Introduction
Breast cancer (BC) is the most common endocrine
tumor and the second leading cause of cancer-related death in women
(1). Approximately 75% of BC cases
is characterized by the presence of hormone receptors (ERs) whose
expression is considered an important prognostic and therapeutic
indicator (2–4). ERs are nuclear receptors that
regulate the transcription of estrogen-responsive genes in target
cells after ligand binding and dimerization (5,6).
Moreover, it became evident that other pathways can be activated by
ERs that involve cytoplasmic signaling proteins, growth factor
receptors or other membrane signaling circuits (7). There are two ER types, ERα and ERβ
which are responsible for both the canonical genomic and the
non-genomic rapid pathways (8). In
2005 Wang et al identified and cloned a novel isoform of
full-length ERα66 with a molecular weight of 36 kDa (ERα36) which
is characterized by the lack of transcriptional activation domains
and retains a partial ligand-binding domain (9). Although the prevalent subcellular
localization of ERα36 (cytosol and cell membrane) and the lack of
intrinsic transcriptional activity associate ERα36 to a non-genomic
rapid estrogen signaling (10),
the interplay between the diverse variants of ERs has not been
fully established so far. It has been demonstrated that ERα36 can
act as a dominant-negative regulator of signaling mediated by ERα66
but, on the other hand, the ERα36 expression seems to be subjected
to an ERα66-dependent negative regulation (11). The presence of ERs in breast cancer
cells allows the employment of ER antagonists, such as
4-hydroxytamoxifen, which are able to block their proliferative
rate (12). However, although
great strides have been made in BC therapy in recent years, 30–50%
of cases of ER-positive BC still displays de novo or
intrinsic resistance to ER antagonists and frequently tumor
recurrence is observed (13,14).
There is substantial evidence that the main cause of
cancer relapse and chemoresistance is the presence of cancer stem
cells (CSCs) in tumor tissues (15). CSCs represent a small population
within the tumor mass that not only drives tumor initiation and
growth, but also mediates tumor metastasis and therapy resistance.
They are very hard to eradicate due to their robust survival
mechanisms. Although many proliferative pathways were identified in
BC stem-like cells obtained from cancer tissues or cell lines, the
role of estrogen signaling has not been fully established and the
expression of ERα66 still remains controversial. Recently, Deng
et al found that stem cells derived from ER-positive BC
cells express high levels of ERα36 that play important roles in
mediating anti-estrogen resistance (16).
It has been largely demonstrated that the malignancy
of tumor cells is also sustained by the ability to escape
immunosurveillance controlled by immune system cells such as
cytotoxic T lymphocytes (CTL) and natural killer (NK) cells that
recognize and kill cancer cells (17). Immune system cells eradicate tumor
cells through two different death pathways: the perforin/granzyme B
and Fas/Fas ligand pathway (18).
In perforin/granzyme B-mediated apoptosis, the recognition of a
target cell triggers the release of granules containing perforin
and the granzyme protease. Perforin promotes granzyme delivery to
the cytosol of target cell and, on entry, this protease triggers
rapid and efficient apoptosis by the direct cleavage of caspase and
non-caspase substrates (19). The
activity of granzyme B, the main effector molecule of CTL and NK
cells, is regulated by PI-9 (proteinase inhibitor 9), a member of
the serine proteinase inhibitor family collectively called serpins
(20). PI-9 is a pseudo-substrate
for granzyme B leading to the irreversible inhibition of protease
upon interaction (20).
We demonstrated that tumorspheres derived from
ERα-positive BC MCF7 cells express a high level of PI-9, supporting
a role for the granzyme B inhibitor in immunosurveillance escape of
BC stem cells.
Materials and methods
Cell culture and reagents
MCF7 cells were purchased from Istituto Scientifico
Tumori (Genoa, Italy) and were maintained in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 5% heat-inactivated fetal
bovine serum (FBS), 2 mM glutamine, 50 U/ml penicillin and 50 μg/ml
streptomycin at 37°C in a humidified atmosphere containing 5%
CO2. For estrogen challenge, 4–5 days before the
experiments medium was replaced with phenol red-free DMEM
containing 5% charcoal-stripped FBS (Life Technologies, Monza,
Italy). All reagents, except where differently reported, were
purchased from Sigma-Aldrich (Milan, Italy).
Tumorsphere production
For tumorsphere preparation, 104 MCF7
cells were seeded in ultra low-attachment 6-well plates in a
conditioned medium bereft of serum and containing phenol red-free
DMEM/F12, supplemented with 1× B27, 20 ng/ml EGF, 20 ng/ml bFGF
(Life Technologies). Growth factors were added every two days
(without removing the old media) and in these conditions cells grew
as non-adherent spherical clusters named tumorspheres. Formed
spheres were visualized under a bright-field Leica DMR inverted
microscope (Wetzlar, Germany) and their sizes and number were
measured and counted using IM50 Leica software (Wetzlar,
Germany).
Tumorsphere passage
For sphere passage experiments, the tumorspheres
were collected by centrifugation and dissociated by means of
enzymatic (0.25% trypsin-EDTA) and mechanical treatment. After
centrifugation to remove the enzyme, pellets were resuspended in
serum starved conditioned medium to allow tumorsphere re-growth.
The dissociated single sphere-forming cells were diluted to a
density of 1,000 cells/ml and culture passaged every 7 days
according to Dontu et al (21). Sphere formation efficiency (SFE)
was calculated dividing the number of tumorspheres by the number of
seeded cells.
Cell viability
To determine cell viability after estrogen
treatments, MTT (3-(4,5-dimethylthiazol-2-yl-2,5-diphenyl
tetrazolium bromide) assay was performed as previously reported
(22). Briefly, 20 μl of MTT (11
mg/ml) was added to the cells and incubation was protracted for 2 h
at 37°C. Then, medium was replaced with 100 μl lysis buffer (20%
SDS, 10% dimethylformamide and 20% acetic acid) to lysate the
cells. After solubilisation, the absorbance was read at 570 and 690
nm. Each condition was tested six times and results were analyzed
for statistical significance.
RNA extraction and qPCR analysis
Total RNA was prepared using Direct-zol™ RNA
MiniPrep kit (Zymo Research, Irvine, CA, USA). Then, after removal
of residual genomic DNA with DNase I (Zymo Research),
oligo(dT)-primed reverse transcription was performed on 1 μg of
total cellular RNA using the iScript™ cDNA Synthesis kit (Bio-Rad
Laboratories Srl, Milan, Italy), following the manufacturer's
instructions. For real-time PCR analyses, each cDNA sample was
amplified by iQTM SYBR Green Supermix (Bio-Rad), using
the following QuantiTect primers (Qiagen, Milan, Italy): Oct3/4
(POU5F1: QT00210840), Sox2 (QT00237601), Nanog (QT01025850), Kfl4
(QT00061033), CXCR4 (QT00223188). As for PI-9 amplification, the
following primers were used: Fw 5′-CGCTGGACTAGGTGGCAG-3′ and Rv
5′-CAGAA CACGTTGTGCGAAGG-3′ (Life Technologies).
All PCR reactions were performed in triplicate in
the iQ5 thermal cycler instrument (Bio-Rad, Laboratories Srl,
Milan, Italy), using a PCR cycling as previously reported (23). The relative quantification of
analyzed genes was calculated using the 2−ΔΔCt method
and data were normalized by using GAPDH (QT01192646; Qiagen) as
endogenous control. Data processing and statistical analysis were
performed by iQ5™ Optical System software (Bio-Rad).
Western blot analyses
The procedure of western blot analysis was performed
as previously described (24).
Briefly, proteins (30 μg) were separated by SDS-polyacrylamide gel
electrophoresis and transferred to a nitrocellulose membrane
(Bio-Rad) for detection with primary antibodies. All antibodies
were from St. Cruz Biotechnology (St. Cruz, CA, USA) except for p38
and phospho-p38 (Cell Signaling Technology, Beverly, MA, USA).
Then, filters were incubated with horseradish-peroxidase-conjugated
secondary antibodies and the detections were developed by ECL Plus™
western blotting reagents (Amersham, GE Healthcare Life Science,
Milan, Italy) according to the manufacturer's instructions. Bands
were visualized and acquired by ChemiDoc XRS System (Bio-Rad,
Hercules, CA, USA).
Densitometric analysis of the bands was performed
using the NIH Image J 1.40 analysis software distributed by
National Institutes of Health (Bethesda, MD, USA). The correct
protein loading was ascertained by both red Ponceau staining and
immunoblotting for β-actin. All the blots shown are representative
of at least three separate experiments.
Statistical analysis
Data, reported as means ± SD from at least three
independent experiments, were analyzed using the Student's t-test.
Differences were considered significant at P<0.05.
Results
Isolation and characterization of
ERα-positive breast cancer tumorspheres
In our study we first isolated and characterized
tumorspheres derived from adherent ERα+ MCF7 breast
cancer cells to individuate new selective targets in BC stem cells.
To this purpose MCF7 cells were seeded in ultra-low attachment
plates and maintained in serum-starved medium to form non-adherent
spherical clusters named tumorspheres. These spheres were already
visible after 4–5 days and, when they reached a diameter >50 μm
(approximately after 7 days), were serially passaged for different
generations. Single cells obtained from sphere dissociation were
propagated forming again new tumorspheres with a sphere formation
efficiency (SFE) ~2%. The morphological and molecular features of
tumorspheres were analyzed after 7 (primary spheres, S1), 14
(secondary spheres, S2) and 21 (tertiary spheres, S3) days,
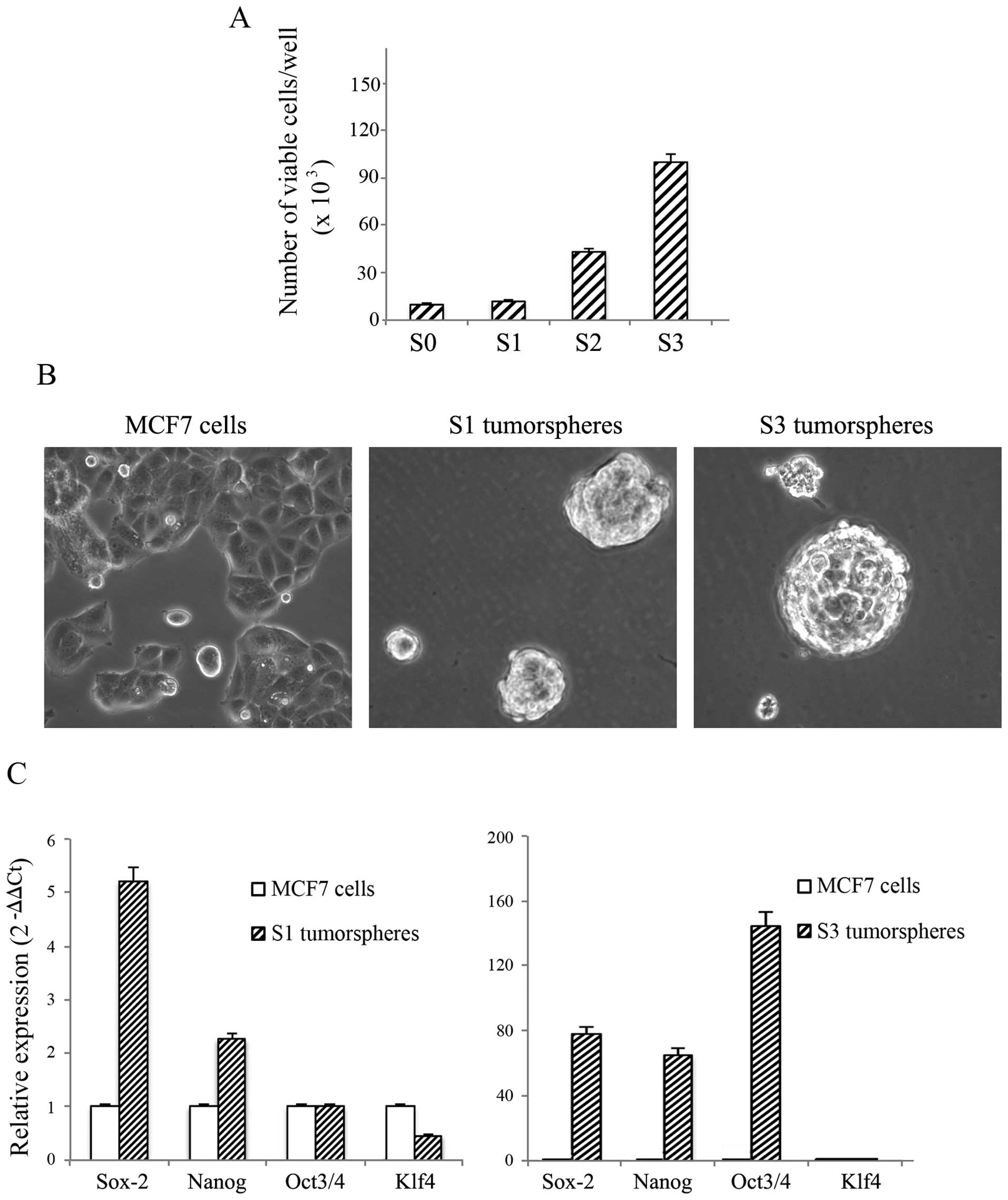
respectively. As shown in Fig. 1A,
the number of viable cells, calculated and plotted against the
equivalent passage number, progressively increased at each
generation. In Fig. 1B the
characteristic morphological aspects of S1 and S3 tumorspheres
grown in anchorage-independent conditions are reported. As shown,
both S1 and S3 tumorspheres grew in spheroidal clusters floating in
the medium and were deprived of the flattened cell morphology,
typical of adherent parental MCF7 cells.
Next, tumorspheres were collected by centrifugation
and characterized by real-time RT-PCR to evaluate changes in the
gene expression of stemness markers such as Sox2, Nanog, Oct3/4,
and Kfl4. The analysis reported in Fig. 1C shows an increase in the levels of
Sox2 and Nanog stemness markers in S1 tumorspheres with respect to
parental MCF7 cells. The gene expression of these markers
consistently increased in S3 tumorspheres where a remarkable
upregulation of Oct3/4 was also observed (Fig. 1C). Our data also provided evidence
that no significant change was present in the content of Kfl4. In
light of these results, since S3 tumorspheres exhibited the highest
content of the examined stemness markers, we performed the next
experiments in these tumorspheres comparing data to parental MCF7
cells.
Effects of 17-β estradiol and genistein
in adherent MCF7 cells and S3 tumorspheres
Since a possible involvement of BC stem cells has
been postulated in anti-estrogen resistance of ER-positive BC
(25), we aimed to clarify the
estrogen responsiveness of BC tumorspheres. At first, we treated S3
tumorspheres with 17-β estradiol (E2) or genistein, a natural
isoflavonoid phytoestrogen that possesses BC preventive properties
(26). The doses (10 nM E2 or 25
μM genistein) employed for these studies were chosen on the basis
of preliminary experiments demonstrating that the incubation of
tumorspheres or parental MCF7 cells with different concentrations
(100 pM-100 μM) of hormones for 48 h increased cell number at low
doses while reduced cell viability at higher concentrations (50–100
μM) (not shown). In light of these data we evaluated the effect of
the hormones in S3 tumorspheres seeded in low attachment conditions
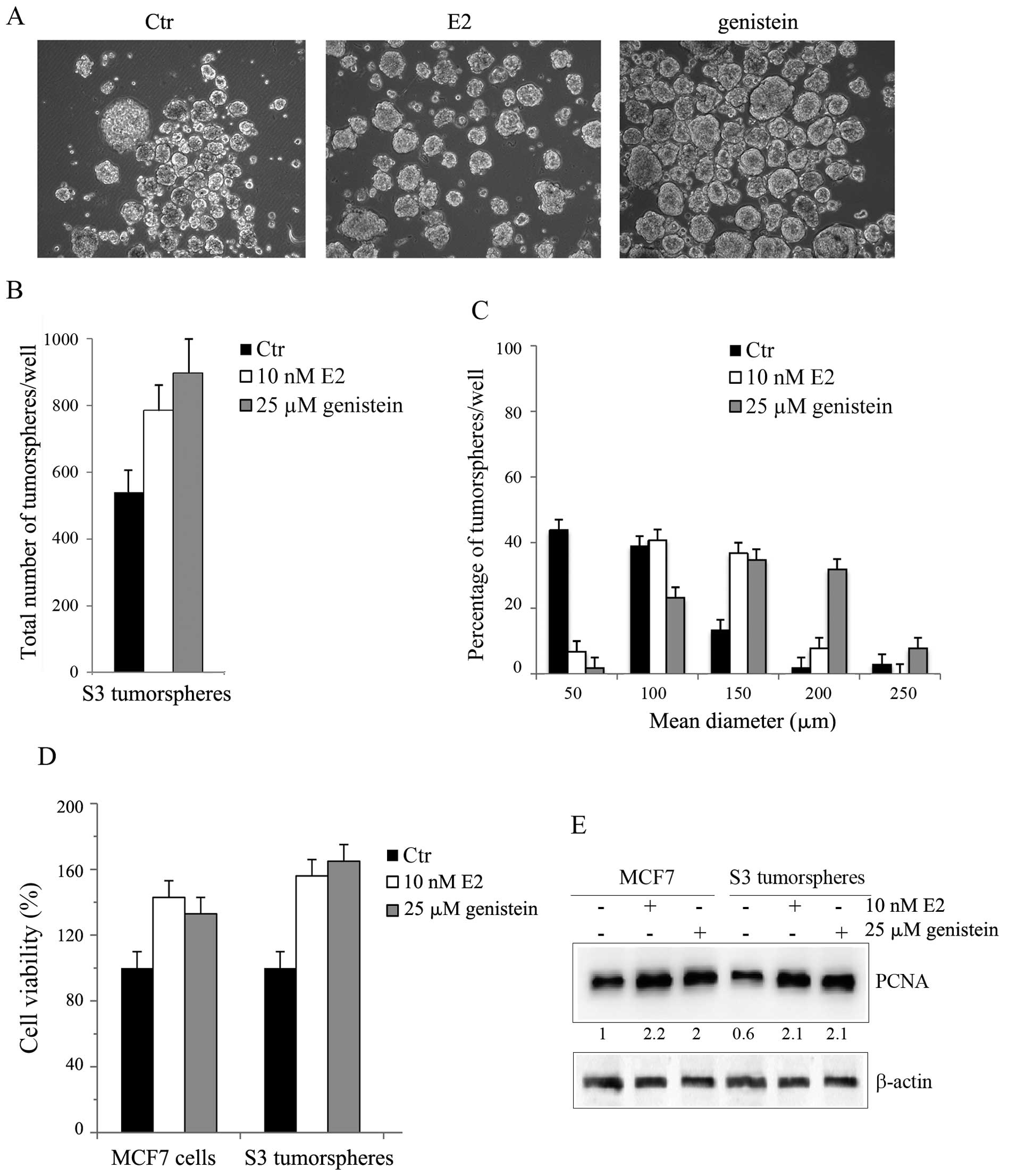
as reported in Materials and methods. As shown in Fig. 2A and B, the treatment increased S3
tumorsphere population by 45% with E2 and by 66% with genistein at
48 h. Moreover, a remarkable increase in tumorsphere sizes
(Fig. 2C) was also observed.
Approximately 86% of E2-treated tumorspheres showed a diameter
>50 μm, most of which with a mean diameter of 100–150 μm. This
percentage amounted to 98% in genistein-treated tumorspheres,
showing an enrichment in the population with a mean diameter of
150–200 μm.
The effect of estrogens on cell viability of S3
tumorspheres was also analyzed by MTT assay. As Fig. 2D reports, the incubation with low
doses of hormones stimulated the growth of S3 tumorspheres by about
50% with E2 and by 63% with genistein, values which were higher
than those obtained in MCF7 cells.
In accordance with these results, western blot
analyses provided evidence that, albeit S3 tumorspheres expressed a
lower level of the proliferating marker PCNA in comparison to
parental MCF7 cells, it significantly increased after E2 or
genisten stimulation (Fig.
2E).
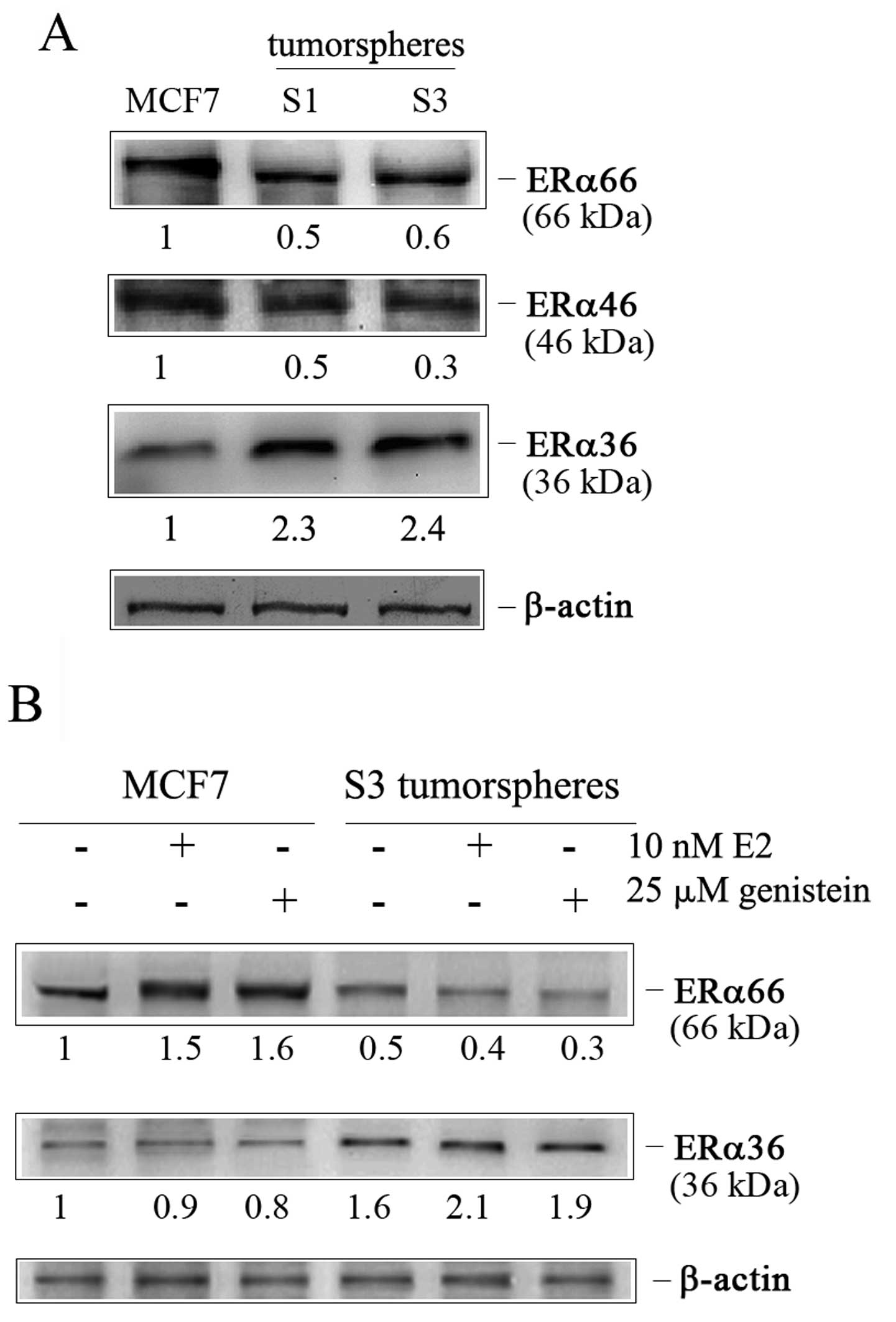
Intrigued by these results we focused on ERα
receptor isoforms which mediate estrogen effects (27). Considering the different
sensitivity of MCF7 cells and S3 tumorspheres to both E2 and
genistein, we wondered whether the observed effects were linked to
a different expression of ER receptors. Thus, we evaluated the
presence of the isoforms of ERα (ERα66, its splice variant ERα46,
and the recently identified variant ERα36) in S1 and S3
tumorspheres in comparison to MCF7 cells. As Fig. 3A shows, tumorspheres expressed a
very low amount of ERα66 and ERα46 isoforms respect to MCF7 cells,
while a remarkable expression of ERα36 isoform was visible.
Moreover, the exposure of S3 tumorspheres to estrogen treatment (E2
and genistein) further increased ERα36 content reducing that of
ERα66. Such an effect was dissimilar to that observed in parental
MCF7 cells (Fig. 3B), where
hormone treatment increased the ERα66 level.
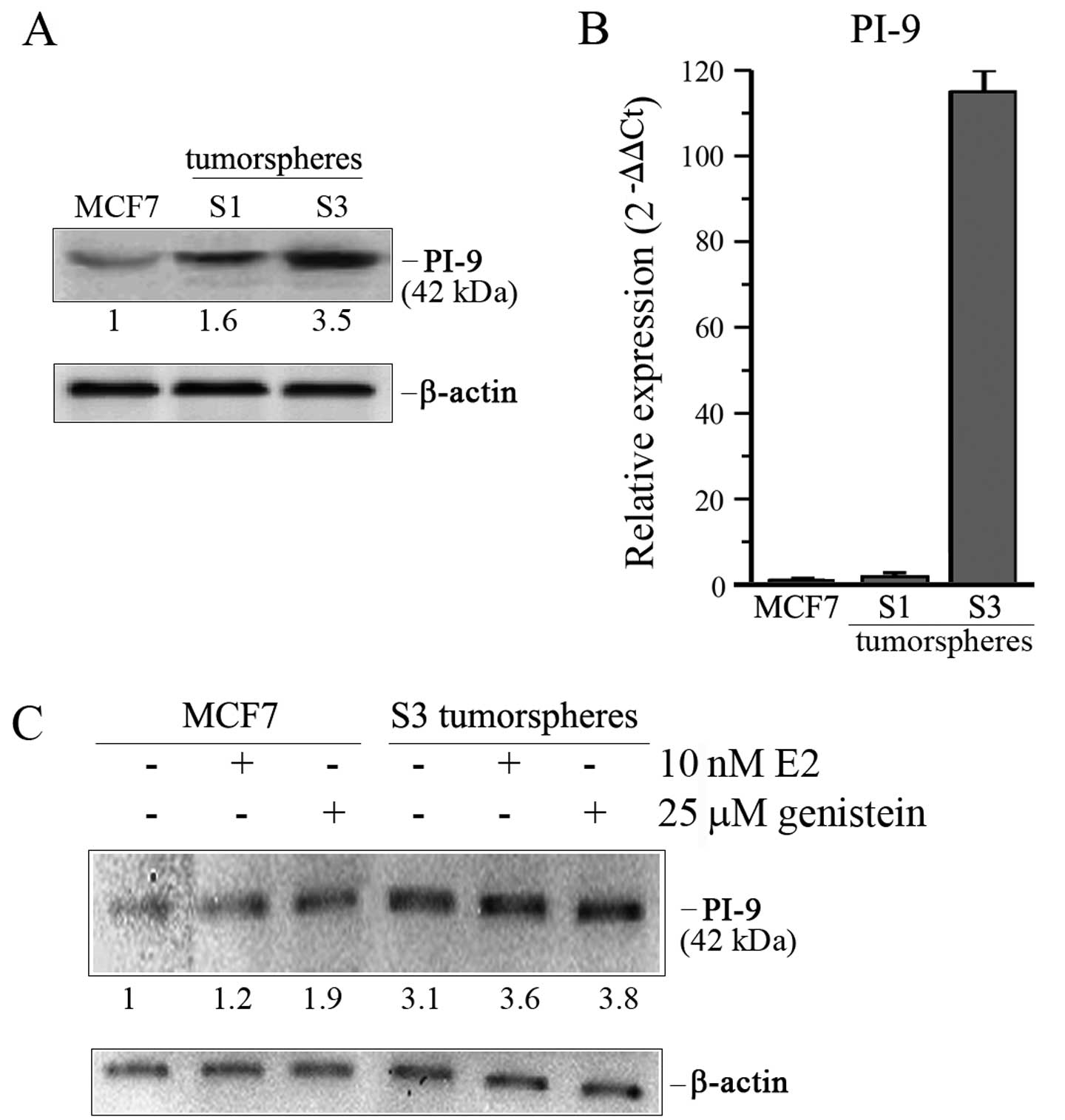
Breast cancer tumorspheres express high
levels of serpin proteinase inhibitor 9
In light of the results obtained on ERα receptor
isoforms, our attention was next focused on PI-9 (proteinase
inhibitor 9), a member of serpin family which is subjected to the
hormonal induction mediated by ERα66-dependent transcriptional
regulation (28). Since S3
tumorspheres showed a higher expression of ERα36 receptor which
lacks transcriptional activity, we expected to find a lower level
of PI-9 in tumorspheres than parental MCF7 cells. Surprisingly,
western blot analysis revealed that the level of PI-9 protein was
1.6 and 3.5-fold higher in S1 and S3 tumorspheres, respectively
(Fig. 4A). Based on these data, we
wondered whether this upregulated level could be ascribed to an
increase in mRNA level. To verify this hypothesis, we performed
comparative analyses by real-time RT-PCR. Data reported in Fig. 4B revealed that S3 tumorspheres also
expressed a pronounced level of PI-9 mRNA which was ~110-fold
higher than that found in adherent MCF7 cells, while a very modest
increase of this factor was observed in S1 tumorspheres.
Interestingly, 17-β estradiol or genistein treatment
further increased the level of PI-9, thus indicating a possible
relationship between PI-9 expression and estrogen-mediated
signaling (Fig. 4C).
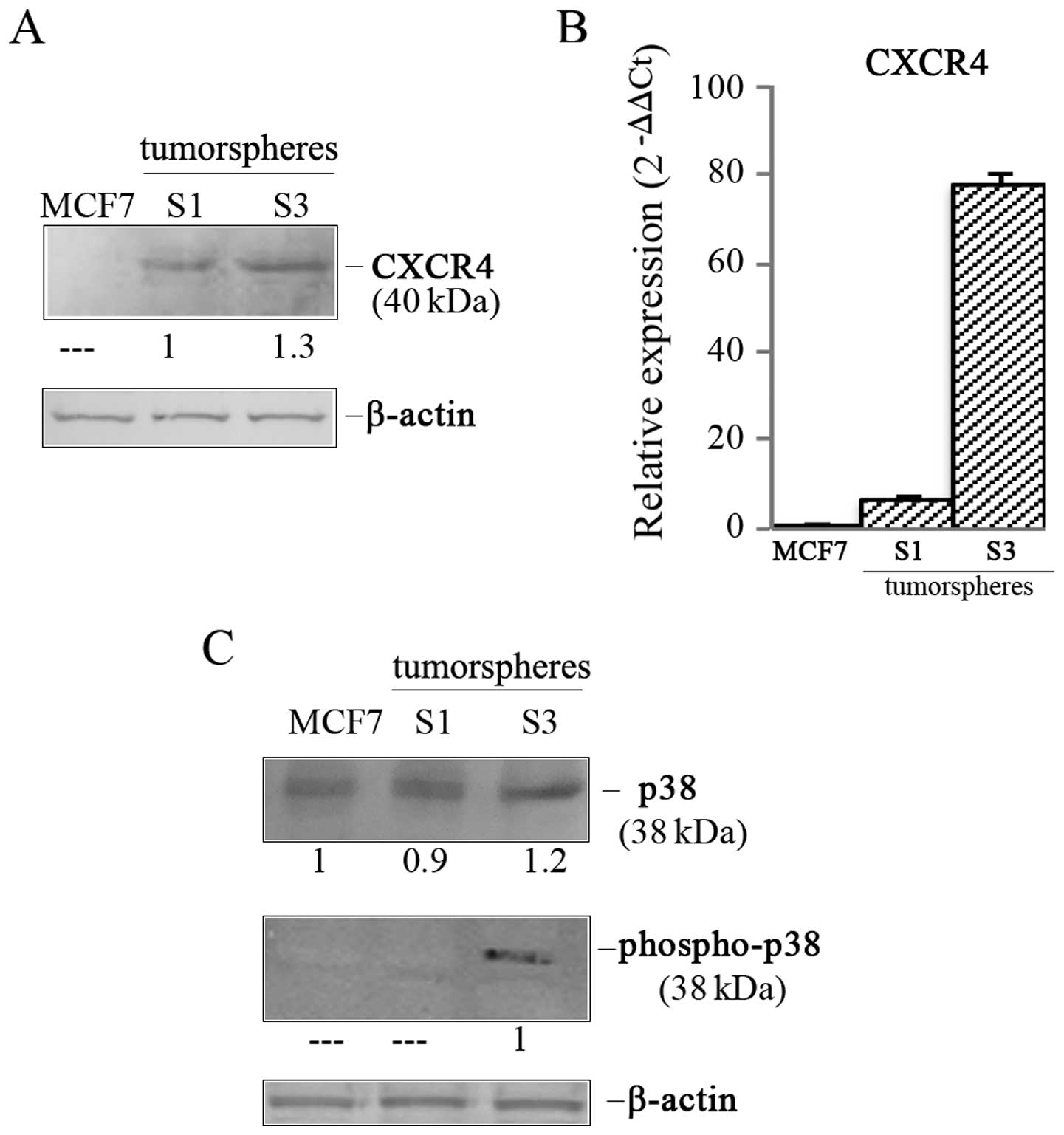
S3 tumorspheres show an active CXCR4
/phospho-p38 proliferative axis
Next, we explored possible molecular scenarios
responsible for PI-9 upregulation in S3 tumorspheres. In this
regard, we focused particular attention on CXCR4, the chemokine
receptor specific for the stromal-derived-factor-1 (29). This receptor represents a
prognostic factor in different types of cancer and plays a role in
chemotaxis, stemness and drug resistance (30,31).
Moreover, recent data have also described this factor as a key
regulator of breast cancer invasion, directing homing of BC cells
to particular sites of metastases (32). In our experiments we observed that
CXCR4 was undetectable in parental MCF7 cells, while it appeared in
tumorspheres increasing from S1 to S3 generation (Fig. 5A). Accordingly, mRNA analysis by
real-time PCR showed a 77.7-fold higher content in S3 tumorspheres
than MCF7 cells (Fig. 5B).
Since Bots et al (33) have shown that PI-9 is induced
through p38 MAP kinase activation in dendritic cells and the
activation of this kinase can be linked to CXCR4, we analyzed the
level of p38 kinase in S3 tumorspheres. As shown in Fig. 5C, we demonstrated that p38 was
present in both parental MCF7 cells and tumorspheres, but its
content was only 1.2-fold higher in S3 tumorspheres than parental
cells. Interestingly, the phosphorylated and active form of the
protein, detected by using an antibody directed against the dual
phosphorylated sites Thr180 and Tyr182, was found only in S3
tumorspheres (Fig. 5C), indicating
the presence of an active p38 MAP kinase pathway in this stem-like
population.
Discussion
Cytotoxic T lymphocytes (CTL) and natural killer
(NK) cells can specifically identify and eliminate tumor cells in a
process termed cancer immunosurveillance that exerts a crucial role
in inhibiting carcinogenesis and maintaining cellular homeostasis
(34). Unfortunately, tumor cells
develop multiple strategies to evade host immune responses,
including reduced expression of major histocompatibility complex
molecules and loss of tumor antigens (35). Recently, it has been suggested that
the inhibition of granzyme B activity in neoplastic cells may
constitute one of the mechanisms that tumors use to escape
immunosurveillance (36,37). Granzyme B belongs to a group of
serine proteases commonly found in the cytotoxic granules of CTL
and NK cells and it mediates apoptosis in target cells together
with the pore forming protein perforin (38). The proteinase inhibitor 9 (PI-9), a
member of family of serine proteinase inhibitors, is a direct
inhibitor of granzyme B in human (39). Its overexpression has been observed
in several types of cancer, such as breast (28), prostate (40) and lung cancer (41) and has been correlated to the
protection of tumor cells from granzyme B-mediated cytotoxicity.
However, no data concerning the status of PI-9 in cancer stem cells
are available so far.
In this study we employed tumorspheres obtained by
us from MCF7 cells serially passaged up to the third generation (S3
tumorspheres) and expressing high levels of stem cell markers.
Interestingly, we show for the first time that ERα+
breast cancer tumorspheres express higher levels of PI-9 protein
than parental MCF7 cells and the content of this serpin proteinase
inhibitor progressively increases with the passaging, resulting
more pronounced in S3 tumorspheres. Real-time RT-PCR also provided
evidence that the increased expression of PI-9 is related to gene
upregulation.
It has been shown that estrogens increase PI-9 level
through an ERα66-dependent transcriptional regulation in breast
cancer cells (28). In particular,
in PI-9 promoter it has been identified a sequence located 200 bp
downstream of the transcription start site and binding the
estrogen/ERα complex (27). These
observations prompted us to evaluate a possible involvement of
estrogens in PI-9 induction in MCF7-derived tumorspheres. After
proving that estrogens (17-β estradiol and genistein) increase both
the number and sizes of tertiary tumorspheres as well as the level
of proliferation marker PCNA, we demonstrated that both the
hormones markedly increase PI-9 level in tumorspheres, suggesting
their involvement in PI-9 upregulation. Estrogens are known to
regulate different cellular processes stimulating both genomic and
non-genomic pathways (3–8). The classical mechanism of estrogen
action involves the binding of the hormones to specific nuclear
receptors (ERα66, its splice variant ERα46, and the closely related
estrogen receptor ERβ) (7,8) which bind to estrogen responsive
elements (EREs) located in the promoter of target genes (8). The experiments described here
demonstrated that the levels of both ERα66 and ERα46 are lower in
tertiary tumorspheres than in parental MCF7 cells. Moreover, both
17-β estradiol and genistein further decreased the level of ERα66
isoform. The divergent trends between the increase in PI-9 and the
decrease in ERα66 observed in tertiary tumorspheres seem to
indicate that ERα66 isoform is not involved in PI-9 upregulation in
ERα-positive BC-derived spheres.
Estrogens were also reported to bind cell surface
receptors to transduce membrane-initiated estrogen signaling
(7). These effects seem to be
related to the expression of ERα36, a variant of ERα66 (9) which lacks both the transcriptional
activation domains (AF-1 and AF-2). Being mainly located near the
plasma membrane and in the cytoplasm, it mediates the activation of
MAPK/ERK and PI3K/AKT signaling pathways (42,43).
Our results showed that ERα36 is expressed at a higher level in
tertiary tumorspheres than MCF7 cells and its content is further
increased by 17-β estradiol or genistein treatment. These results
are in accordance with the observation of Deng et al
(44) showing that ERα36
expression is markedly increased in stem cells derived from
ER-positive breast cancer cells where it plays an important role in
expanding breast cancer stem-like cell population. Thus, we
postulated that the activation of non-genomic signaling involving
estrogen/ERα36 could be responsible for PI-9 increased expression
in tertiary tumorspheres. This hypothesis is also corroborated by
the observation that ERα36 stimulation is involved in the
activation of MAPK/ERK signaling pathway (45) and that the expression of PI-9 is
regulated via the p38/MAPK pathway in human dendritic cells
(33). Interestingly, we
demonstrate that both parental MCF7 cells and tertiary tumorspheres
express p38, while the phosphorylated and active form of this
protein only occurs in tumorspheres of third generation, thus
suggesting that this event could be related to the increase in PI-9
content found in tertiary tumorspheres.
Recently, several lines of evidence suggested that
the biological effects of CXCR4, the chemokine receptor specific
for the stromal-derived-factor-1 (SDF-1), are mediated by the
activation of MAPK family members (46). The SDF-1-CXCR4 signaling axis is a
known mediator of metastasis in breast cancer (32) as demonstrated by CXCR4 expression
observed in human breast cancer specimens (47). A recent study has also shown that
CXCR4 maintains a cancer progenitor population in
tamoxifen-resistant breast cancer cells (48). Data reported herein showing that
tertiary tumorspheres express high levels of both CXCR4 mRNA and
protein suggest that the activation of this receptor could be also
involved in p38 MAPK activation.
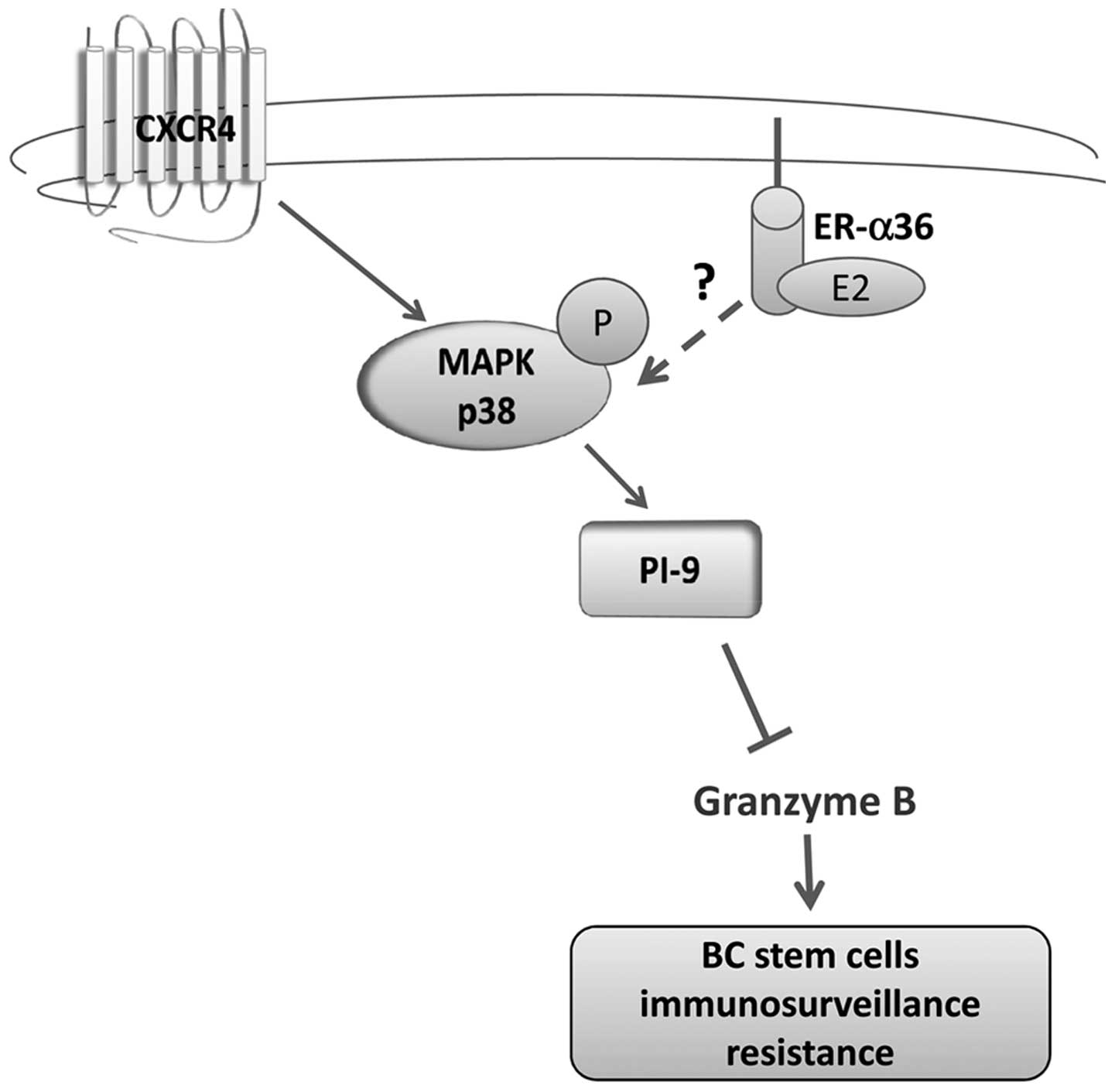
Taken together, our results demonstrate for the
first time that ERα+ breast cancer stem-like cells
express a high content of granzyme B inhibitor PI-9 and that this
event could be ascribed to the activation of a CXCR4-p38 axis.
Considering that ERα36 has been reported to play a role in MAPK
signaling activation (43), it is
plausible to hypothesize an involvement of ERα36 isoform which is
overexpressed in tertiary tumorspheres in this signaling circuit. A
possible molecular mechanism controlling this pathway in BC
tumorspheres is illustrated in Fig.
6.
In conclusion, we consider that PI-9 overexpression
found in mammary tumorspheres could cause a short-circuit in
immunosurveillance's signaling preserving these cells from
cytotoxic T lymphocytes and NK cell-mediated apoptosis. Therefore,
this experimental system might represent a suitable in vitro
model to study breast cancer-initiating cells and to explore the
response of these tumorspheres to granzyme B and agents molecularly
interfering with immunosurveillance system.
Acknowledgements
This study was supported by a grant from: European
Regional Development Fund, European Territorial Cooperation
2007–2013, CCI 2007 CB 163 PO 037, OP Italia-Malta 2007–2013; Dr D.
Carlisi was supported by a grant by ‘Italian Ministry of Education,
University and Research (MIUR)’. Dr C. Cernigliaro was financially
supported by a research contract of the OP Italia-Malta 2007–2013
grant.
Abbreviations:
|
BC
|
breast cancer
|
|
ER
|
estrogen receptor
|
|
CTL
|
cytotoxic T lymphocytes
|
|
NK
|
natural killer
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
PI-9
|
proteinase inhibitor 9
|
References
|
1
|
Forouzanfar MH, Foreman KJ, Delossantos
AM, Lozano R, Lopez AD, Murray CJ and Naghavi M: Breast and
cervical cancer in 187 countries between 1980 and 2010: A
systematic analysis. Lancet. 378:1461–1484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Key T, Appleby P, Barnes I and Reeves G;
Endogenous Hormones and Breast Cancer Collaborative Group.
Endogenous sex hormones and breast cancer in postmenopausal women:
reanalysis of nine prospective studies. J Nat Cancer Inst.
94:606e6162002.
|
|
3
|
Welboren WJ, Sweep FC, Span PN and
Stunnenberg HG: Genomic actions of estrogen receptor alpha: What
are the targets and how are they regulated? Endocr Relat Cancer.
16:1073–1089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robertson JF: Oestrogen receptor: A stable
phenotype in breast cancer. Br J Cancer. 73:5–12. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nilsson S, Mäkelä S, Treuter E, Tujague M,
Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M and
Gustafsson JA: Mechanisms of estrogen action. Physiol Rev.
81:1535–1565. 2001.PubMed/NCBI
|
|
6
|
Katzenellenbogen BS and Katzenellenbogen
JA: Estrogen receptor transcription and transactivation: Estrogen
receptor alpha and estrogen receptor beta: regulation by selective
estrogen receptor modulators and importance in breast cancer.
Breast Cancer Res. 2:335–344. 2000. View
Article : Google Scholar
|
|
7
|
Björnström L and Sjöberg M: Mechanisms of
estrogen receptor signaling: Convergence of genomic and nongenomic
actions on target genes. Mol Endocrinol. 19:833–842. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deroo BJ and Buensuceso AV: Minireview:
Estrogen receptor-beta: mechanistic insights from recent studies.
Mol Endocrinol. 24:1703–1714. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Zhang X, Shen P, Loggie BW, Chang
Y and Deuel TF: Identification, cloning, and expression of human
estrogen receptor-alpha36, a novel variant of human estrogen
receptor-alpha66. Biochem Biophys Res Commun. 336:1023–1027. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chaudhri RA, Schwartz N, Elbaradie K,
Schwartz Z and Boyan BD: Role of ERα36 in membrane-associated
signaling by estrogen. Steroids. 81:74–80. 2014. View Article : Google Scholar
|
|
11
|
Zou Y, Ding L, Coleman M and Wang Z:
Estrogen receptor-alpha (ER-alpha) suppresses expression of its
variant ER-alpha 36. FEBS Lett. 583:1368–1374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Osborne CK: Tamoxifen in the treatment of
breast cancer. N Engl J Med. 339:1609–1618. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clarke R, Leonessa F, Welch JN and Skaar
TC: Cellular and molecular pharmacology of antiestrogen action and
resistance. Pharmacol Rev. 53:25–71. 2001.PubMed/NCBI
|
|
14
|
Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY,
Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, et al: Antiestrogen
resistance in breast cancer and the role of estrogen receptor
signaling. Oncogene. 22:7316–7339. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Charafe-Jauffret E, Ginestier C, Iovino F,
Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F,
Dutcher J, et al: Breast cancer cell lines contain functional
cancer stem cells with metastatic capacity and a distinct molecular
signature. Cancer Res. 69:1302–1313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng H, Yin L, Zhang XT, Liu LJ, Wang ML
and Wang ZY: ER-α variant ER-α36 mediates antiestrogen resistance
in ER-positive breast cancer stem/progenitor cells. J Steroid
Biochem Mol Biol. 144B:417–426. 2014. View Article : Google Scholar
|
|
17
|
Massari F, Santoni M, Ciccarese C and
Santini D: The immunocheckpoints in modern oncology: The next 15
years. Expert Opin Biol Ther. 15:917–921. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mori S, Jewett A, Murakami-Mori K,
Cavalcanti M and Bonavida B: The participation of the Fas-mediated
cytotoxic pathway by natural killer cells is tumor-cell-dependent.
Cancer Immunol Immunother. 44:282–290. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goping IS, Barry M, Liston P, Sawchuk T,
Constantinescu G, Michalak KM, Shostak I, Roberts DL, Hunter AM,
Korneluk R, et al: Granzyme B-induced apoptosis requires both
direct caspase activation and relief of caspase inhibition.
Immunity. 18:355–365. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fritsch K, Finke J and Grüllich C:
Suppression of granzyme B activity and caspase-3 activation in
leukaemia cells constitutively expressing the protease inhibitor 9.
Ann Hematol. 92:1603–1609. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D'Anneo A, Carlisi D, Lauricella M, Puleio
R, Martinez R, Di Bella S, Di Marco P, Emanuele S, Di Fiore R,
Guercio A, et al: Parthenolide generates reactive oxygen species
and autophagy in MDA-MB231 cells. A soluble parthenolide analogue
inhibits tumour growth and metastasis in a xenograft model of
breast cancer. Cell Death Dis. 4:e8912013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Di Fiore R, Drago-Ferrante R, Pentimalli
F, Di Marzo D, Forte IM, D'Anneo A, Carlisi D, De Blasio A,
Giuliano M, Tesoriere G, et al: MicroRNA-29b-1 impairs in vitro
cell proliferation, self-renewal and chemoresistance of human
osteosarcoma 3AB-OS cancer stem cells. Int J Oncol. 45:2013–2023.
2014.PubMed/NCBI
|
|
24
|
Carlisi D, D'Anneo A, Martinez R, Emanuele
S, Buttitta G, Di Fiore R, Vento R, Tesoriere G and Lauricella M:
The oxygen radicals involved in the toxicity induced by
parthenolide in MDA-MB-231 cells. Oncol Rep. 32:167–172.
2014.PubMed/NCBI
|
|
25
|
Arif K, Hussain I, Rea C and El-Sheemy M:
The role of Nanog expression in tamoxifen-resistant breast cancer
cells. Onco Targets Ther. 8:1327–1334. 2015.PubMed/NCBI
|
|
26
|
Bilal I, Chowdhury A, Davidson J and
Whitehead S: Phytoestrogens and prevention of breast cancer: The
contentious debate. World J Clin Oncol. 5:705–712. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Krieg AJ, Krieg SA, Ahn BS and Shapiro DJ:
Interplay between estrogen response element sequence and ligands
controls in vivo binding of estrogen receptor to regulated genes. J
Biol Chem. 279:5025–5034. 2004. View Article : Google Scholar
|
|
28
|
Jiang X, Ellison SJ, Alarid ET and Shapiro
DJ: Interplay between the levels of estrogen and estrogen receptor
controls the level of the granzyme inhibitor, proteinase inhibitor
9 and susceptibility to immune surveillance by natural killer
cells. Oncogene. 26:4106–4114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Furusato B, Mohamed A, Uhlén M and Rhim
JS: CXCR4 and cancer. Pathol Int. 60:497–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Trautmann F, Cojoc M, Kurth I, Melin N,
Bouchez LC, Dubrovska A and Peitzsch C: CXCR4 as biomarker for
radioresistant cancer stem cells. Int J Radiat Biol. 90:687–699.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lourenco S, Teixeira VH, Kalber T, Jose
RJ, Floto RA and Janes SM: Macrophage migration inhibitory
factor-CXCR4 is the dominant chemotactic axis in human mesenchymal
stem cell recruitment to tumors. J Immunol. 194:3463–3474. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rhodes LV, Short SP, Neel NF, Salvo VA,
Zhu Y, Elliott S, Wei Y, Yu D, Sun M, Muir SE, et al: Cytokine
receptor CXCR4 mediates estrogen-independent tumorigenesis,
metastasis, and resistance to endocrine therapy in human breast
cancer. Cancer Res. 71:603–613. 2011. View Article : Google Scholar :
|
|
33
|
Bots M, de Bruin E, Rademaker-Koot MT and
Medema JP: Proteinase inhibitor-9 expression is induced by
maturation in dendritic cells via p38 MAP kinase. Hum Immunol.
68:959–964. 2007. View Article : Google Scholar
|
|
34
|
Lakshmi Narendra B, Eshvendar Reddy K,
Shantikumar S and Ramakrishna S: Immune system: A double-edged
sword in cancer. Inflamm Res. 62:823–834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krasnova Y, Putz EM, Smyth MJ and
Souza-Fonseca-Guimaraes F: Bench to bedside: NK cells and control
of metastasis. Clin Immunol. 15:30050–30054. 2015.
|
|
36
|
Medema JP, de Jong J, Peltenburg LT,
Verdegaal EM, Gorter A, Bres SA, Franken KL, Hahne M, Albar JP,
Melief CJ, et al: Blockade of the granzyme B/perforin pathway
through overexpression of the serine protease inhibitor PI-9/SPI-6
constitutes a mechanism for immune escape by tumors. Proc Natl Acad
Sci USA. 98:11515–11520. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Igney FH and Krammer PH: Immune escape of
tumors: Apoptosis resistance and tumor counterattack. J Leukoc
Biol. 71:907–920. 2002.PubMed/NCBI
|
|
38
|
Martínez-Lostao L, Anel A and Pardo J: How
do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res.
21:5047–5056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cunningham TD, Jiang X and Shapiro DJ:
Expression of high levels of human proteinase inhibitor 9 blocks
both perforin/granzyme and Fas/Fas ligand-mediated cytotoxicity.
Cell Immunol. 245:32–41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ray M, Hostetter DR, Loeb CR, Simko J and
Craik CS: Inhibition of Granzyme B by PI-9 protects prostate cancer
cells from apoptosis. Prostate. 72:846–855. 2012. View Article : Google Scholar :
|
|
41
|
Soriano C, Mukaro V, Hodge G, Ahern J,
Holmes M, Jersmann H, Moffat D, Meredith D, Jurisevic C, Reynolds
PN, et al: Increased proteinase inhibitor-9 (PI-9) and reduced
granzyme B in lung cancer: Mechanism for immune evasion? Lung
Cancer. 77:38–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Levin ER: Cellular functions of plasma
membrane estrogen receptors. Steroids. 67:471–475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee LM-J, Cao J, Deng H, Chen P, Gatalica
Z and Wang ZY: ER-alpha36, a novel variant of ER-alpha, is
expressed in ER-positive and -negative human breast carcinomas.
Anticancer Res. 28B:479–483. 2008.
|
|
44
|
Deng H, Zhang XT, Wang ML, Zheng HY, Liu
LJ and Wang ZY: ER-α36-mediated rapid estrogen signaling positively
regulates ER-positive breast cancer stem/progenitor cells. PLoS
One. 9:e880342014. View Article : Google Scholar
|
|
45
|
Zhang X, Deng H and Wang ZY: Estrogen
activation of the mitogen-activated protein kinase is mediated by
ER-α36 in ER-positive breast cancer cells. J Steroid Biochem Mol
Biol. 143:434–443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mao L, Yuan L, Slakey LM, Jones FE, Burow
ME and Hill SM: Inhibition of breast cancer cell invasion by
melatonin is mediated through regulation of the p38
mitogen-activated protein kinase signaling pathway. Breast Cancer
Res. 12:R1072010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang M, Liu HX, Teng XD, Wang HB, Cui J,
Jia SS, Gu XY and Li ZG: The differences in CXCR4 protein
expression are significant for the five molecular subtypes of
breast cancer. Ultrastruct Pathol. 36:381–386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dubrovska A, Hartung A, Bouchez LC, Walker
JR, Reddy VA, Cho CY and Schultz PG: CXCR4 activation maintains a
stem cell population in tamoxifen-resistant breast cancer cells
through AhR signalling. Br J Cancer. 107:43–52. 2012. View Article : Google Scholar : PubMed/NCBI
|