K27me3 on
CTDSPL/miR-26a-1 and CTDSP2/miR-26a-2 promoters. The result
indicates that miR-26a is repressed by EZH2-mediated H3K27
methylation.
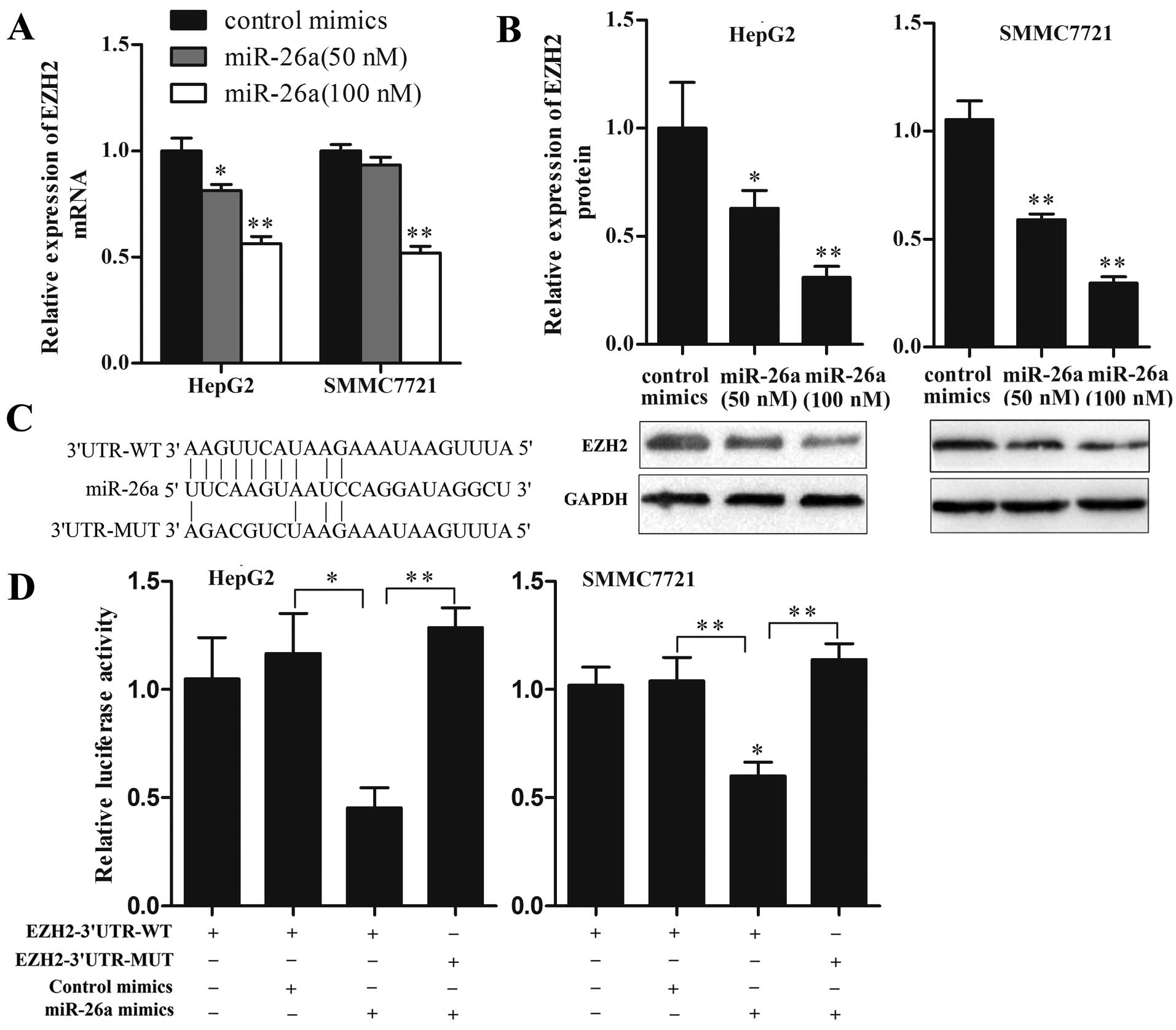
EZH2 is a direct target of miR-26a in HCC
cells
As EZH2 has been proposed to be a direct target of
miR-26a in several types of cancers, it is of interest to
investigate whether this relationship also exists in HCC. To
investigate whether miR-26a could regulate EZH2 expression in HCC
cells, we transfected HepG2 and SMMC7721 cells with miR-26a mimics
at different concentrations to upregulate miR-26a expression and
then detected EZH2 expression levels. As shown in Fig. 4A and B, ectopic expression of
miR-26a significantly decreased both mRNA and protein levels of
EZH2 in a dose-dependent manner. At the dose of 100 nM, the mRNA
and protein levels of EZH2 were decreased by ~50 and 70%,
respectively (P<0.01). Furthermore, luciferase reporter assay
was performed to determine whether miR-26a could bind to the 3'UTR
of EZH2 mRNA. Luciferase reporter plasmids containing wild-type or
mutant EZH2 3'UTR were constructed (Fig. 4C) and cotransfected with miR-26a
mimics or control mimics into HepG2 and SMMC7721 cells. As shown in
Fig. 4D, miR-26a but not control
mimics, significantly decreased the luciferase activity of
EZH2-3'UTR-WT reporter, and EZH2-3'UTR-MUT reporter was not
affected by miR-26a. These results suggest that EZH2 is a direct
target of miR-26a in HCC cells.
miR-26a and EZH2 form a double-negative
feedback loop in HCC cells
The above results suggested that EZH2 and miR-26a
may reciprocally regulate each other, forming a double-negative
feedback loop. Here, we performed a self-induction experiment of
miR-26a. HCC cells were transfected with exogenous miR-26a mimics,
and the expression of endogenous miR-26a was detected. Because the
detection of endogenous miR-26a could be disturbed by exogenous
miR-26a mimics, we detected its host genes (CTDSPL and CTDSP2)
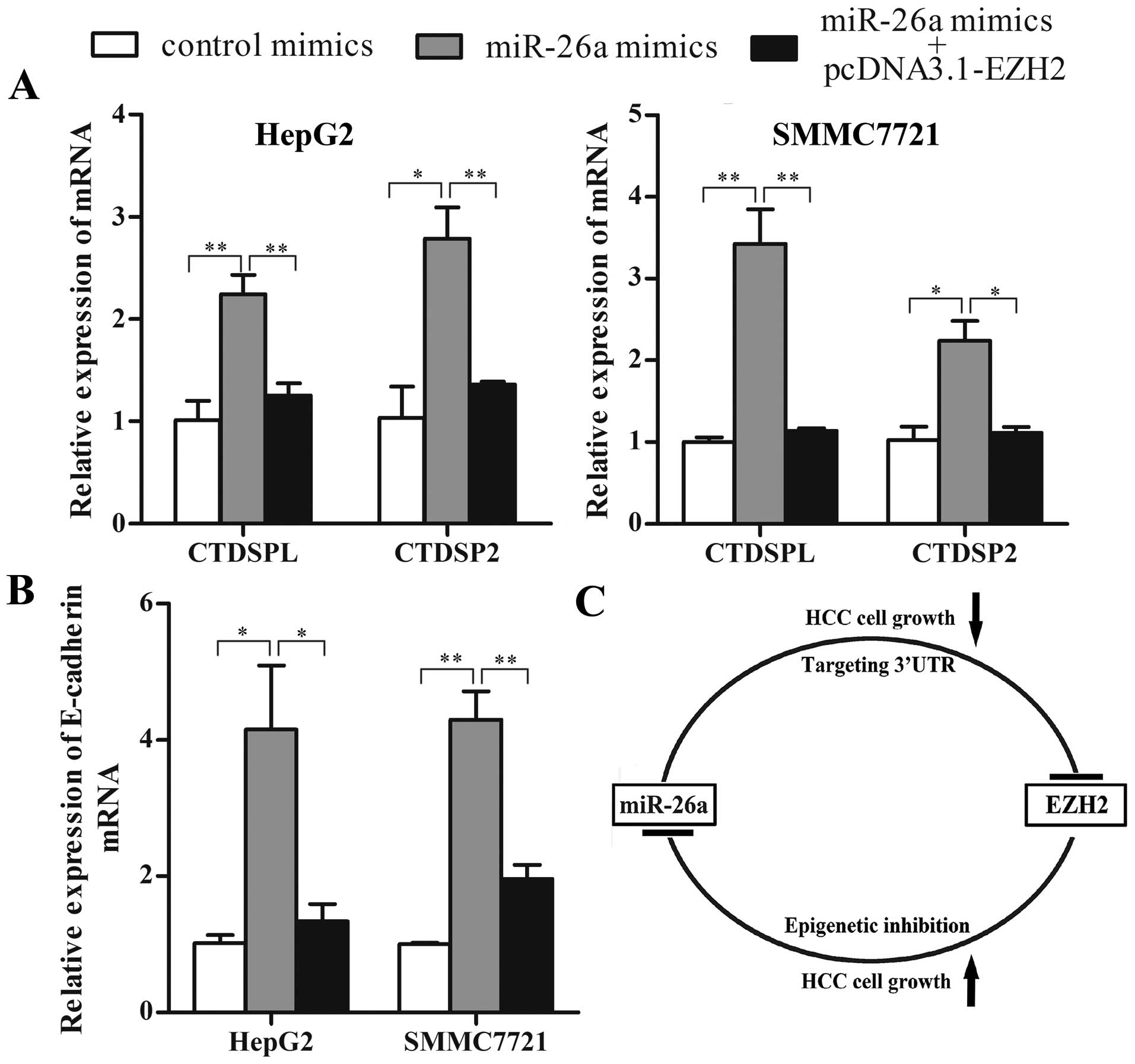
instead. As shown in Fig. 5A,
compared with control, miR-26a mimics increased the mRNA expression
of CTDSPL and CTDSP2 in both HepG2 and SMMC7721 cells. Ectopic
expression of EZH2 abrogated miR-26a induction of CTDSPL and
CTDSP2. These results benefit the existence of the double-negative
feedback loop between EZH2 and miR-26a in HCC. Moreover, we
analyzed the expression of E-cadherin, which has been reported to
be a target of PRC2. The result showed that miR-26a significantly
increased E-cadherin mRNA expression, and the induction was
abrogated by EZH2 overexpression (Fig.
5B).
The imbalance of double-negative feedback
loop between EZH2 and miR-26a contributes to HCC cell growth
As the double-negative feedback loop between EZH2
and miR-26a is imbalanced in HCC cells, in the present study we
restored the balance and investigated the effect of double-negative
feedback loop on HCC cell growth. First, we evaluated whether
miR-26a restoration could induce growth inhibition of HCC cells,
and whether ectopic expression of EZH2 could rescue the growth
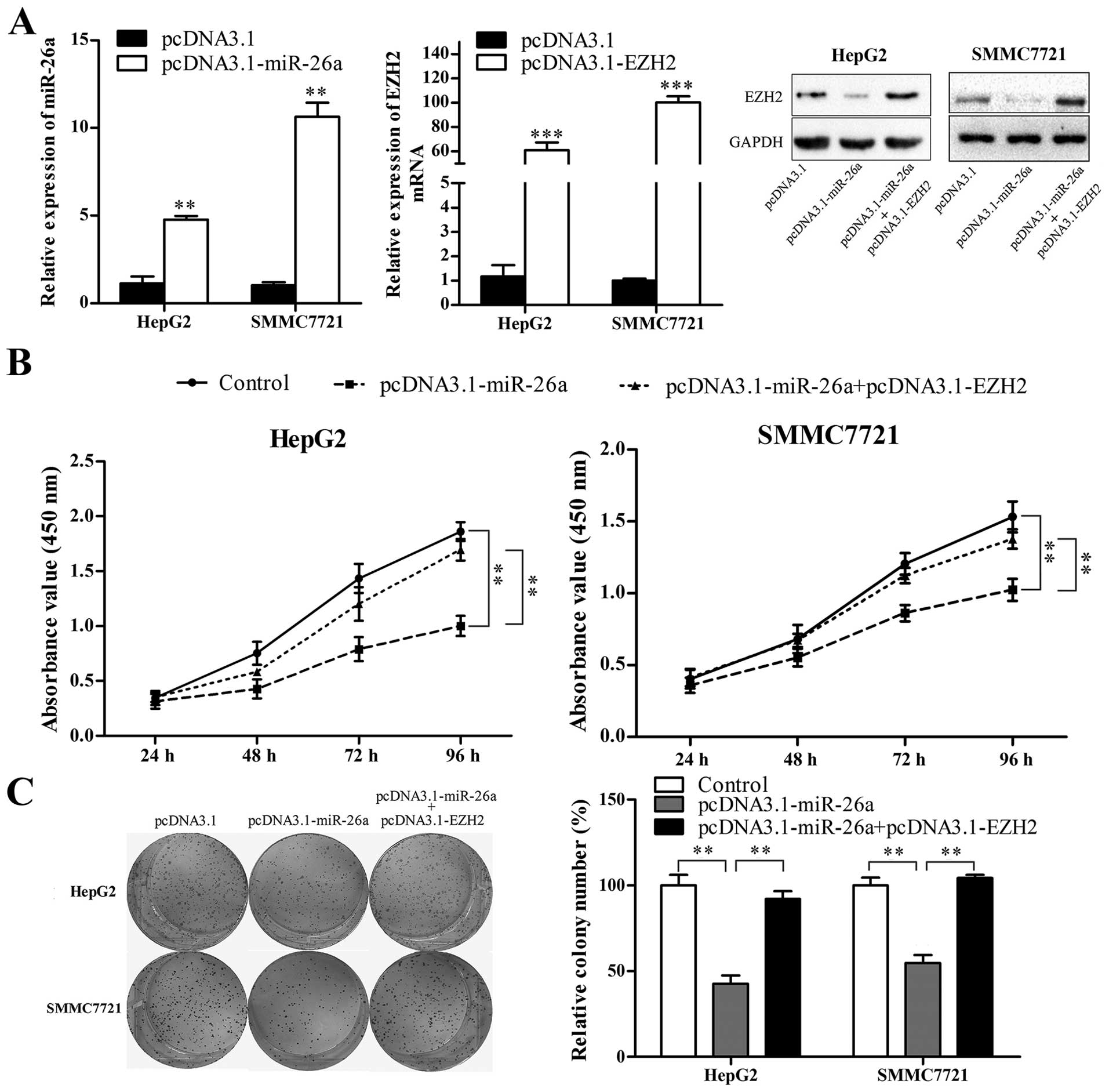
inhibition effect of miR-26a. miR-26a expression vector
(pcDNA3.1-miR-26a) and EZH2 expression vector (pcDNA3.1-EZH2,
without 3'UTR) were constructed and confirmed to be effective by
qRT-PCR and western blot analysis (Fig. 6A). HepG2 and SMMC7721 cells were
transfected with pcDNA3.1 or pcDNA3.1-miR-26a, or cotransfected
with pcDNA3.1-miR-26a and pcDNA3.1-EZH2, and then CCK-8 and colony
formation assays were performed. As shown in Fig. 6B and C, miR-26a restoration
significantly suppressed HCC cell proliferation and colony
formation (P<0.01), and EZH2 abrogated miR-26a-induced cell
proliferation inhibition and suppression of colony formation
(P<0.01).
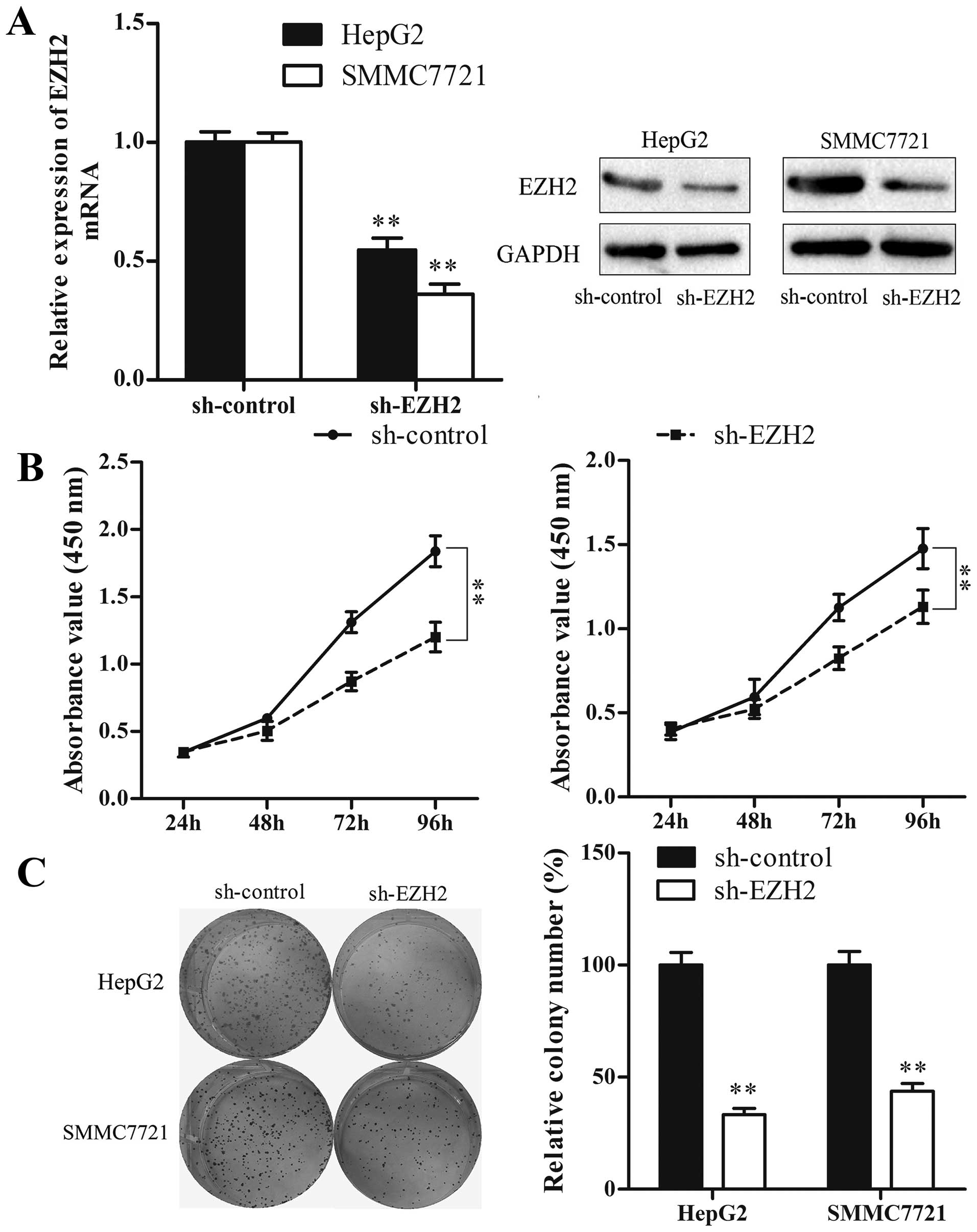
Subsequently, we used the sh-EZH2 vector to suppress
EZH2 expression, and investigated whether reduction of EZH2 could
mimic the suppressive effect of miR-26a restoration. The result
showed that, similar to miR-26a restoration, knockdown of EZH2
significantly inhibited HCC cell proliferation and colony formation
(P<0.01) (Fig. 7). Taken
together, our results suggest that the double-negative feedback
loop between EZH2 and miR-26a regulates HCC cell growth.
Discussion
Accumulating evidence demonstrates the important
roles of miRNAs in tumor development and progression (32), but the regulatory mechanisms of
miRNA expression are still largely unknown. PRC2 is an epigenetic
regulator and plays an important role in gene regulation (9,10).
Growing number of studies suggest that, similar to protein coding
genes, miRNAs could also be regulated by epigenetic mechanisms
(21,33,34).
As the key component of PRC2, EZH2 has been shown to participant in
the epigenetic regulation of miRNAs in a variety of cancers. Zhang
et al (27) demonstrated
that EZH2 suppressed miR-31 expression by regulating H3K27
trimethylation on miR-31 promoter in prostate cancer. Wang et
al (20) reported that EZH2
coordinated with c-Myc epigenetically silencing miR-101 expression
during hepatocarcinogenesis, showing that EZH2 was a target of
miR-101, thus creating a double-negative feedback loop that
regulated the process of hepatocarcinogenesis. In the present
study, we report that reduction of EZH2 caused a significant
increase of miR-26a expression. This effect was accompanied by an
increase in transcriptional activity of miR-26a promoter. CHIP
assay indicated that EZH2 regulated H3K27 trimethylation on miR-26a
promoter. These results are consistent with a previous study by
Zhao et al (35), which
reported an enrichment of EZH2 on miR-26a promoter, and that
reduction of EZH2 increased miR-26a expression in aggressive B-cell
lymphomas. Moreover, another study by Borno et al (36) showed that both the expression and
the promoter methylation status of miR-26a were associated with
EZH2 expression in prostate cancer tissues and cell lines. It seems
that epigenetic regulation of miR-26a by EZH2 may be a common
mechanism in malignancies.
CTDSPL and CTDSP2, the host genes of miR-26a, have
been reported as tumor suppressors. It has been demonstrated that
CTDSPL and CTDSP2 dephosphorylated the ppRb protein and induce cell
cycle arrest in HCC (3) and renal
carcinoma (37). Moreover, CTDSPL
is reported to be downregulated and frequently methylated in
non-small cell lung cancer (38),
renal cell carcinoma (39) and
ovarian cancer (40).
Consistently, the bioinformatics analysis revealed that the
promoter region of CTDSPL and CTDSP2 contained many CpG islands. As
EZH2 can directly regulate DNA methylation (41), it is possible that EZH2 suppresses
miR-26a by regulating both H3K27 trimethylation and DNA
methylation.
miR-26a has been previously reported to function as
a tumor suppressor in a variety of cancers by targeting multiple
oncogenes (5–8,28–30)
including EZH2, but its relationship with EZH2 is not fully
elucidated in HCC. A recent study by Wang et al (42) reported that miR-26a suppressed EZH2
expression in HepG2 cells. In the present study, we demonstrated
that miR-26a suppressed EZH2 expression in a dose-dependent manner,
which was consistent with Wang et al (42). We further confirmed that EZH2 was a
direct target of miR-26a in HCC cells by luciferase reporter assay.
Moreover, we showed that exogenous miR-26a mimics could efficiently
induce the endogenous expression of CTDSPL and CTDSP2, and that
overexpression of EZH2 disrupted this induction. These data
indicate that EZH2 and miR-26a form a double-negative feedback
loop, and that overexpression of EZH2 contributes to the imbalance
of the double-negative feedback loop, resulting in deregulation of
miR-26a in HCC. These results also suggest that, besides directly
targeting oncogenes, ectopic expression of miR-26a could indirectly
upregulate tumor suppressor genes through suppressing EZH2
expression. Thus, miR-26a may function as a master tumor
suppressor.
Moreover, we restored the balance of the
double-negative feedback loop and found that, similar to miR-26a
restoration, knockdown of EZH2 induced cell growth inhibition, and
overexpression of EZH2 rescued the growth inhibition effect of
miR-26a in HCC cells. These data suggest that the imbalance of
double-negative feedback loop between EZH2 and miR-26a may
contribute to HCC cell growth.
In conclusion, our data suggest that EZH2 and
miR-26a reciprocally regulate each other in HCC, forming a
double-negative feedback loop, which contributes to miR-26a
deregulation and regulates tumor cell growth. Our findings provide
new insights into the regulatory mechanism of miR-26a expression in
HCC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81372259).
References
|
1
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Y, Lu Y, Zhang Q, Liu JJ, Li TJ, Yang
JR, Zeng C and Zhuang SM: MicroRNA-26a/b and their host genes
cooperate to inhibit the G1/S transition by activating the pRb
protein. Nucleic Acids Res. 40:4615–4625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kato M, Goto Y, Matsushita R, Kurozumi A,
Fukumoto I, Nishikawa R, Sakamoto S, Enokida H, Nakagawa M,
Ichikawa T, et al: MicroRNA-26a/b directly regulate La-related
protein 1 and inhibit cancer cell invasion in prostate cancer. Int
J Oncol. 47:710–718. 2015.PubMed/NCBI
|
|
5
|
Yang X, Liang L, Zhang XF, Jia HL, Qin Y,
Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, et al: MicroRNA-26a
suppresses tumor growth and metastasis of human hepatocellular
carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology.
58:158–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fu X, Meng Z, Liang W, Tian Y, Wang X, Han
W, Lou G, Wang X, Lou F, Yen Y, et al: miR-26a enhances miRNA
biogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth
and metastasis. Oncogene. 33:4296–4306. 2014. View Article : Google Scholar
|
|
7
|
Zhang X, Cheng SL, Bian K, Wang L, Zhang
X, Yan B, Jia LT, Zhao J, Gammoh N, Yang AG, et al: MicroRNA-26a
promotes anoikis in human hepatocellular carcinoma cells by
targeting alpha5 integrin. Oncotarget. 6:2277–2289. 2015.
View Article : Google Scholar :
|
|
8
|
Kota J, Chivukula RR, O'Donnell KA,
Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P,
Torbenson M, Clark KR, et al: Therapeutic microRNA delivery
suppresses tumorigenesis in a murine liver cancer model. Cell.
137:1005–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sparmann A and van Lohuizen M: Polycomb
silencers control cell fate, development and cancer. Nat Rev
Cancer. 6:846–856. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu Y, Zhang L, Zhang L, Wang Y, Li H, Ren
X, Wei F, Yu W, Liu T, Wang X, et al: Long non-coding RNA HOTAIR
promotes tumor cell invasion and metastasis by recruiting EZH2 and
repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol.
46:2586–2594. 2015.PubMed/NCBI
|
|
11
|
Chinaranagari S, Sharma P and Chaudhary J:
EZH2 dependent H3K27me3 is involved in epigenetic silencing of ID4
in prostate cancer. Oncotarget. 5:7172–7182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang MD, Chen WM, Qi FZ, Sun M, Xu TP, Ma
P and Shu YQ: Long non-coding RNA TUG1 is up-regulated in
hepatocellular carcinoma and promotes cell growth and apoptosis by
epigenetically silencing of KLF2. Mol Cancer. 14:1652015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Simon JA and Lange CA: Roles of the EZH2
histone methyl-transferase in cancer epigenetics. Mutat Res.
647:21–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Völkel P, Dupret B, Le Bourhis X and
Angrand PO: Diverse involvement of EZH2 in cancer epigenetics. Am J
Transl Res. 7:175–193. 2015.PubMed/NCBI
|
|
15
|
Xu L, Beckebaum S, Iacob S, Wu G, Kaiser
GM, Radtke A, Liu C, Kabar I, Schmidt HH, Zhang X, et al:
MicroRNA-101 inhibits human hepatocellular carcinoma progression
through EZH2 downregulation and increased cytostatic drug
sensitivity. J Hepatol. 60:590–598. 2014. View Article : Google Scholar
|
|
16
|
Gao SB, Zheng QF, Xu B, Pan CB, Li KL,
Zhao Y, Zheng QL, Lin X, Xue LX and Jin GH: EZH2 represses target
genes through H3K27-dependent and H3K27-independent mechanisms in
hepatocellular carcinoma. Mol Cancer Res. 12:1388–1397. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng AS, Lau SS, Chen Y, Kondo Y, Li MS,
Feng H, Ching AK, Cheung KF, Wong HK, Tong JH, et al: EZH2-mediated
concordant repression of Wnt antagonists promotes
β-catenin-dependent hepatocarcinogenesis. Cancer Res. 71:4028–4039.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao SB, Xu B, Ding LH, Zheng QL, Zhang L,
Zheng QF, Li SH, Feng ZJ, Wei J, Yin ZY, et al: The functional and
mechanistic relatedness of EZH2 and menin in hepatocellular
carcinoma. J Hepatol. 61:832–839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Juan AH, Kumar RM, Marx JG, Young RA and
Sartorelli V: Mir-214-dependent regulation of the polycomb protein
Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell.
36:61–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Zhang X, Jia LT, Hu SJ, Zhao J,
Yang JD, Wen WH, Wang Z, Wang T, Zhao J, et al: c-Myc-mediated
epigenetic silencing of MicroRNA-101 contributes to dysregulation
of multiple pathways in hepatocellular carcinoma. Hepatology.
59:1850–1863. 2014. View Article : Google Scholar
|
|
21
|
Cao Q, Mani RS, Ateeq B, Dhanasekaran SM,
Asangani IA, Prensner JR, Kim JH, Brenner JC, Jing X, Cao X, et al:
Coordinated regulation of polycomb group complexes through
microRNAs in cancer. Cancer Cell. 20:187–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ning X, Shi Z, Liu X, Zhang A, Han L,
Jiang K, Kang C and Zhang Q: DNMT1 and EZH2 mediated methylation
silences the microRNA-200b/a/429 gene and promotes tumor
progression. Cancer Lett. 359:198–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tao T, Liu D, Liu C, Xu B, Chen S, Yin Y,
Ang L, Huang Y, Zhang X and Chen M: Autoregulatory feedback loop of
EZH2/miR-200c/E2F3 as a driving force for prostate cancer
development. Biochim Biophys Acta. 1839:858–865. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang B, Liu Y, Luo F, Xu Y, Qin Y, Lu X,
Xu W, Shi L, Liu Q and Xiang Q: Epigenetic silencing of
microRNA-218 via EZH2-mediated H3K27 trimethylation is involved in
malignant transformation of HBE cells induced by cigarette smoke
extract. Arch Toxicol. Dec 20–2014.Epub ahead of print. View Article : Google Scholar
|
|
25
|
Zhang Q, Zhao W, Ye C, Zhuang J, Chang C,
Li Y, Huang X, Shen L, Li Y, Cui Y, et al: Honokiol inhibits
bladder tumor growth by suppressing EZH2/miR-143 axis. Oncotarget.
6:37335–37348. 2015.PubMed/NCBI
|
|
26
|
Liu H, Liu Y, Liu W, Zhang W and Xu J:
EZH2-mediated loss of miR-622 determines CXCR4 activation in
hepatocellular carcinoma. Nat Commun. 6:84942015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Q, Padi SK, Tindall DJ and Guo B:
Polycomb protein EZH2 suppresses apoptosis by silencing the
proapoptotic miR-31. Cell Death Dis. 5:e14862014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu J, He ML, Wang L, Chen Y, Liu X, Dong
Q, Chen YC, Peng Y, Yao KT, Kung HF, et al: MiR-26a inhibits cell
growth and tumorigenesis of nasopharyngeal carcinoma through
repression of EZH2. Cancer Res. 71:225–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dang X, Ma A, Yang L, Hu H, Zhu B, Shang
D, Chen T and Luo Y: MicroRNA-26a regulates tumorigenic properties
of EZH2 in human lung carcinoma cells. Cancer Genet. 205:113–123.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koh CM, Iwata T, Zheng Q, Bethel C,
Yegnasubramanian S and De Marzo AM: Myc enforces overexpression of
EZH2 in early prostatic neoplasia via transcriptional and
post-transcriptional mechanisms. Oncotarget. 2:669–683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Xie J, Xu X, Wang J, Ao F, Wan Y and
Zhu Y: MicroRNA-548 down-regulates host antiviral response via
direct targeting of IFN-λ1. Protein Cell. 4:130–141. 2013.
View Article : Google Scholar
|
|
32
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, He D, Dong XD, Dong F, Wang J,
Wang L, Tang J, Hu DN, Yan D and Tu L: MicroRNA-124a is
epigenetically regulated and acts as a tumor suppressor by
controlling multiple targets in uveal melanoma. Invest Ophthalmol
Vis Sci. 54:2248–2256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lujambio A, Ropero S, Ballestar E, Fraga
MF, Cerrato C, Setién F, Casado S, Suarez-Gauthier A,
Sanchez-Cespedes M, Git A, et al: Genetic unmasking of an
epigenetically silenced microRNA in human cancer cells. Cancer Res.
67:1424–1429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao X, Lwin T, Zhang X, Huang A, Wang J,
Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E and Tao J:
Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell
lymphoma survival and clonogenicity. Leukemia. 27:2341–2350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Börno ST, Fischer A, Kerick M, Fälth M,
Laible M, Brase JC, Kuner R, Dahl A, Grimm C, Sayanjali B, et al:
Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative
prostate cancers implicate an EZH2-dependent mechanism with miR-26a
hypermethylation. Cancer Discov. 2:1024–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kashuba VI, Li J, Wang F, Senchenko VN,
Protopopov A, Malyukova A, Kutsenko AS, Kadyrova E, Zabarovska VI,
Muravenko OV, et al: RBSP3 (HYA22) is a tumor suppressor gene
implicated in major epithelial malignancies. Proc Natl Acad Sci
USA. 101:4906–4911. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Senchenko VN, Anedchenko EA, Kondratieva
TT, Krasnov GS, Dmitriev AA, Zabarovska VI, Pavlova TV, Kashuba VI,
Lerman MI and Zabarovsky ER: Simultaneous down-regulation of tumor
suppressor genes RBSP3/CTDSPL, NPRL2/G21 and RASSF1A in primary
non-small cell lung cancer. BMC Cancer. 10:752010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dmitriev AA, Rudenko EE, Kudryavtseva AV,
Krasnov GS, Gordiyuk VV, Melnikova NV, Stakhovsky EO, Kononenko OA,
Pavlova LS, Kondratieva TT, et al: Epigenetic alterations of
chromosome 3 revealed by NotI-microarrays in clear cell renal cell
carcinoma. BioMed Res Int. 2014:7352922014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kashuba V, Dmitriev AA, Krasnov GS,
Pavlova T, Ignatjev I, Gordiyuk VV, Gerashchenko AV, Braga EA,
Yenamandra SP, Lerman M, et al: NotI microarrays: Novel epigenetic
markers for early detection and prognosis of high grade serous
ovarian cancer. Int J Mol Sci. 13:13352–13377. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Viré E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden
JM, et al: The Polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar
|
|
42
|
Wang G and Sun Y, He Y, Ji C, Hu B and Sun
Y: miR-26a promoted by interferon-alpha inhibits hepatocellular
carcinoma proliferation and migration by blocking EZH2. Genet Test
Mol Biomarkers. 19:30–36. 2015. View Article : Google Scholar
|