Introduction
Traditional chemotherapy and radiotherapy of cancer
treatments suffer from severe side effects, development of drug
resistance and cross-resistance, cancer migration and recurrence
(1). In light of recent
breakthroughs in molecular oncology, targeted therapies using
monoclonal antibodies to mutated cell surface receptors and small
molecule agents inhibiting tyrosine kinases, serine/threonine
kinases, small GTP-binding proteins and other oncogenic proteins in
the proliferation-driving signaling pathways have become standard
in the current treatment of cancer (2). However, due to the genetic
heterogeneous nature of cancer cells, particularly for solid
tumors, resistance to these targeted agents can develop rapidly,
and thus limit the overall efficacy (3). Novel cancer therapies are needed. One
potential immuno-surveillance mechanism for therapy is apoptosis
induced by cytokines produced by immune cells such as T and natural
killer (NK) cells.
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL), discovered two decades ago by Wiley et al
(4) and Pitti et al
(5), attracted enthusiastic
attention worldwide as a potential cancer therapy because of its
capacity to specifically induce cancer cell death, but not the
death of normal and healthy cells (6). TRAIL produced from immune NK cells
(7), can induce apoptosis of
cancer cells upon binding to the cell surface death receptors (DR,
TRAIL receptor), DR4 (or TRAIL R1) and/or DR5 (or TRAIL R2). In
addition, TRAIL recruits the adaptor Fas-associated death domain
(FADD) and procaspase-8 to form death-inducing signaling complexes
(DISC), which results in the activation of the initiator caspase-8,
leading to the activation of extrinsic and intrinsic apoptotic
signaling downstream of caspase-3 (4,8).
Recently, several phase 2 clinical studies based on the use of
recombinant human TRAIL or agonistic monoclonal antibodies against
DR4/5 have failed to show clinical efficacy, even when combined
with traditional chemotherapy (9,10).
Thus, enthusiasm has greatly dampened for cancer therapies based on
TRAIL-induced apoptosis. Moreover, in the past decade, studies have
demonstrated that only a small portion of cancer cells are
sensitive to TRAIL, while most tumors were TRAIL-resistant
(11,12). This property limits the potential
of TRAIL-based cancer therapy.
Currently, inhibitors of the apoptosis proteins,
cellular FLICE-like inhibitory protein (c-FLIP) and inhibitors of
apoptosis protein (IAPs, including XIAP) are considered to be
responsible for cellular TRAIL resistance. The utility of
TRAIL-based therapy is dependent on mitigating this TRAIL
resistance. IAPs bind to downstream executor caspases-3/6/7/9 to
inhibit their activities and prevent the execution of apoptosis
(13,14). To overcome this obstacle, IAPs
antagonists with excellent activity in vivo have been
developed, and several of these antagonist (e.g., AT406) are
currently under clinical investigation (15–18).
These IAP antagonists are second mitochondria-derived activator of
caspase (Smac) mimetics. c-FLIP, a procaspase-8 homologue, can
compete with procaspase-8 to bind to the death effective domain
(DED) of FADD and block the apoptotic signal from upstream of the
apoptosis pathway (19). In
vitro studies with some cytotoxic anticancer agents revealed
that the downregulation of c-FLIP induced by these agents was
partly responsible for their pro-apoptotic effects (20).
However, there is no specific antagonist available
for c-FLIP (21). Downregulating
the expression of c-FLIP through specific siRNA sensitized
resistant melanoma cells to TRAIL-induced apoptosis (22). Rocaglamide, a natural product
isolated from Aglaia species, is a translational inhibitor
of de novo c-FLIP synthesis (23,24).
Previous studies showed that a c-FLIP inhibitor and a XIAP
inhibitor cooperatively sensitized TRAIL-mediated apoptosis in
Hodgkin's lymphoma cells (25).
However, no studies have shown that a triple combination can be
effective in other solid tumors. Recent genetic analysis for
various tumor cells revealed the extremely heterogeneous nature of
cancers (1). The results in a
single cancer cell line cannot be generalized to other types of
cancer cells without empirical evidence. Furthermore, there is no
safety testing on normal cells for this combination treatment. In
our investigation, a combination of AT406 (A) a pan-antagonist of
IAPs, rocaglamide (R) or c-FLIP-siRNA and TRAIL (T) (ART triple
combination) was used to evaluate its possible broad spectrum
activities on selected 17 solid cancer cell lines (from different
tissues or organs), three glioma cell lines and two normal cells
(pulp cells and MRC5). In addition, various combination effects
were assessed. Our study showed that the ART-triple combination may
be applied as a broad-spectrum antitumor therapeutic approach for
cancer treatment. We also confirmed that our triple combination
treatment had no harmful effects on normal cells tested, similar to
TRAIL-only treatment. These features provide a theoretical and
experimental basis for the TRAIL-induced apoptosis pathway as a
potential target for cancer treatment.
Materials and methods
Cell lines and culture conditions
The cancer cell lines U87, SW480, U251 and U373 were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). HCT116, HT29, LOVO, H460, SK-OV-3,
MDA-MB-231, A549, MCF7, SK-BR-3, T-47D, BT474, U2OS, HeLa, HepG2,
MDA-MB-468, Vcap, and MRC5 were purchased from ATCC (MD, USA).
HCT116, HT29, LOVO, H460, SK-OV-3, MDA-MB-231, A549, U87, MCF7,
SK-BR-3, T-47D, BT474 and SW480 were maintained in RPMI-1640
(Hyclone, USA). U2OS, HeLa, HepG2, MDA-MB-468, Vcap, U251 and U373
were cultured in Dulbecco's modified minimal essential medium
(DMEM) growth medium (Hyclone). MRC5 cells (human embryonic lung
cells) were maintained in MEM growth medium (Hyclone). All culture
media were supplemented with 10% fetal bovine serum (Hyclone). All
cancer cells were maintained in a humidified incubator at 37°C with
5% CO2, and passaged with 0.25% trypsin-EDTA when ~80%
confluence was reached. The pulp cells were isolated and cultured
according to a previously described method (26).
Antibodies and chemicals
The antibodies for immunoblotting were from the
following sources: mouse anti-caspase-8 p55/53/43/41/18 (#9746),
rabbit anti-PARP p118/89 (#9532) and mouse anti-caspase-3 p35/19/17
(#9668) were from Cell Signaling Technology (Beverly, MA, USA);
mouse anti-FADD (#F8053)was from Sigma (St. Louis, MO, USA);rabbit
anti-DR5 was from Abcam (Cambridge, MA, USA); mouse anti-c-FLIP
(clone 7F10, #ALX-804-961-0100) was from Enzo Life (New York, NY,
USA) and mouse anti-GAPDH (sc-365062) was from Santa Cruz
Biotechnology (Dallas, TX, USA). Recombinant hTRAIL (#310-04) was
from R&D Systems (Minneapolis, MN, USA), rocaglamide
(#350-121-C100) was purchased from Enzo Life, AT406 (#S2754) was
from Selleckchem (Houston, TX, USA) and valproic acid was from
J&K Scientific.
Cell viability assay
Cells were seeded in 96-well cell culture plates and
treated the next day with the given agents for the indicated times.
The cells were then incubated with 100 μg/well of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
at 37°C for 4 h. Finally, the medium was discarded carefully and
150 μl of dimethyl sulfoxide (DMSO) was added to solubilize the
formazan crystals. The absorbance was measured using a Microplate
Reader (Perkin-Elmer 2030) at a wavelength of 490 nm. The
experiments were performed in triplicate.
Caspase activity
Cells were seeded in 96-well cell culture plates
(white/black walled) and treated the next day with the given agents
for the indicated times, then manipulated according to the
technical bulletin of Caspase-Glo 3/7 assay (Promega).
Western blotting
For each sample, 1×106 cells were lysed
using a solubilizing solution [20 mM Tris-HCl (pH 7.4), 150 mM
NaCl, 1% NP-40, 1 mM PMSF, 0.02% NaN3, protease
inhibitor cocktail tablet; Roche, Mannheim, Germany]. Protein
concentration was determined using a Bio-Rad Protein assay kit
(Hercules, CA, USA). An equal quantity (10–30 μg) of proteins was
separated by 10–15% SDS-PAGE and transferred onto a PVDF membrane
(Millipore Corp., Billerica, MA, USA). The membrane was blocked in
10% skim milk (in TBS, pH 7.2, containing 0.1% Tween-20) overnight
at 4°C, then incubated with primary antibodies followed by
peroxidase-conjugated anti-mouse or anti-rabbit IgG (Thermo Fisher,
Inc., Rockford, IL, USA). The epitope was detected using an ECL
western blot detection kit (Millipore Corp.). GADPH was used as an
internal control for all western blots.
Data analysis
Data were expressed as the means ± SD values for
western blots or mean ± SEM for cell viability MTT assays.
Statistical analysis was performed by using Graphpad Prism 5
software. The IC50 values were evaluated by SPSS version
19. The one-way ANOVA followed by a post hoc multiple comparison
test was used to compare control (positive control) and treated
groups. A p-value <0.05 was considered statistically
significant. The cell viability and blot experiments were performed
in triplicate. Densitometry analysis was performed by using ImageJ
software.
Results
Inhibition of c-FLIPS/L and
IAPs can increase the sensitivity of U2OS to TRAIL-induced
apoptosis
By evaluating TRAIL promotion of tumor cell
apoptosis, we found that U2OS is resistant to TRAIL-induced
apoptosis. Because c-FLIP is a crucial apoptotic resistance factor,
we used the c-FLIP-siRNA overexpression plasmid which was reported
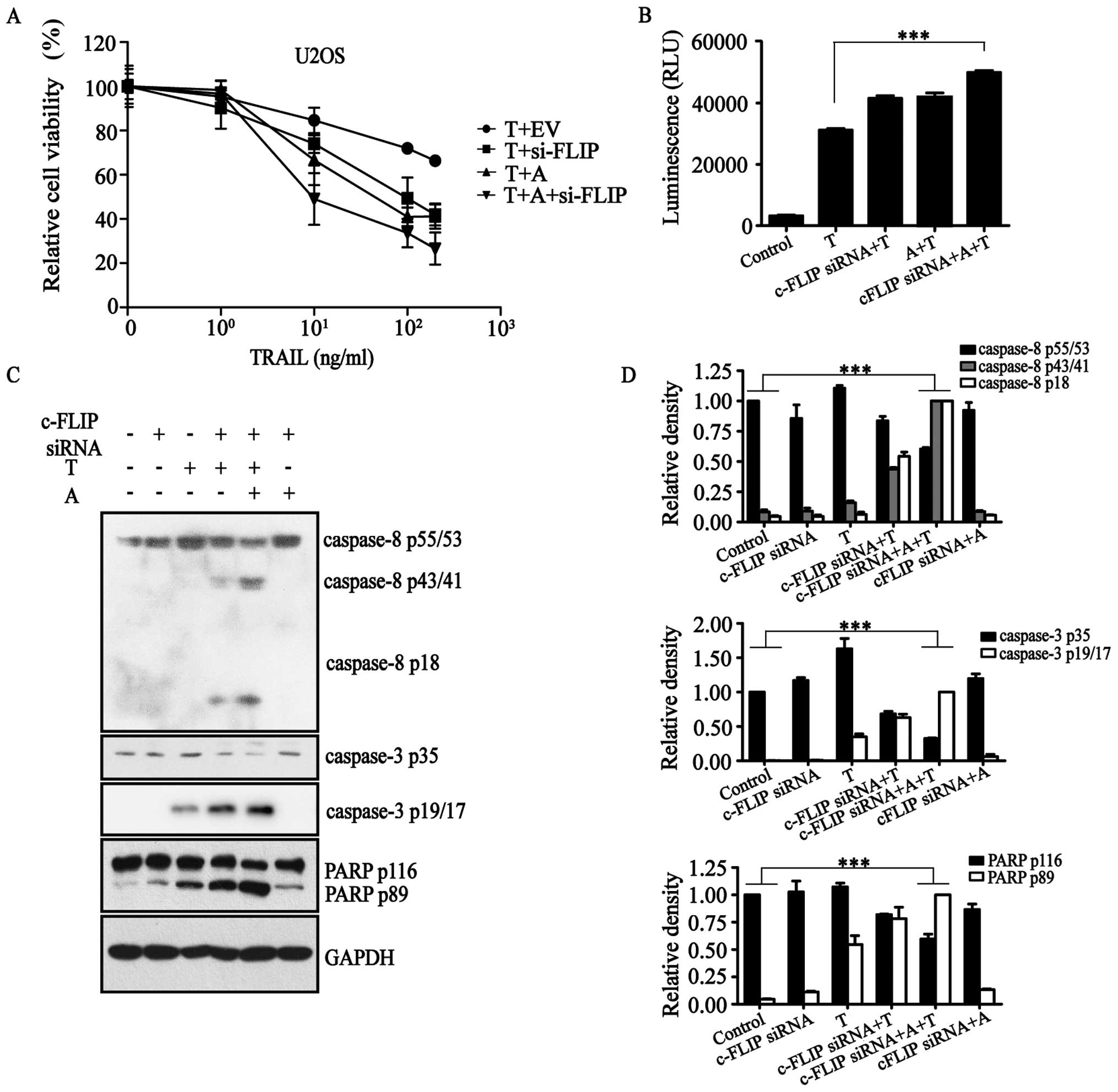
in our previous study to inhibit the expression of c-FLIP (Fig. 1A) (27). Furthermore, we determined the
effective concentrations of AT406 (SM406), a synthetic Smac mimetic
that is a pan-antagonist of IAPs, including XIAP (Fig. 2A, left). The results of an MTT
assay showed that in combination with TRAIL with or without
c-FLIP-siRNA, AT406 could increase the cell death rate of U2OS
(Fig. 1A).
Results of the Caspase-Glo 3/7 assay (Fig. 1B) confirmed that c-FLIP-siRNA in
the presence of AT406 activated caspase proteins in the
TRAIL-activated apoptotic pathway. That is, apoptosis initiator
protein caspase-8, apoptosis executor protein caspase-3 and
apoptosis substrate poly(ADP-ribose) polymerase (PARP) were all in
their cleaved states to various degrees (Fig. 1C and D). All of these results
demonstrated that c-FLIP-siRNA and AT406 together activated the
extrinsic apoptotic pathway in the presence of TRAIL to induce
apoptosis.
Triple combination of
TRAIL/AT406/rocaglamide functions via an extrinsic apoptosis
pathway
The c-FLIP-siRNA, mentioned above, cannot be
transfected successfully to most cell lines. Rocaglamide is a
natural product isolated from Aglaia species and has been
previously shown to downregulate the expression of c-FLIP in
leukemic T cells (23) and
Hodgkin's lymphoma cells (25).
Therefore, we tested whether rocaglamide could replace the siRNA.
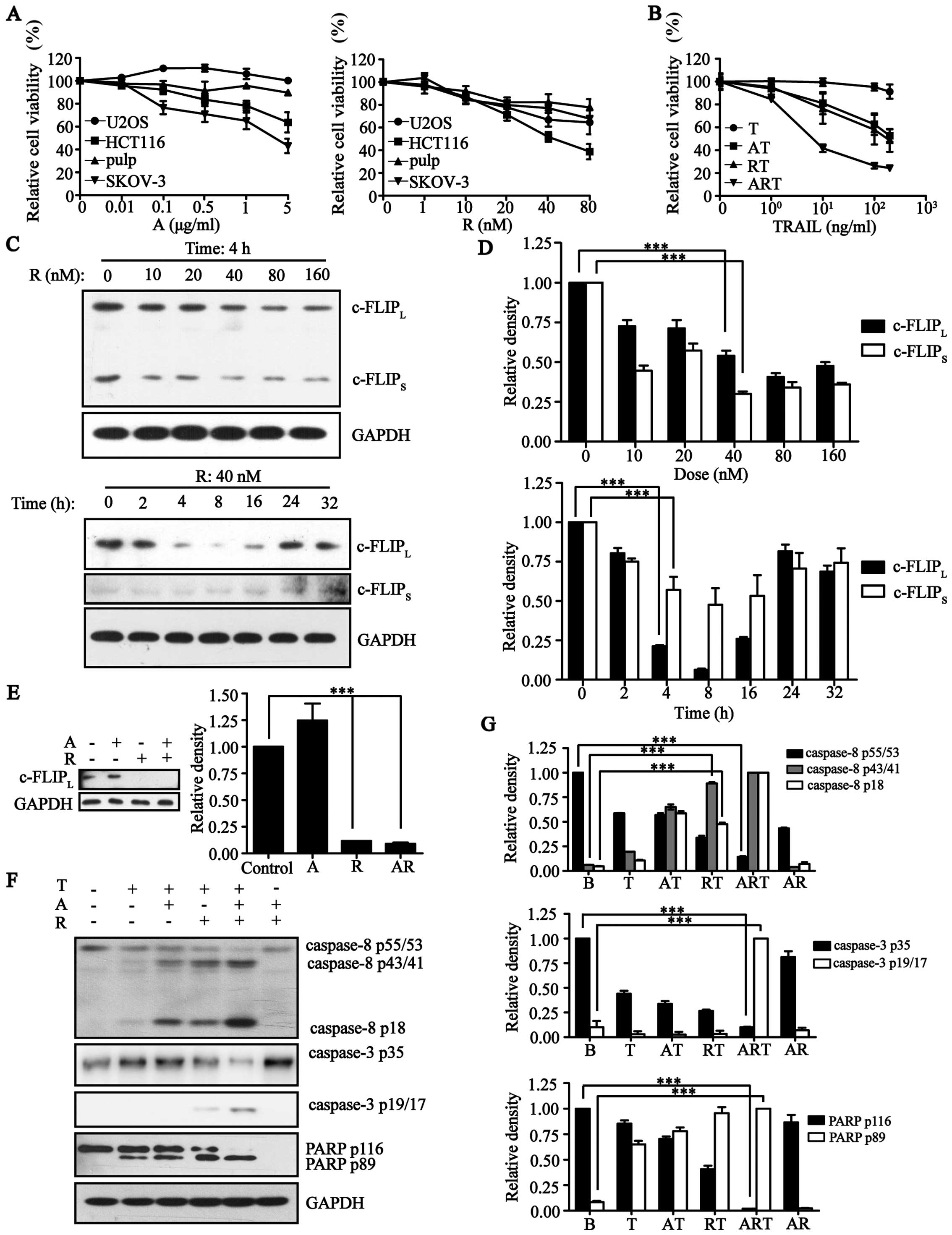
After determining the scope of drug safety of rocaglamide (Fig. 2A), we found that rocaglamide could
also inhibit the expression of c-FLIP effectively (Fig. 2C and D). Because AT406 did not
interfere with rocaglamide (Fig.
2E), we combined AT406 and rocaglamide with TRAIL. This
combination could also increase the cell death rate of U2OS cells
(Fig. 2B) and similarly activated
caspase-8, caspase-3 and PARP by increasing the cleaved and active
forms of these caspases (Fig. 2F and
G). This result showed that, rocaglamide could inhibit c-FLIP
as well as c-FLIP-siRNA, and further activated the extrinsic
apoptotic pathway in combination with AT406 in the presence of
TRAIL.
The actions of the triple combination
overcame the resistance of most solid tumor cell lines to
TRAIL-induced apoptosis
We collected 20 tumor cell lines that originated
from colorectal cancers (HCT116, HT29, LOVO, SW480), lung cancers
(H460, A549), ovarian cancer (SK-OV-3), osteosarcoma (U2OS), breast
cancers (MDA-MB-231, SK-BR-3, T-47D, BT474, MDA-MB-468, MCF7),
cervical cancer (HeLa), liver cancer (HepG2), prostate cancer
(Vcap) and gliomas (U87, U251, U373) to test for apoptotic effects
induced by TRAIL alone (Table I).
Among these twenty tumor cell lines, only two cell lines, HCT116
and LOVO, responded to TRAIL; the remaining 18 tumor cell lines
were highly resistant.
 | Table IThe sensitivity to TRAIL-induced
apoptosis of 20 cancer cell lines with or without pre-treatment of
AT406 (A) or rocaglamide (R) or A + R combination. |
Table I
The sensitivity to TRAIL-induced
apoptosis of 20 cancer cell lines with or without pre-treatment of
AT406 (A) or rocaglamide (R) or A + R combination.
| | IC50
(ng/ml) | Imax
(%) |
|---|
| |
|
|
|---|
| Entry | Cell line | T | AT | RT | ART | T | ART-24 h | ART-72 h |
|---|
| 1 | LOVO | 4.2±1.4 | 0.3±0.9 | 7.4±7.9 | 0.5±0.3 | 22±7 | 33±8 | 6±0.3 |
| 2 | HCT116 | 54.2±19 | 12±2.9 | 5.4±5.1 | 2.7±3.3 | 22±8 | 17±6 | 2±0.1 |
| 3 | HT29 | >200 | 48.7±21 | 106.8±56 | 3.4±1.1 | 91±3 | 14±3 | 7±0.5 |
| 4 | H460 | >200 | 158±40.7 | 9±2.9 | 4.3±0.9 | 51±7 | 34±6 | 6±0.6 |
| 5 | BT474 | >200 | >200 | 89.3±26 | 26.2±7.8 | 97±3 | 20±1 | 3±0,7 |
| 6 | SK-BR-3 | >200 | >200 | 122.3±119 | 21.9±2.1 | 97±6 | 29±1 | 5±0.2 |
| 7 | U2OS | >200 | >200 | >200 | 11.3±12 | 91±8 | 24±2 | 3±0.5 |
| 8 | T-47D | >200 | >200 | >200 | 35.8±13.2 | 102±1 | 32±3 | 7±0.3 |
| 9 | MDA-MB-231 | >200 | >200 | >200 | 17.8±7.3 | 81±6 | 33±2 | 7±0.4 |
| 10 | SK-OV-3 | >200 | >200 | >200 | 56.6±32.8 | 83±7 | 41±2 | 13±1.8 |
| 11 | Vcap | >200 | >200 | >200 | 52.5±20 | 100±2 | 43±3 | 15±1.2 |
| 12 | SW480 | >200 | >200 | >200 | >200 | 88±10 | 62±8 | 14±5.3 |
| 13 | MDA-MB-468 | >200 | >200 | >200 | >200 | 93±4 | 71±2 | 7±2.4 |
| 14 | HeLa | >200 | >200 | >200 | >200 | 101±3 | 57±4 | 27±0.4 |
| 15 | Hep G2 | >200 | >200 | >200 | >200 | 70±6 | 76±7 | 17±7.9 |
| 16 | A549 | >200 | >200 | >200 | >200 | 81±8 | 68±12 | 32±10.9 |
| 17 | U87 | >200 | >200 | >200 | >200 | 85±9 | 73±8 | 36±2.7 |
| 18 | MCF7 | >200 | >200 | >200 | >200 | 104±9 | 93±7 | 48±13.2 |
| 19 | U251 | >200 | >200 | >200 | >200 | 108±7 | 101±12 | 81±3.4 |
| 20 | U373 | >200 | >200 | >200 | >200 | 94±4 | 102±4 | 93±4.4 |
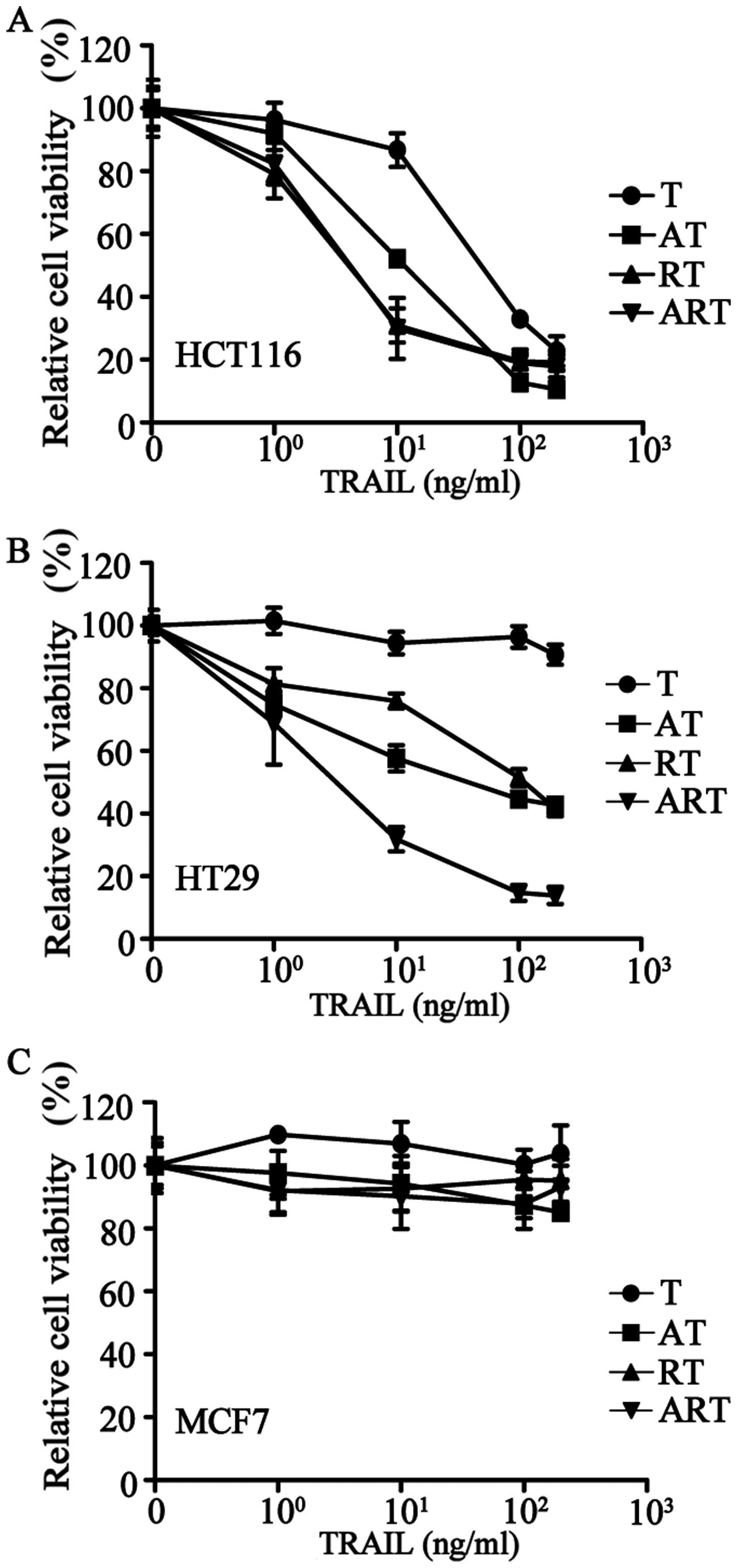
The addition of AT406 to HCT116 (a TRAIL sensitive
cell line) or HT29 (a TRAIL-resistant cell line) to antagonize IAPs
significantly improved the apoptotic effects of TRAIL (Fig. 3A and B). Pre-treating these two
cell lines with rocaglamide to downregulate the expression of
c-FLIP showed similar effects (Fig. 3A
and B). Furthermore, after a combined pre-treatment of
rocaglamide and AT406, TRAIL-resistant HT29 cells became highly
sensitive to TRAIL (Fig. 3B).
Next, we treated the 20 cancer cell lines with a
combination of AT406 (1 μg/ml), rocaglamide (40 nM) and TRAIL at
various concentrations (ART combination) (Table I) for 24 h of treatment. Our
results showed that these 20 tumor cell lines could be divided into
the following three groups: group 1, highly sensitive (<50%
relative cell viability), 11 cell lines (e.g., HCT116) (Fig. 3A); group 2, sensitive (50–80%
relative cell viability), 6 cell lines (e.g., HT29) (Fig. 3B); and group 3, resistant (>80%
relative cell viability), 3 cell lines (e.g., MCF7) (Fig. 3C). This ART-induced cell death can
be further improved over time by extending the treatment to a 72-h
incubation. Only three cancer cell lines were resistant to ART even
after 72-h treatment (Table I),
i.e., the breast cancer cell line, MCF7, and brain cancer cell
lines, U373 and U251.
Inhibition of IAPs or c-FLIP alone is not
sufficient to achieve the remarkable cell death induced with an ART
triple combination
To confirm that an ART triple combination is
necessary for inducing profound cell death, we determined the cell
viability inhibition concentration (IC50 values) of
TRAIL on cancer cells with or without AT406 or rocaglamide
pre-treatment (Table I). When
cells were treated with TRAIL alone, IC50 values could
be obtained only on TRAIL-sensitive LOVO and HCT116 cells. After
combination with either AT406 or rocaglamide pre-treatment (AT or
RT combination), the potency of TRAIL increased >4-fold (lower
IC50 values) in these two cell lines (entries 1 and 2),
and IC50 values could be obtained on other cancer cells
(8 cell lines for AT, 11 cell lines for RT). ART triple combination
further enhanced the apoptosis-inducing effects of TRAIL. For the
11 highly sensitive cancer cell lines, >50% apoptosis was
observed after 24-h treatment (entries 1–11). IC50
values using TRAIL on these cells were markedly lower than those
values using AT or RT combinations, and 7 of them were <10
ng/ml. When ART exposure was extended to 72 h, the relative cell
viability (Imax values) further decreased to <10% for
10 cell lines, and 10–40% for 7 cell lines (column of ART-72 h).
Only three cells remained resistant to ART (entries 18–20).
On a protein level, cleaved caspases and PARP were
barely detected when U2OS cells were treated with TRAIL alone.
However, ART completely activated the extrinsic apoptotic pathway
and resulted in cleavage of all related proteins (Fig. 2F and G).
Triple combination is safe for normal
cells
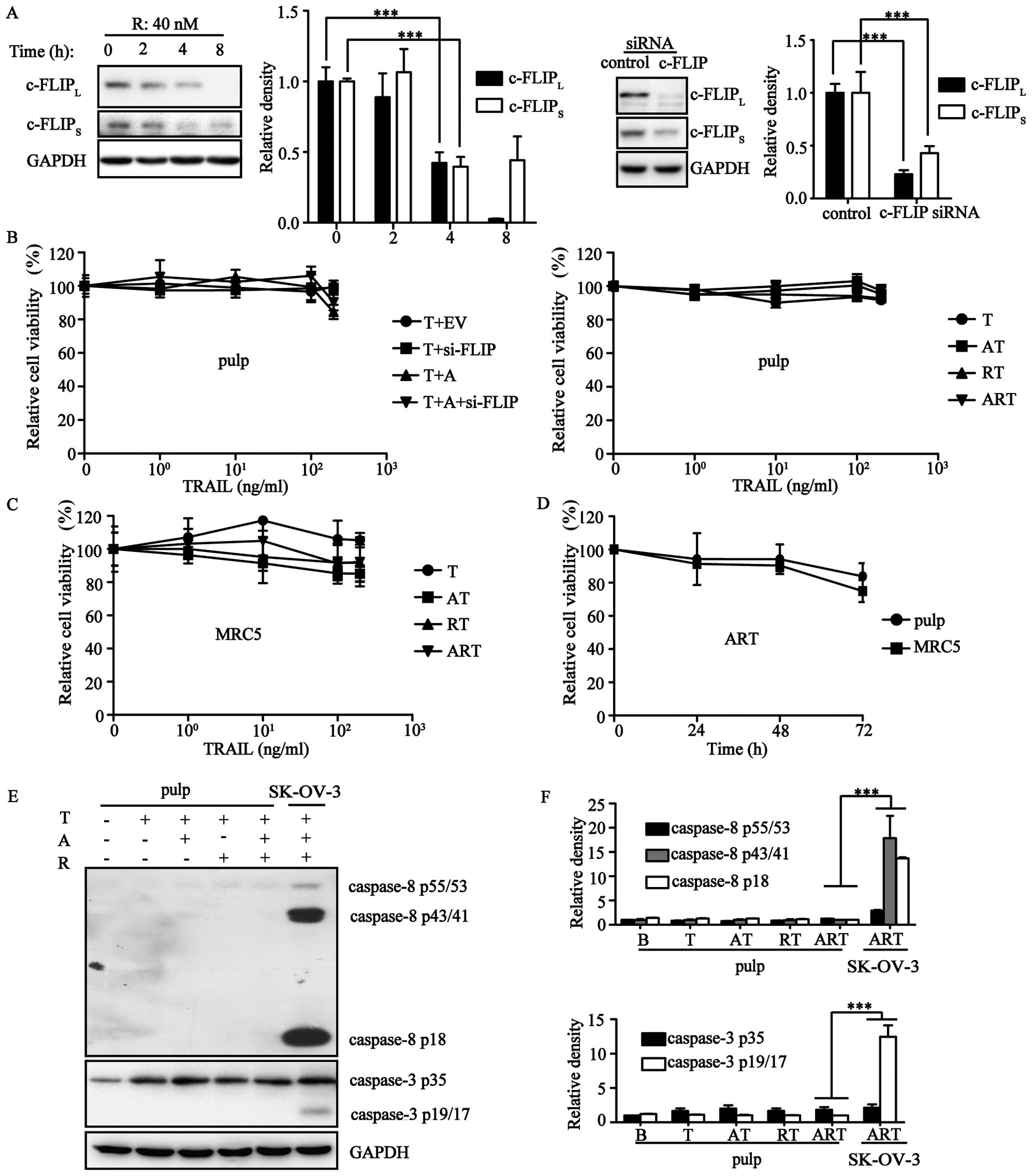
Mounting evidence has demonstrated that TRAIL does
not hurt normal cells. To assess the safety of ART, cell viability
assays were carried out using pulp and MRC5 cells. Our results
showed that both ART and its equivalent AT406/c-FLIP-siRNA/TRAIL
combination could not induce cell death in these normal cells
(Fig. 4A–C). Both of these normal
cells were resistant to ART even after a 72-h treatment (Fig. 4D). Next, we evaluated the variation
of protein expression levels during TRAIL-induced extrinsic
apoptosis. Pulp expression of procaspase-3 and procaspase-8 were
much lower than expression in the ovarian cancer cell line SK-OV-3.
After stimulation by A/R/T in different combination patterns, we
did not observe cleavage of the apoptotic proteins procaspase-3/8,
but rather, only a slight upregulation of these two apoptotic
proteins (Fig. 4E and F).
ART-resistance may attribute to the low
expression of procaspases
To investigate the molecular mechanism of TRAIL- and
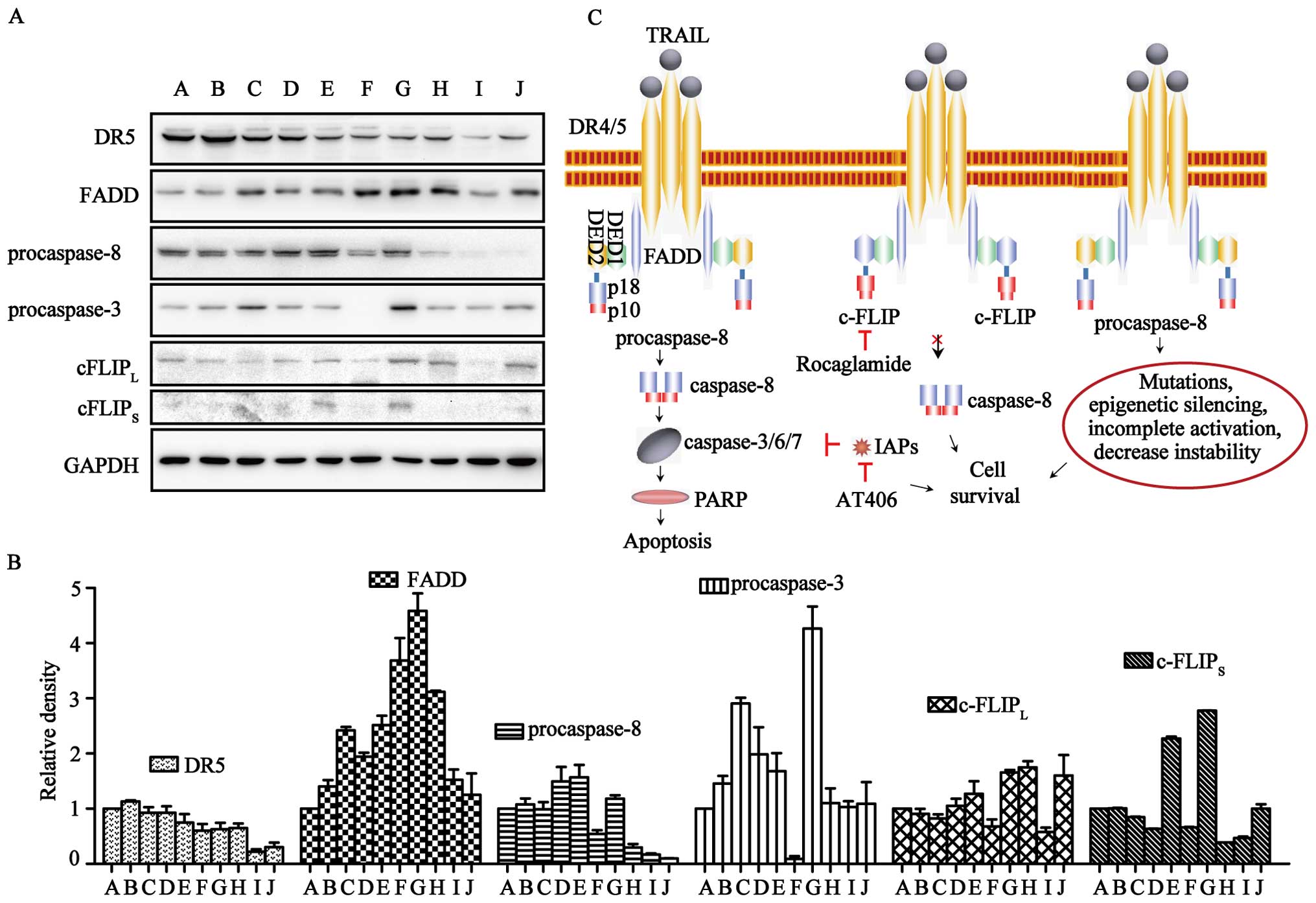
ART-resistance in normal and cancer cells, western blot analysis
was carried out for the key proteins involved in the TRAIL
apoptotic pathway, i.e., DR5, FADD, procaspase-3/-8 and
c-FLIPL/S (Fig. 5A and
B). DR5 was expressed in all of the cell lines, but levels in
TRAIL-resistant cells were lower. FADD was expressed in all cells
at levels unrelated to TRAIL- or ART-resistance. The expression of
c-FLIPL was found in all ten cell lines, although it was
relatively higher in U87, U251 and pulp cells. c-FLIPS could be
clearly observed in HT29, U87 and pulp cells, and it was difficult
to detect in other cells. Notably, procaspase-8 was expressed at
relatively high levels in ART-sensitive cells (LOVO, HCT116, HT29,
U2OS and SK-OV-3 cells) and at significantly lower levels in
ART-resistant cells (U251, U373 and pulp cells). In contrast,
procaspase-3 in ART-resistant MCF7 cells was nearly invisible.
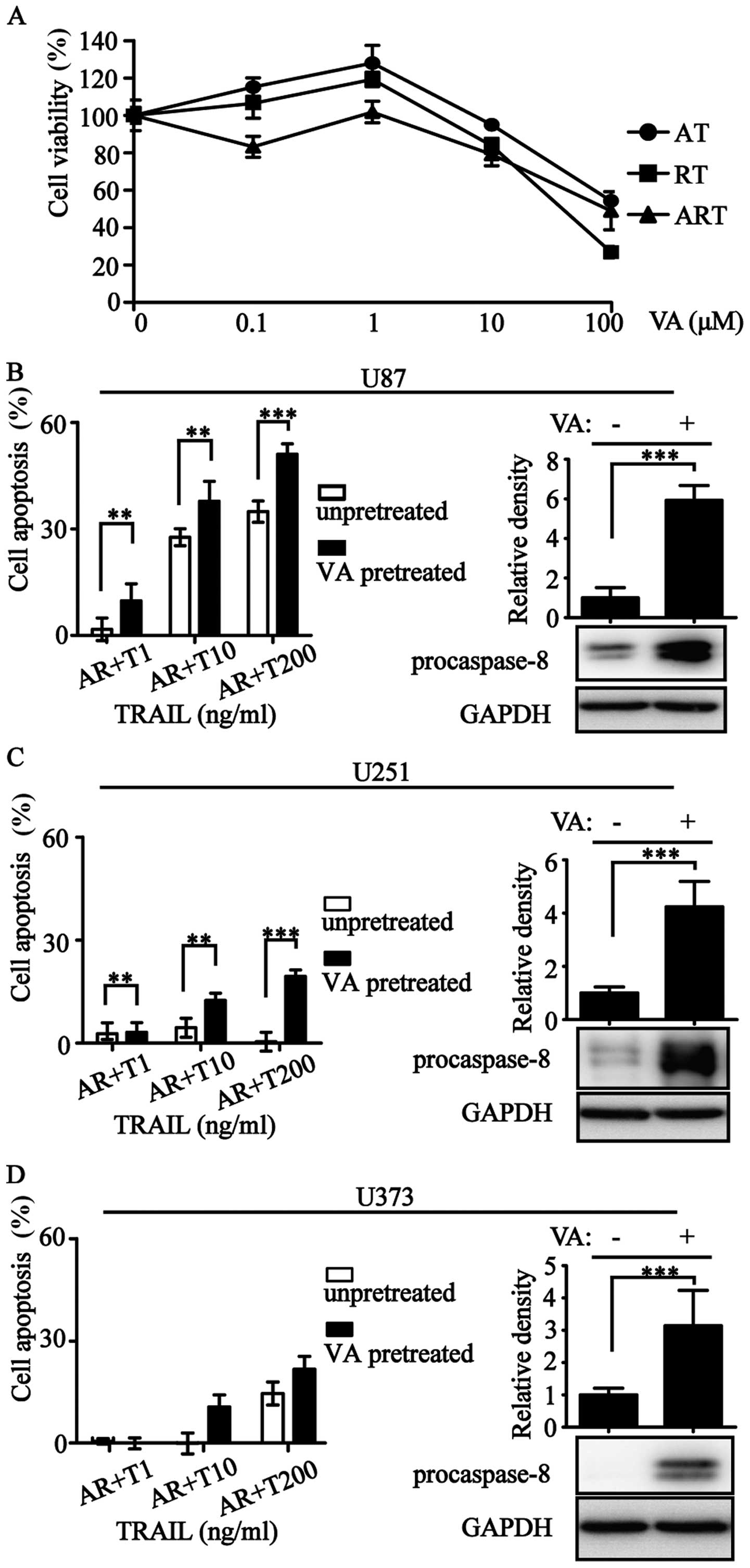
Valproic acid (VA) is an antiepileptic drug with histone
deacetylase inhibitory activity (28). We used VA at an appropriate
concentration to increase procaspase-8 expression (Fig. 6). The improvement of procaspase-8
expression resulted in the enhancement of apoptotic rate (Fig. 6B–D). Overall, levels of
procaspase-3/-8 correlated with ART-sensitivity.
Discussion
Recombinant human TRAIL and several agonistic
monoclonal antibodies of DR4/5 have been developed and used in
clinical trials (28–30). However, their development as a
cancer therapy is hampered by the resistance observed in most
cancer cells (31,32). Although synergy has been described
for combinations of TRAIL with a variety of cytotoxic agents,
including etoposide, 5-FU, oxamflatin, sorbitol, staurosporine,
MG132, bortezomib, doxorubicin, azacitidine and sorafenibin, these
synergistic effects were apparent in only TRAIL-sensitive tumor
cells (33). However, in resistant
tumor cells, a combination of TRAIL or DR4/5 agonist with
chemotherapy showed no profound effect on cell death.
In this study, we hypothesized that TRAIL resistance
could be overcome by simultaneously attacking three apoptosis
inhibiting factors. First, we demonstrated that a higher
concentration of TRAIL (200 ng/ml) would be necessary for
competitive binding of DcR1/2 (34). Second, we showed that the
apoptosis-inhibiting function of IAPs can be blocked by adding
AT406, a small molecule IAPs inhibitor in clinical trials. Third,
we showed that the cytosolic apoptotic inhibitor
c-FLIPL/S can be downregulated by transient transfection
of a plasmid containing c-FLIP-siRNA or by adding rocaglamide, a
known c-FLIP expression suppressor (Fig. 5C). We firmly believe that only with
the elimination of the main inhibitory factors of an extrinsic
apoptotic pathway, can a drug combination based on TRAIL achieve
the strongest and broadest therapeutic effects. This may also be
the reason for failure of clinical trials of TRAIL or agonist
monoclonal antibodies of death receptors and cytotoxic agent
combinations on different cancer types. Additionally, in
vitro culture systems are limited in making in vivo
predictions because TRAIL is quickly degraded in vivo
(35,36).
A double combination of either AT or RT only
slightly improved the TRAIL-induced apoptosis on cancer cells. In
contrast, powerful combined effects were observed for ART on most
of the resistant cancer cells. Among the 18 TRAIL-resistant cancer
cells, after a 72-h triple combination treatment, 8 became highly
sensitive (<10% relative cell viability), and 7 became sensitive
(10–50% relative cell viability). The normal pulp and MRC5 cells
remained highly resistant to an ART triple combination treatment
(Fig. 3 and Table I). These results clearly
demonstrate that the ART triple combination generates effects that
specifically activates an extrinsic apoptotic pathway in solid
tumor cells. The profound apoptotic effects of ART triple
combination on cell lines originally from solid tumors also
suggests that c-FLIPs and IAPs contribute to the high resistance of
these cancer cells to TRAIL-induced apoptosis. Therefore, a
combination of DR 4/5 agonist, IAPs antagonist and a c-FLIP
antagonist, i.e., the ART combination and its equivalents, is
likely a better cancer therapy than TRAIL or agonistic monoclonal
antibodies of the death receptors alone. These results also suggest
that simultaneous activation of death receptors, inhibition of
c-FLIP and antagonization of IAPs are minimally required for
induction of profound apoptosis of solid tumor cells.
It is worth noting that ART triple combination does
not impair normal cells. Previous research suggests that
TRAIL-resistant cancer cells are resistant due to the expression of
only one anti-apoptotic protein and that these cells have lost the
redundancy in resistance mechanisms observed in non-transformed
cells (37). In contrast, our
study suggests that most of these highly resistant cancer cells
rely on multiple tolerance mechanisms rather than only one
resistance mechanism. We suggest that TRAIL-resistance mechanisms
of normal cells are far more complicated than cancer cells and at
the same time, confirm that ART triple combination is safe for
normal primary culture cells from adults or normal passage cells
from the human embryo.
Among the 20 cancer cell lines used in this study,
the breast cancer cell line MCF7 and two brain cancer cell lines
remained highly resistant to ART triple combination, even after a
72-h exposure (Table I and
Fig. 3). Western blot analysis
revealed that c-FLIP is expressed in these cell lines; however, the
levels of procaspase-8 in these cell lines are lower than in the
other cell lines. In addition, MCF7 showed extremely low, nearly
undetectable, levels of procaspase-3 (Fig. 5). These results suggest that low
expression or loss of procaspase-8 and -3 is a likely mechanism for
resistance to the ART combination. MCF-7 is deficient of caspase-3
and is relatively insensitive to many traditional chemotherapeutic
agents (38–40). Because of the indispensable role of
the extrinsic apoptotic pathway, deficiency of caspase-3 in MCF-7
results in high TRAIL-resistance during exposure to the ART
combination.
Eggert et al (41) reported that only one out of 18
neuroblastoma cell lines showed sensitivity to TRAIL-induced
apoptosis. This number of TRAIL sensitive cell lines increased to
five by adding cycloheximide (CHX, a protein synthesis inhibitor)
to cultured cells to inhibit the synthesis of c-FLIP. The remaining
13 TRAIL-resistant neuroblastoma cell lines (70%) showed a loss of
procaspase-8 expression, correlating with resistance to
TRAIL-induced apoptosis. These results suggest that in addition to
c-FLIP, the loss of procaspase-8 plays a major role in TRAIL
resistance in brain cancer cells that originated from a
neuroblastoma. In our present study, similarly, all three glioma
cell lines and normal pulp cells showed high resistance to TRAIL,
as well as ART triple combination treatment (Fig. 3 and Table I). Two (U373 and U251) of these
glioma cell lines and pulp cells also expressed at very low levels
of procaspase-8 and variable levels of c-FLIPL/S,
suggesting that the resistance of these cells to TRAIL and ART are
the results of low level expression of procaspase-8 and the
presence of anti-apoptosis factors, such as c-FLIP and/or IAPs. The
underlying mechanisms of TRAIL resistance caused by decreased
expression of the caspase-8 initiator have been attributed to
epigenetic silencing, such as DNA methylation, histone acetylation
modification (42–45). Indeed, when histone deacetylase
inhibitor was used on three glioma cell lines, the procaspase-8
expression increased. There is more cell apoptosis under ART
treatment (Fig. 6). Decreased
caspase-8 activities in some cancer cells are related to
procaspase-8 gene mutation (41,46–48),
decrease in stability, and incomplete activation (42–46,49,50)
(Fig. 5C). The loss of
procaspase-8 expression is particularly prevalent in both
neuroblastoma (39) and glioma
brain cancer cells. These results may explain the poor prognosis of
patients suffering from malignant brain cancers regardless of the
therapy.
Recently reported unsuccessful clinical phase 2
studies of rhTRAIL or agonistic monoclonal antibodies to death
receptors using a randomly unselected patient population, with and
without traditional chemotherapy (10,11),
suggest that future clinical studies should consider the
aforementioned mechanisms of TRAIL resistance in tumors. For
peripheral solid tumors, it appears that a majority of tumor cells
are sensitive to ART triple combination therapy; however, a small
number of these tumors may have mutations in the procaspase-8 gene
rendering caspase-8 inactive (50). Future clinical protocol designs and
prognosis analysis based on death receptors should exclude patients
with abnormal expression of procaspase-8 (51).
Indeed, the TRAIL resistance of cancer cell lines
was successfully reversed with ART in the majority of peripheral
solid tumor cells with the exception of brain cancer cells. While
being used alone, AT406 has only a limited effect on cancer cell
death (Fig. 2A). For rocaglamide,
certain degrees of cytotoxicity were observed on both cancer cell
lines and normal cells (Fig. 2A).
The fact that rocaglamide is cytotoxic to normal cells could raise
concerns for the future application of ART combination therapy. To
study the cause of this cytotoxicity, we carried out additional
experiments in which rocaglamide was replaced with c-FLIP-siRNA.
However, similar results were obtained using c-FLIP-siRNA alone or
in combination with TRAIL and AT406, confirming that this
cytotoxicity is the result of c-FLIP downregulation and the
suppression of the anti-apoptotic functions of c-FLIP (52,53).
Therefore, it is highly desirable to develop a specific disruptor
or antagonist of c-FLIP-FADD interactions to avoid the
cyto-toxicity caused by changes in the cellular levels of c-FLIP in
normal cells.
Acknowledgements
This study was supported by Chinese National Natural
Science Foundation, grant no. 81360162, 81260351 and U1302225.
References
|
1
|
Tannock IF: Cancer: Resistance through
repopulation. Nature. 517:152–153. 2015. View Article : Google Scholar
|
|
2
|
Santarpia L, Lippman SM and El-Naggar AK:
Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.
Expert Opin Ther Targets. 16:103–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sriraman SK, Aryasomayajula B and
Torchilin VP: Barriers to drug delivery in solid tumors. Tissue
Barriers. 2:e295282014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashkenazi A, Pai RC, Fong S, Leung S,
Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert
A, et al: Safety and antitumor activity of recombinant soluble Apo2
ligand. J Clin Invest. 104:155–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki
N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H and Okumura K:
Involvement of tumor necrosis factor-related apoptosis-inducing
ligand in surveillance of tumor metastasis by liver natural killer
cells. Nat Med. 7:94–100. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lemke J, von Karstedt S, Zinngrebe J and
Walczak H: Getting TRAIL back on track for cancer therapy. Cell
Death Differ. 21:1350–1364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holland PM: Death receptor agonist
therapies for cancer, which is the right TRAIL? Cytokine Growth
Factor Rev. 25:185–193. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koschny R, Walczak H and Ganten TM: The
promise of TRAIL - potential and risks of a novel anticancer
therapy. J Mol Med Berl. 85:923–935. 2007. View Article : Google Scholar
|
|
13
|
Salvesen GS and Duckett CS: IAP proteins:
Blocking the road to death's door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fulda S: Inhibitor of apoptosis proteins
as targets for anticancer therapy. Expert Rev Anticancer Ther.
7:1255–1264. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai Q, Sun H, Peng Y, Lu J,
Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R,
et al: A potent and orally active antagonist (SM-406/AT-406) of
multiple inhibitor of apoptosis proteins (IAPs) in clinical
development for cancer treatment. J Med Chem. 54:2714–2726. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fulda S, Wick W, Weller M and Debatin KM:
Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced
apoptosis and induce regression of malignant glioma in vivo. Nat
Med. 8:808–815. 2002.PubMed/NCBI
|
|
17
|
Fakler M, Loeder S, Vogler M, Schneider K,
Jeremias I, Debatin KM and Fulda S: Small molecule XIAP inhibitors
cooperate with TRAIL to induce apoptosis in childhood acute
leukemia cells and overcome Bcl-2-mediated resistance. Blood.
113:1710–1722. 2009. View Article : Google Scholar
|
|
18
|
Vogler M, Walczak H, Stadel D, Haas TL,
Genze F, Jovanovic M, Bhanot U, Hasel C, Möller P, Gschwend JE, et
al: Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis
and antitumor activity in preclinical models of pancreatic
carcinoma. Cancer Res. 69:2425–2434. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Irmler M, Thome M, Hahne M, Schneider P,
Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C,
et al: Inhibition of death receptor signals by cellular FLIP.
Nature. 388:190–195. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aoudjit F and Vuori K: Integrin signaling
in cancer cell survival and chemoresistance. Chemother Res Pract.
2012:2831812012.PubMed/NCBI
|
|
21
|
Safa AR and Pollok KE: Targeting the
anti-apoptotic protein c-FLIP for cancer therapy. Cancers (Basel).
3. pp. 1639–1671. 2011, View Article : Google Scholar
|
|
22
|
Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs
BS, Lindner DJ and Borden EC: Downregulation of Bcl-2, FLIP or IAPs
(XIAP and survivin) by siRNAs sensitizes resistant melanoma cells
to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 11:915–923.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bleumink M, Köhler R, Giaisi M, Proksch P,
Krammer PH and Li-Weber M: Rocaglamide breaks TRAIL resistance in
HTLV-1-associated adult T-cell leukemia/lymphoma by translational
suppression of c-FLIP expression. Cell Death Differ. 18:362–370.
2011. View Article : Google Scholar :
|
|
24
|
Zhu JY, Giaisi M, Köhler R, Müller WW,
Mühleisen A, Proksch P, Krammer PH and Li-Weber M: Rocaglamide
sensitizes leukemic T cells to activation-induced cell death by
differential regulation of CD95L and c-FLIP expression. Cell Death
Differ. 16:1289–1299. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Giaisi M, Kohler R, Fulda S, Krammer PH
and Li-Weber M: Rocaglamide and a XIAP inhibitor cooperatively
sensitize TRAIL-mediated apoptosis in Hodgkin's lymphomas. Int J
Cancer. 131:1003–1008. 2012. View Article : Google Scholar
|
|
26
|
Gronthos S, Mankani M, Brahim J, Robey PG
and Shi S: Postnatal human dental pulp stem cells (DPSCs) in vitro
and in vivo. Proc Natl Acad Sci USA. 97:13625–13630. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang YP, Kong QH, Huang Y, Wang GL and
Chang KJ: Inhibition of c-FLIP by RNAi enhances sensitivity of the
human osteogenic sarcoma cell line U2OS to TRAIL-induced apoptosis.
Asian Pac J Cancer Prev. 16:2251–2256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ziauddin MF, Yeow WS, Maxhimer JB, Baras
A, Chua A, Reddy RM, Tsai W, Cole GW Jr, Schrump DS and Nguyen DM:
Valproic acid, an antiepileptic drug with histone deacetylase
inhibitory activity, potentiates the cytotoxic effect of
Apo2L/TRAIL on cultured thoracic cancer cells through
mitochondria-dependent caspase activation. Neoplasia. 8:446–457.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Micheau O, Shirley S and Dufour F: Death
receptors as targets in cancer. Br J Pharmacol. 169:1723–1744.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wiezorek J, Holland P and Graves J: Death
receptor agonists as a targeted therapy for cancer. Clin Cancer
Res. 16:1701–1708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Graves JD, Kordich JJ, Huang TH, Piasecki
J, Bush TL, Sullivan T, Foltz IN, Chang W, Douangpanya H, Dang T,
et al: Apo2L/TRAIL and the death receptor 5 agonist antibody AMG
655 cooperate to promote receptor clustering and antitumor
activity. Cancer Cell. 26:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fulda S: Safety and tolerability of TRAIL
receptor agonists in cancer treatment. Eur J Clin Pharmacol.
71:525–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sayers TJ: Targeting the extrinsic
apoptosis signaling pathway for cancer therapy. Cancer Immunol
Immunother. 60:1173–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Menke C, Bin L, Thorburn J, Behbakht K,
Ford HL and Thorburn A: Distinct TRAIL resistance mechanisms can be
overcome by proteasome inhibition but not generally by synergizing
agents. Cancer Res. 71:1883–1892. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Daniel PT, Wieder T, Sturm I and
Schulze-Osthoff K: The kiss of death: Promises and failures of
death receptors and ligands in cancer therapy. Leukemia.
15:1022–1032. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schneider P, Olson D, Tardivel A, Browning
B, Lugovskoy A, Gong D, Dobles M, Hertig S, Hofmann K, Van Vlijmen
H, et al: Identification of a new murine tumor necrosis factor
receptor locus that contains two novel murine receptors for tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol
Chem. 278:5444–5454. 2003. View Article : Google Scholar
|
|
37
|
Wu GS, Burns TF, Zhan Y, Alnemri ES and
El-Deiry WS: Molecular cloning and functional analysis of the mouse
homologue of the KILLER/DR5 tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res.
59:2770–2775. 1999.PubMed/NCBI
|
|
38
|
van Dijk M, Halpin-McCormick A, Sessler T,
Samali A and Szegezdi E: Resistance to TRAIL in non-transformed
cells is due to multiple redundant pathways. Cell Death Dis.
4:e7022013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jänicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang XH, Sladek TL, Liu X, Butler BR,
Froelich CJ and Thor AD: Reconstitution of caspase 3 sensitizes
MCF-7 breast cancer cells to doxorubicin- and etoposide-induced
apoptosis. Cancer Res. 61:348–354. 2001.PubMed/NCBI
|
|
41
|
Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt
BR, Ho R, Ikegaki N and Brodeur GM: Resistance to tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in neuroblastoma cells correlates with a loss of caspase-8
expression. Cancer Res. 61:1314–1319. 2001.PubMed/NCBI
|
|
42
|
Hopkins-Donaldson S, Ziegler A, Kurtz S,
Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U and Stahel
R: Silencing of death receptor and caspase-8 expression in small
cell lung carcinoma cell lines and tumors by DNA methylation. Cell
Death Differ. 10:356–364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duiker EW, Meijer A, van der Bilt ARM,
Meersma GJ, Kooi N, van der Zee AG, de Vries EG and de Jong S:
Drug-induced caspase 8 upregulation sensitises cisplatin-resistant
ovarian carcinoma cells to rhTRAIL-induced apoptosis. Br J Cancer.
104:1278–1287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jia SH, Parodo J, Kapus A, Rotstein OD and
Marshall JC: Dynamic regulation of neutrophil survival through
tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol
Chem. 283:5402–5413. 2008. View Article : Google Scholar
|
|
45
|
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC,
Lill JR and Ashkenazi A: Cullin3-based polyubiquitination and
p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis
signaling. Cell. 137:721–735. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bellail AC, Olson JJ, Yang X, Chen ZJ and
Hao C: A20 ubiquitin ligase-mediated polyubiquitination of RIP1
inhibits caspase-8 cleavage and TRAIL-induced apoptosis in
glioblastoma. Cancer Discov. 2:140–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fulda S, Küfer MU, Meyer E, van Valen F,
Dockhorn-Dworniczak B and Debatin KM: Sensitization for death
receptor- or drug-induced apoptosis by re-expression of caspase-8
through demethylation or gene transfer. Oncogene. 20:5865–5877.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Casciano I, De Ambrosis A, Croce M, Pagnan
G, Di Vinci A, Allemanni G, Banelli B, Ponzoni M, Romani M and
Ferrini S: Expression of the caspase-8 gene in neuroblastoma cells
is regulated through an essential interferon-sensitive response
element (ISRE). Cell Death Differ. 11:131–134. 2004. View Article : Google Scholar
|
|
49
|
Fulda S and Debatin KM: IFNgamma
sensitizes for apoptosis by upregulating caspase-8 expression
through the Stat1 pathway. Oncogene. 21:2295–2308. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Crowder RN and El-Deiry WS: Caspase-8
regulation of TRAIL-mediated cell death. Exp Oncol. 34:160–164.
2012.PubMed/NCBI
|
|
51
|
Soung YH, Lee JW, Kim SY, Sung YJ, Park
WS, Nam SW, Kim SH, Lee JY, Yoo NJ and Lee SH: Caspase-8 gene is
frequently inactivated by the frameshift somatic mutation
1225_1226delTG in hepatocellular carcinomas. Oncogene. 24:141–147.
2005. View Article : Google Scholar
|
|
52
|
Shirley S and Micheau O: Targeting c-FLIP
in cancer. Cancer Lett. 332:141–150. 2013. View Article : Google Scholar
|
|
53
|
Wajant H, Haas E, Schwenzer R, Muhlenbeck
F, Kreuz S, Schubert G, Grell M, Smith C and Scheurich P:
Inhibition of death receptor-mediated gene induction by a
cycloheximide-sensitive factor occurs at the level of or upstream
of Fas-associated death domain protein (FADD). J Biol Chem.
275:24357–24366. 2000. View Article : Google Scholar : PubMed/NCBI
|