Introduction
Cervical cancer, as the most common malignant
gynecological tumor and the second leading cause of cancer
mortality, is a major health concern world-wide (1,2).
Majority of cervical carcinoma is associated with the presence of
human papilloma virus (HPV). The viral encoded E6 and E7 proteins
play key roles in cervical carcinoma progression (3). Multiple other signaling pathways,
such as Notch, Wnt, COX2, NF-κB, p53 and RhoC, are also believed to
be involved in the occurrence and development of cervical cancer
(4–6). Notch signaling especially has been
found to play a critical role in cervical cancer development
(6,7).
Notch signaling is highly conserved and is critical
for the determination of cell proliferation, differentiation and
apoptosis. Moreover, Notch receptors and their ligands are often
found aberrantly expressed in many types of cancers and plays an
important role in cancer progression (8–10).
The above led to increasing investigation of Notch signaling in the
cancer progression and the development of potential therapeutic
strategies targeting Notch signaling (11–13).
However, Notch activity is complicated and highly cell- and
context-specific, and differs due to the tumor microenvironment and
crosstalk with other signaling pathways (14,15).
In cervical cancer, up regulation of Notch signaling is observed in
the early stage and a downregulation in the late stage (16,17).
Even in the same cervical cell models, Notch signaling exhibits
controversial effects by independent studies. Activation of Notch1
signaling suppressed the growth of cervical cancer cells such as
HeLa, SiHa, and CaSki cells (18,19).
While the opposite effect of Notch signaling was also reported in
the same cancer cells. Blocking of Notch1 signaling was found to
result in cell growth arrest in HeLa and CaSki cells (20). Thus, the precise molecular
mechanisms of Notch signaling in cervical cancer are not completely
known and still need further study.
Histone deacetylase (HDAC) is reported to contribute
to tumor progression via silencing of tumor suppressor genes
(21). Overexpression of HDAC2 is
observed in cervical cancer (22).
The HDAC inhibitor VPA is currently being investigated for its
anticancer effect on several cancers (14,23,24).
Recent studies also showed that VPA could induce cell arrest and
apoptosis via activating Notch signaling in small cell lung cancer
and neuroblastoma (25,26). Our previous study demonstrated the
VPA anticancer efficacy in cervical cancer HeLa cells (27). In the present study, we further
investigated the VPA effects in three human cervical cancer cell
lines (HeLa, CaSki, HT-3) that differed in their ICN1-expression or
HPV integration statues, expecting to compare the difference of
VPA's effects on cervical cancer cells with different genetic
background. We also attempted to clarify the role of Notch
signaling in VPA-induced tumor suppression and the involvement of
other signaling pathways.
Materials and methods
Materials
Valproic Acid (VPA) was purchased from Sigma (cat.
no. PHR1061-1G). LY294002 (cat. no. HY-10108) was purchased from
Medchem Express, while DAPT (cat. no. INO1001-0005MG) was from
Jinpu Bio-Technology. Antibody to cleaved Notch1 (Val1744) (cat.
no. 4147) was from Cell Signaling Technology, and β-actin (cat. no.
Ab101-01) from Vazyme Biotechnology.
Cell lines and cell culture
Human cervical cancer HeLa, CaSki and HT-3 cells
were cultured in DMEM, RPMI-1640 and McCoy's 5A medium (Gibco)
respectively, supplemented with 10% fetal bovine serum (Gibco) and
1% penicillin/streptomycin (Gibco). All cell lines were obtained
from Cell Bank of State Key Laboratory of Genetic Engineering,
Fudan University. HeLa and CaSki cells are HPV18 and
HPV16-positive, respectively, while HT-3 cells are HPV negative.
VPA, DAPT and LY294002 were dissolved in DMSO with the final
concentration of DMSO in the culture medium <1%. DMSO was used
as a control when these compounds were used to treat cells.
RT-PCR and real-time PCR
Total RNA was extracted with RNeasy Mini kit
(Qiagen) as described in the protocol. The cDNA was synthesized
using PrimeScript II 1st strand cDNA Synthesis kit (Takara) from 2
μg of total RNA. Reverse transcription was run for one cycle of
30°C for 10 min, 42°C for 60 min, followed by 5 min at 95°C for
inactivation and held at 4°C. The primers for real-time PCR assays
are shown in Table I. The
real-time assays were performed for 40 cycles of 95°C for 100 sec,
60°C for 20 sec, and 72°C for 10 sec on a Roche LightCycler 480.
Assays were set up using SYBR Premix Ex Taq (Takara). PCR reactions
were run on a Veriti 96-well Thermal Cycler (Applied Biosystems).
β-actin was used as the internal control and expression levels for
each target gene were calculated by applying 2−ΔΔCT
methods. The experiments were done separately three times.
 | Table IPrimer sequences for gene
amplification. |
Table I
Primer sequences for gene
amplification.
| Genes | Primer (5′-3′) | PCR products
(bp) | Genebank no. |
|---|
| β-actin | F:
CATGTACGTTGCTATCCAGGC
R: CTCCTTAATGTCACGCACGAT | 250 | NM_001101 |
| Notch1 | F:
GGCCACCTGGGCCGGAGCTTC
R: GCGATCTGGGACTGCATGCTG | 365 | NM_017617 |
| HES1 | F:
TCAACACGACACCGGATAAAC
R: GCCGCGAGCTATCTTTCTTCA | 153 | NM_005524 |
| p53 | F:
CAGCACATGACGGAGGTTGT
R: TCATCCAAATACTCCACACGC | 125 | NM_001126118 |
| PCNA | F:
CCTGCTGGGATATTAGCTCCA
R: CAGCGGTAGGTGTCGAAGC | 109 | NM_002592 |
| SST | F:
ACCCAACCAGACGGAGAATGA
R: GCCGGGTTTGAGTTAGCAGA | 108 | NM_001048 |
| SSTR2 | F:
TCTGGGGCTTGGTACACAG
R: GATGGACACCATTCGGGTGA | 180 | NM_001050 |
| Casp3 | F:
TGCTTCTGAGCCATGGTGAA
R: TCTGTTGCCACCTTTCGGTT | 388 | NM_032991 |
| Ki-67 | F:
ACGCCTGGTTACTATCAAAAGG
R: CAGACCCATTTACTTGTGTTGGA | 209 | NM_002417 |
| Snail1 | F:
TATGCTGCCTTCCCAGGCTTG
R: ATGTGCATCTTGAGGGCACCC | 143 | NM_005985 |
| HPV16 E6 | F:
ACTTTGCTTTTCGGGATTTATGC
R: AGGACACAGTGGCTTTTGACAGTT | 206 | KP965162 |
| HPV18 E6 | F:
GGATCCAACACGGCGACCCTA
R: GGATTCAACGGTTTCTGGCACGCG | 350 | M14710.1 |
Western blotting
Cells were harvested and mixed with loading buffer
and heated for 5 min in boiling water. Supernatants were loaded
onto 10% Tris-glycine gel after centrifugation at 12,000 x g for 10
min and then transferred onto a PVDF membrane (Millipore) by
electroblotting. Membrane was then blocked with 5% fat-free milk,
washed three times with TBST and incubated with antibodies against
cleaved Notch1 (Val1744) and β-actin, respectively, at 4°C
overnight. Membrane was incubated with HRP-conjugated secondary
antibodies for 1 h after washing. Signals were detected by ECL kit
(GE Healthcare).
Cell proliferation assay
The cell proliferation assay was performed using the
WST-1 Cell proliferation and cytotoxicity assay kit (Beyotime) to
evaluate effects of VPA on the in vitro proliferation of
human cervical cancer HeLa, CaSki and HT-3 cells. Briefly, 100 μl
of the indicated cell stock (1×105 cells/ml in media)
was added to 96-well plates. Medium was replaced 8 h later with new
medium containing different concentrations of compounds and plates
were incubated at 37°C in a CO2 incubator for 72 h. All
compound concentrations were tested in triplicate. Following the
incubation, 10 μl of WST-1 reagent was added to each well and
incubated for an additional 3 h. The absorbance at 450 nm was
measured by Thermal MultiSkan FC (Thermal Scientific). The
experiments were done separately three times.
Cell apoptosis and cell cycle
analysis
Cell apoptosis and cell cycle was analyzed by flow
cytometry. Cells treated with VPA were harvested and washed twice
with PBS. Apoptosis assay was performed using the Annexin V-FITC
Apoptosis Detection kit (BD Bioscience). Cells (1×106)
were suspended in 1 ml 1X Binding buffer. Annexin V (5 μl)
conjugate and 5 μl of PI were added to 100 μl of the solution and
then incubated for 15 min in the dark. Cells were analyzed with a
BD FACS Calibur. For cell cycle analysis, cells were fixed in 70%
ethanol overnight and washed twice with PBS. After resuspended in
0.5 ml PBS with 30 μg PI (Sangon Biotech) and 25 μg DNase-free
RNase A (Tiangen Biotech), cells were incubated for 1 h at room
temperature and tested by flow cytometry. Experiments were repeated
three times.
Results
Effects of VPA on the proliferation of
cervical cancer cell lines
In our previous study, we demonstrated that VPA
could suppress cell proliferation in HeLa cells (27). VPA were further analyzed in this
study for its effects on proliferation in three human cervical
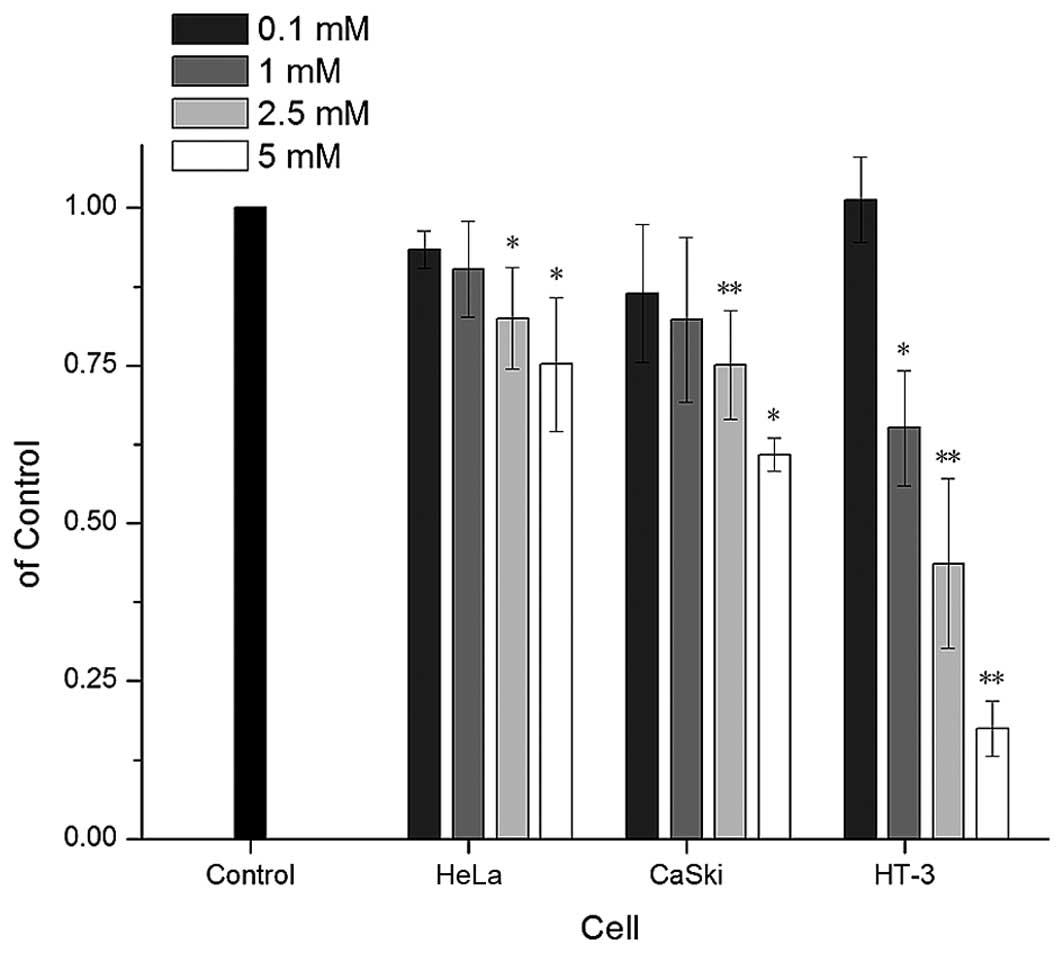
cancer cell lines. As expected, VPA suppressed proliferation in all
these tested cell lines in a dose-depend manner (Fig. 1). HT-3 cells, which were
HPV-negative and ICN1-overexpressing, exhibited the most
significant inhibition with the inhibitory rates of 82.5%. However,
HeLa, as a HPV-positive cell line with a low ICN1 background, were
less sensitive towards VPA. There was also a decrease of the cell
proliferation marker PCNA (-1.19, -1.26, -3.65-fold) and Ki-67
(-3.26, -1.34, -1.99-fold) assayed by real-time PCR in all the cell
lines after VPA treatment (Table
II).
| Table IIExpression of certain genes in
cervical cancer cells via real-time PCR analysis. |
Table II
Expression of certain genes in
cervical cancer cells via real-time PCR analysis.
| Genes | Gene
description | HeLa | CaSki | HT-3 |
|---|
| Notch1 | | −1.44±1.18a | 1.61±1.00 | −1.21±0.61 |
| HES1 | Target gene of
Notch1 | 2.14±0.80 | 1.68±0.35 | 1.15±0.18 |
| p53 | Tumor
suppressor | −2.81±0.71 | −1.16±0.08 | −3.65±1.91 |
| PCNA | Cell
differentiation marker | −1.19±0.24 | −1.26±0.08 | −1.66±0.52 |
| SST | Somatostatin | 2.01±0.39 | 4.32±1.47 | 5.34±2.74 |
| SSTR2 | SST receptor 2 | 1.68±1.96 | 1.17±0.39 | 1.51±2.03 |
| Casp3 | Cell apoptosis
marker | −1.38±0.57 | 1.29±0.18 | −1.02±0.43 |
| Ki-67 | Cell proliferation
marker | −3.26±1.37 | −1.34±0.12 | −1.99±0.36 |
| Snail1 | Key transcription
factors of EMT | 13.90±4.22 | 10.29±2.27 | 98.36±58.47 |
| E6 | Human papilloma
virus encoded gene | 2.05±0.85 | −29.45±4.97 | / |
VPA induces cell apoptosis and cell cycle
arrest
We further compared the effects of VPA on cell
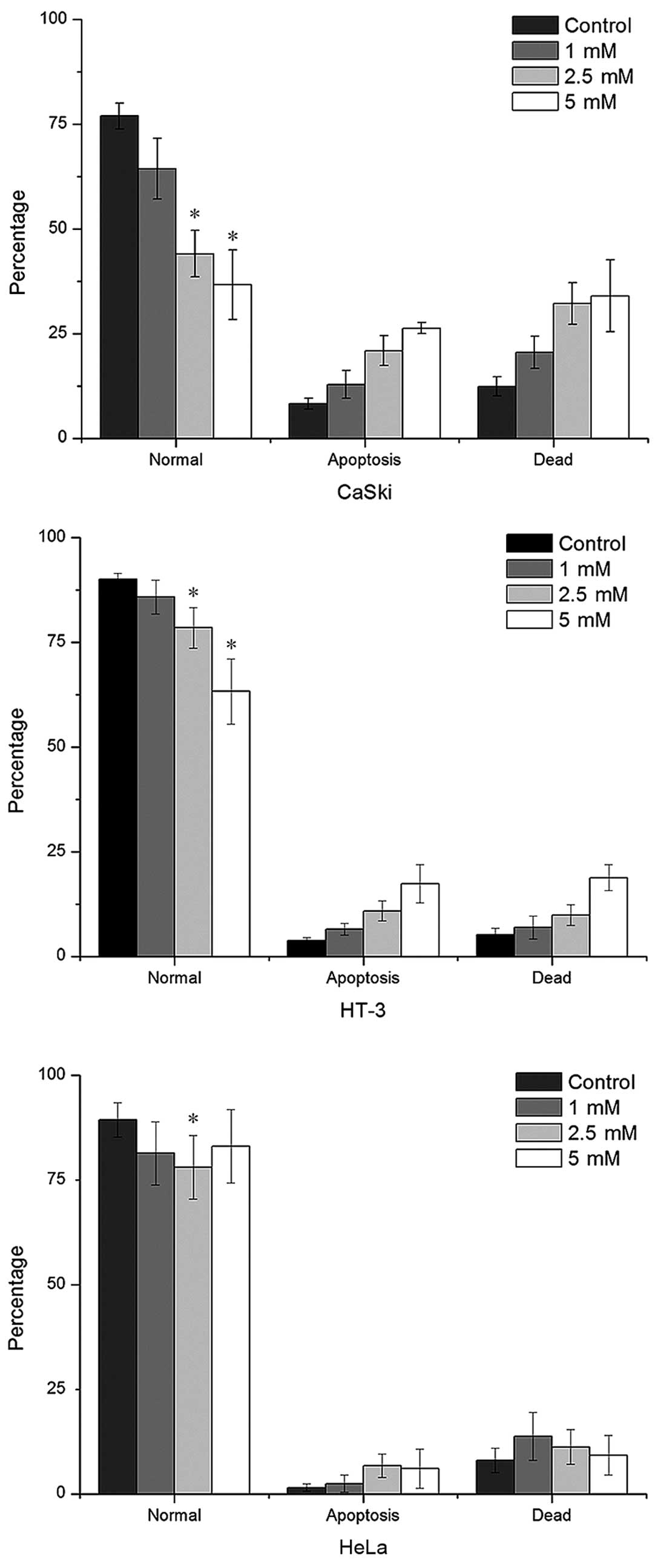
apoptosis in cervical cancer cell lines using FACS. VPA induced the
increase of both early apoptosis and necro-apoptosis in a
dose-depend manner in CaSki and HT-3 cells. However, only slightly
apoptosis was observed in HeLa cells at low VPA concentration
(Fig. 2). V-FITC-negative cells
decreased the most significantly in CaSki cells, with a decline of
50% compared to the control at 5 mM and a 72 h incubation. The
decrease of V-FITC-negative HT-3 cells was 29% at the same
condition. However, changes of the apoptotic marker caspase-3 were
only upregulated in CaSki cells (1.29-fold) assayed by real-time
PCR (Table II).
We also investigated the effects of VPA on cell
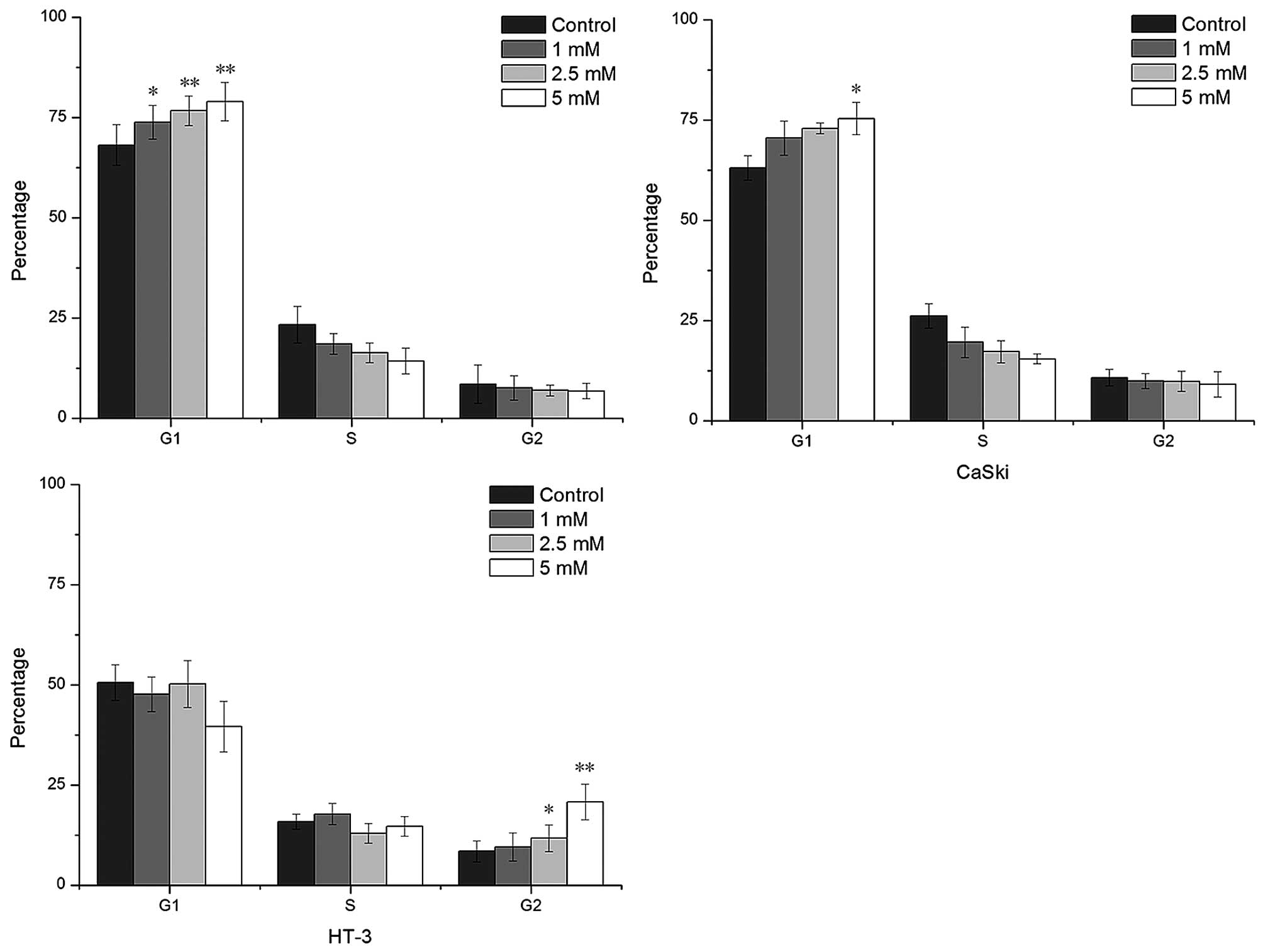
cycle progression, cell cycle distribution was analyzed at 72 h.
The results showed that VPA induced cell cycle arrest at phase G1
in HeLa and CaSki cells, and at phase G2 in HT-3 cells (Fig. 3). VPA, at 5 mM, enhanced the
percentage of G1-phase cells from 68 to 79% and 63 to 75% in HeLa
and CaSki cells respectively compared to the control. The
percentage of G2-phase cells was elevated from 12 to 25% in HT-3
cells in the same VPA concentration.
VPA acts as a Notch signaling
activator
VPA is believed to upregulate Notch signaling in
various cancers (28–30). Therefore, we evaluated VPA on the
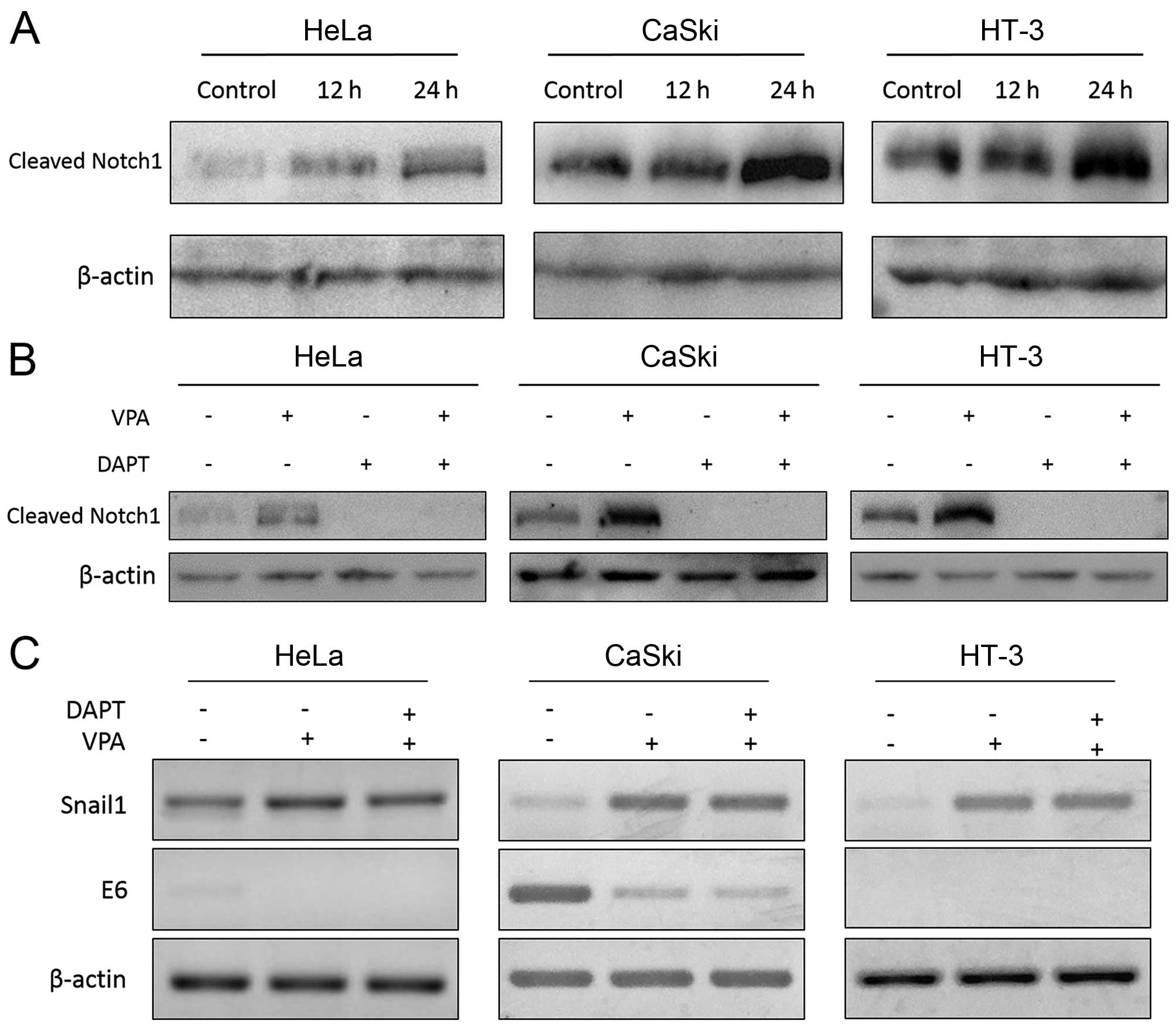
expression of Notch1. We first investigated the expressional
profiles of Notch signaling in these tested cell lines. We found
that expression of ICN1 (the active form of Notch1), was relatively
higher in CaSki and HT-3 cells than that in HeLa cells at the
protein level. This was identical to other research in which ICN1
was insignificant in HeLa cells compared with CaSki cells (31). Moreover, VPA in a time-dependent
manner induced an increase of ICN1 in all the tested cell lines
(Fig. 4A). The Notch downstream
target gene HES1 was also increased after VPA treatment (Table II). Despite the elevation of ICN1
at the protein level, no obvious change of Notch1 at the mRNA level
was detected by RT-PCR (data not shown) and Real-time analysis
(Table II). In our previous
study, we found that Notch1 directly plays an anti-oncogenic role
via inducing cell growth arrest in HeLa cells (26). Thus, VPA functions as a tumor
suppressor probably via activating Notch1 signaling.
In this study, Notch pathway inhibitor DAPT was used
to investigate the role of Notch signaling in VPA-induced tumor
suppression and could eliminate ICN1 with or without the appearance
of VPA after a 12 h incubation (Fig.
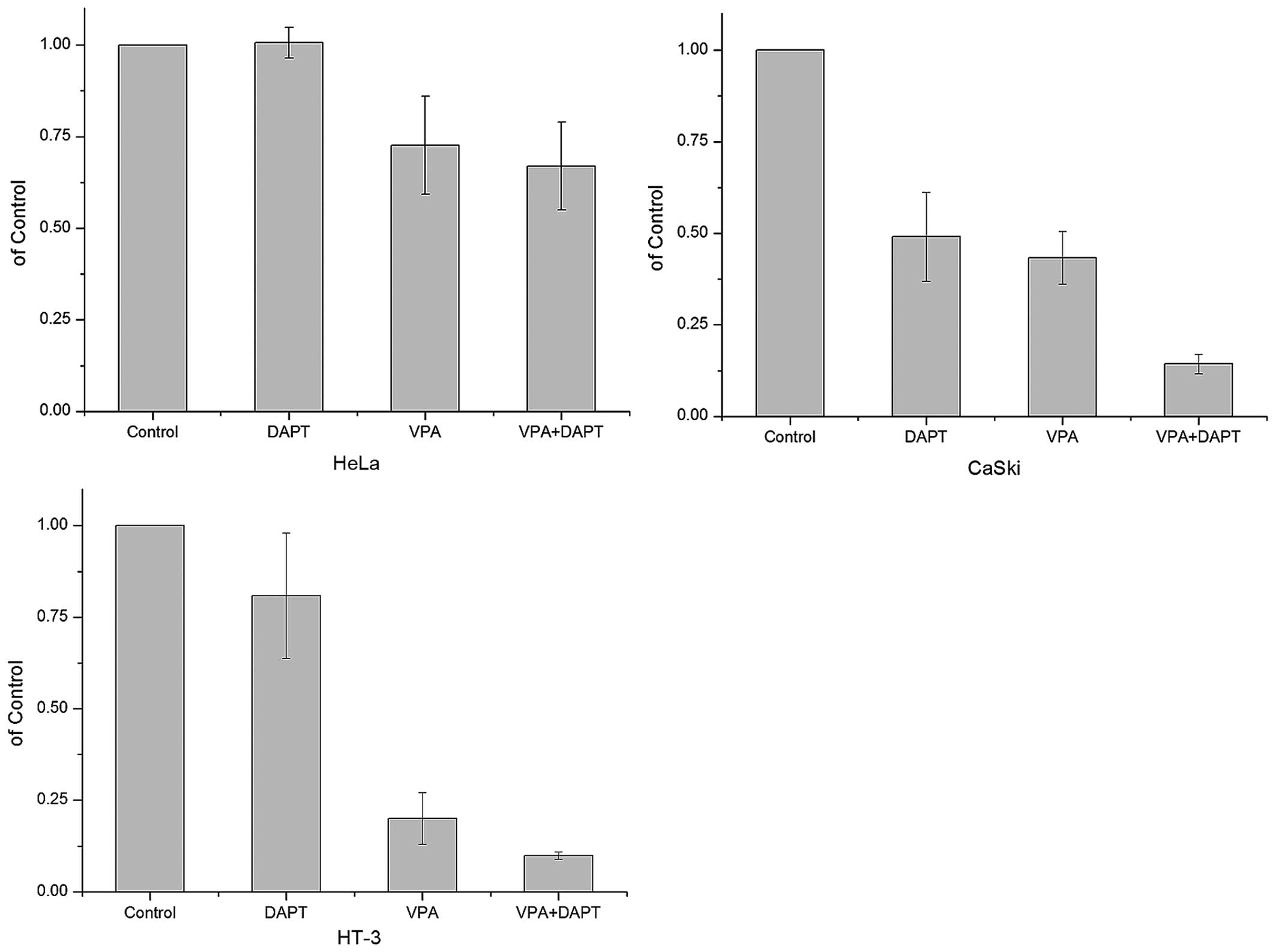
4B). DAPT suppressed cell proliferation individually and
enhanced suppression effect of VPA in CaSki and HT-3 cells. While
HeLa cells, which have a lower expression of ICN1, were not
affected by DAPT (Fig. 5).
Furthermore, VPA could still suppress cell proliferation after DAPT
treatment, indicating that VPA could also suppress tumor
progression via Notch-independent pathways.
Effects of VPA on the expression of HPV
E6 gene
We evaluated the VPA effects on the expression of
HPV E6 gene. The result by real-time PCR showed that VPA treatment
for 20 h significantly downregulated the expression of E6 in CaSki
cells (Table II), whilst in HeLa
cells, E6 was only downregulated in early stage (data not shown).
DAPT showed no effect on the VPA-induced inhibition of E6 by RT-PCR
(Fig. 4C) or real-time PCR (data
not shown), indicating that VPA-induced E6 downregulation may
possibly be Notch-independent. It is known that E6 acts as a tumor
suppressor in cervical cancer (26). E6 was highly expressed in CaSki
cells, so cell apoptosis and caspase-3 upregulation induced by VPA
may be explained by E6 inhibition. However, p53, which is
negatively regulated by E6, also decreased slightly, differing from
the expected (Table II).
Effects of VPA on cell morphological
change and the expression of Snail1
One of the key transcription factors of EMT, Snail1,
was upregulated by VPA in HeLa cells in our previous study. Snail1
was further confirmed to be activated in all the three cervical
cell lines (Table II).
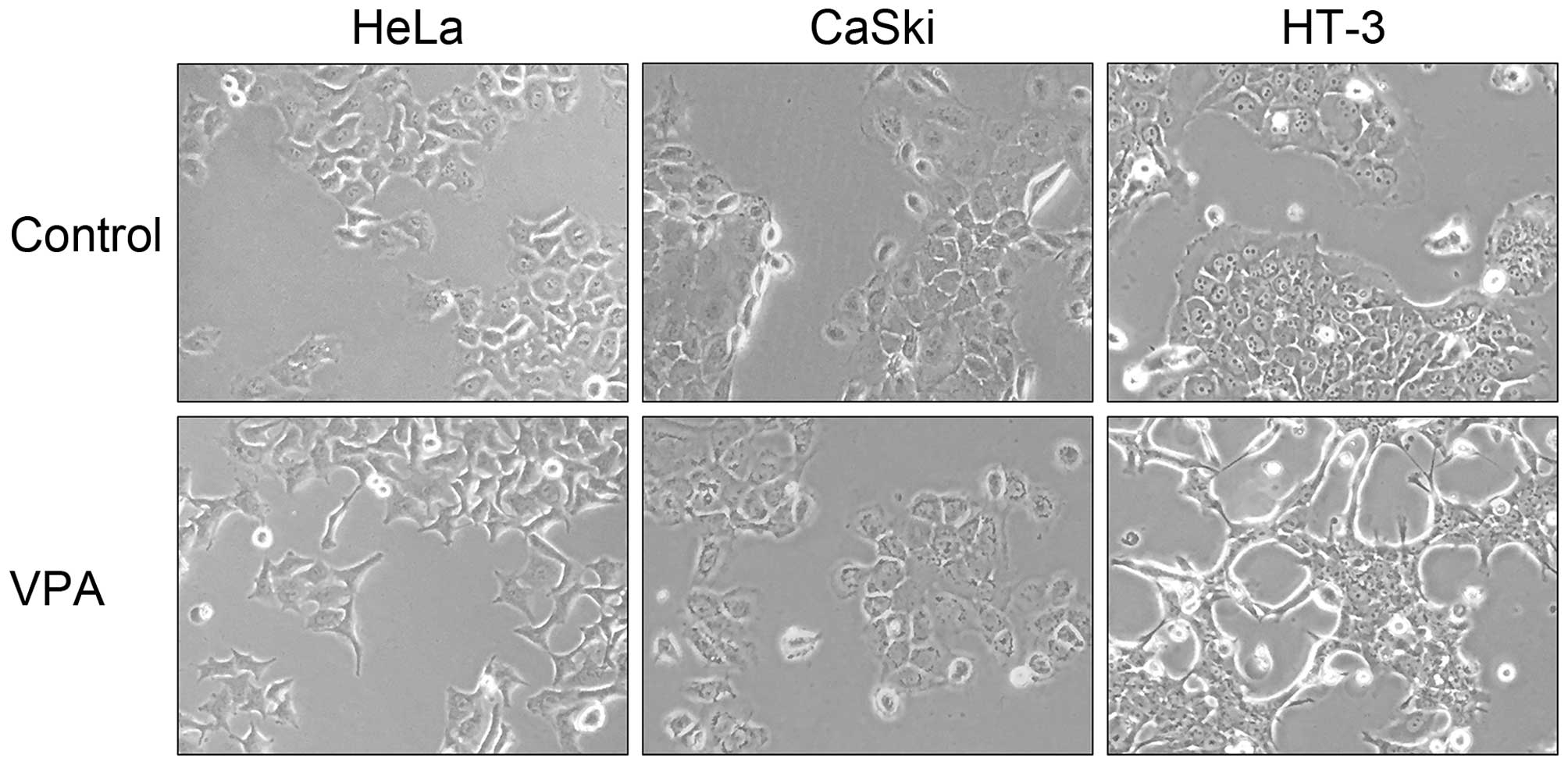
Morphological changes were observed in HeLa and HT-3 cells after
treatment of VPA at 5 mM, but not in CaSki cells despite the
upregulation of Snail1 (Fig. 6).
DAPT could partly reverse the increase of Snail1 induced by VPA in
HeLa cells, while it showed no effect in HT-3 cells (Fig. 4C). These findings indicated that
VPA-induced upregulation of Snail1 may probably be Notch-dependent
in HeLa cells, and some other pathways may also be involved in this
progression.
PI3K/Akt pathway is involved in
VPA-induced Snail1 expression and EMT
PI3K/Akt pathway participated in EMT and mediated
expression and stabilization of Snail (32,33).
Recent studies showed that VPA activated PI3K/Akt pathway by
increasing the phosphorylation levels of Akt and GSK-3β (23,34).
Thus, we further investigated whether PI3K/Akt was involved in
VPA-induced EMT in cervical cancer cells. HeLa cells were treated
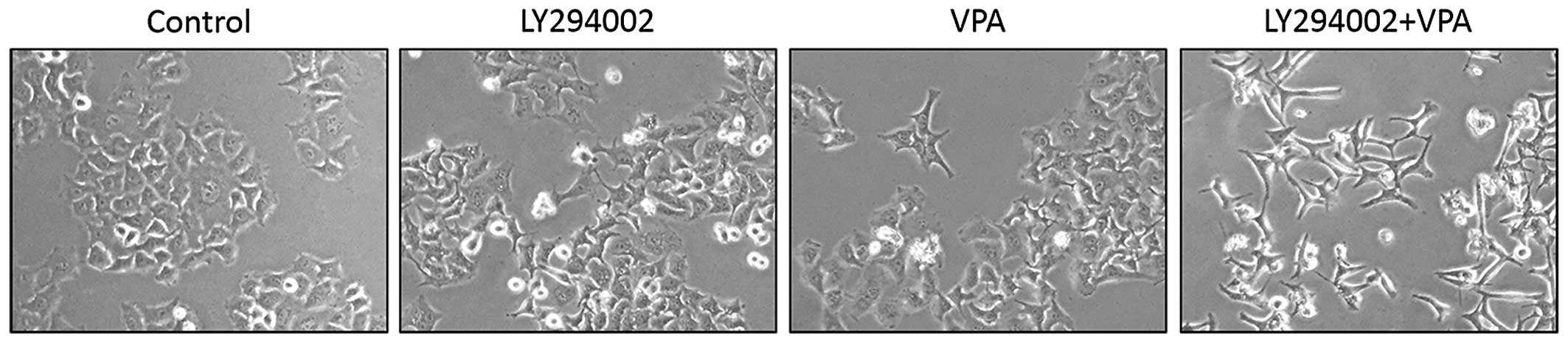
with LY294002, the inhibitor of PI3K/Akt, prior to exposure to VPA.
The results revealed that LY294002 itself induced slightly EMT and
could significantly enhance VPA-induced EMT (Fig. 7). These findings indicated that
PI3k/Akt pathway is involved in VPA-induced EMT. However, the
mechanism behind this enhancement is unknown and needed further
research.
Discussion
VPA, as a HDAC inhibitor, is currently under
investigation for its anticancer activities in many different types
of cancers (14,29,35).
Our previous study showed that VPA may act as a potential tumor
suppressor in cervical cancer. In this study, we compared the VPA
effects on cell proliferation, apoptosis, cell cycle and
differentiation in three HPV-positive and negative cervical cancer
cell lines. VPA exhibited proliferation inhibition in all the test
cells, especially in CaSki and HT-3 cells. Both early apoptosis and
necro-apoptosis increased significantly in a dose-depend manner
after VPA treatment. However, VPA was able to induce slight
apoptosis in HeLa cells, but only at low concentration. Cell cycle
arrest was observed in all the cell lines. Noteworthy, the effects
of VPA on the cell cycle were different. That is to say, HeLa and
CaSki cells exhibited G1 phase arrest, while G2 phase arrest was
induced in HT-3 cells. These distinct results in different cell
lines indicated that the effect of VPA may depend on the molecular
and genetic background of the cells.
Notch signaling is reported involved in the
pathogenesis of many human cancers and is highly cell-specific
(14,36). Notch1 activation via ICN1 and VPA
directly suppressed cervical cancer HeLa cells both in vitro
and in vivo in our previous study (26). We wonder if VPA directly modulate
Notch signaling. In this study, the effects of VPA were further
confirmed not only in HeLa but also in cervical cancer CaSki and
HT-3 cell lines. VPA could elevate the expression of ICN1, but had
no effect on Notch1 at mRNA level. VPA was reported to modulate
γ-secretase cleavage of β-amyloid precursor protein in mouse brain
cells (37). Therefore, VPA may
upregulate γ-secretase cleavage of Notch1 in cervical cancer cells
and release ICN1. Several studies showed that ICN1 overexpression
by ICN1-transfection suppress cell growth in HeLa and CaSki cells
(18,26). Thus, upregulation of ICN1 may
contribute to the VPA growth inhibitory effects. Of note, DAPT also
suppressed cell proliferation and enhanced suppression of VPA in
CaSki and HT-3 cells. This was possibly caused by the complicity of
Notch signaling. It is reported that both ICN1 transfection and RNA
interference resulted in cell growth arrest in CaSki cells
(3,31). Therefore, Notch signaling may act
as either a tumor suppressor or an oncogene in the same cell model.
We speculated that overexpression of ICN1 activates several tumor
suppressors and exhibit anticancer effects. While endogenous ICN1
is necessary for survival of ICN1-overexpressing cells, leading to
a cell growth arrest after full suppression of Notch signaling in
these cell lines. Moreover, VPA suppressed cell growth with the
appearance of Notch signaling inhibitor DAPT, suggesting that
another pathway is involved in VPA-induced growth suppression.
The oncogene E6 is necessary for HPV-induced
cervical cancer malignancy (5,18,38).
Notch1 activation was reported to downregulate HPV E6 expression
and resulted in cell arrest in HPV-positive cervical cancer cells
(18,39). E6 was also found to be
downregulated by VPA in this study, especially in CaSki cells.
However, DAPT failed to reverse VPA-induced E6 suppression,
suggesting that VPA suppress E6 gene in a Notch-independent
pathway. Several studies have revealed that E6 protects cells from
apoptosis via accelerating the degradation of several key proteins
in apoptotic signaling, such as caspase 8 and p53 (40,41).
CaSki cells exhibited high expression of E6 and the most
significant apoptosis induced by VPA. Thus, VPA may induce strong
apoptosis in cells with a high E6 expression via E6
suppression.
VPA was reported to induce cell morphological change
(or EMT) in cervical cancer HeLa cells in our previous study
(27). EMT was also observed in
HeLa and HT-3 cells after VPA treatment, coupled with a significant
upregulation of the transcription factor Snail1. Notch signaling
regulates EMT directly and indirectly through various signaling
pathways (34). Our study showed
that DAPT partly reversed VPA-induced Snail1 upregulation in HeLa
cells, suggesting that VPA upregulated Snail1 at least partly via
Notch signaling activation in cervical cancer. PI3K/Akt pathway,
which was reported to be activated by VPA, participated in EMT and
mediated expression and stabilization of Snail (23,24,32,33).
PI3K/Akt pathway inhibitor LY294002 was reported to inhibit
VPA-induced upregulation of Snail1 and EMT in colorectal cancer
(34). However, LY294002 showed no
effect on VPA-induced Snail1 upregulation in our study. Oppositely,
LY294002 enhanced VPA-induced EMT. These results indicated that
PI3K/Akt pathway may be involved in VPA-induced EMT. However,
further research is needed to understand its precise mechanism.
In conclusion, VPA could induce tumor suppression
via either Notch signaling activation or acting as a HDAC inhibitor
in cervical cancers. The effects of VPA depend on the molecular and
genetic background of specific cells. This provides an access to
precision medicine when VPA is used as a cervical cancer
therapeutic. We also found the oncogene E6 and EMT transcription
factor Snail1 were regulated by VPA, and PI3K/Akt may be involve in
VPA-induced EMT. However, the specific mechanism still need further
study.
Acknowledgements
This study is supported by the Open Research Funds
of the State Key Laboratory of Genetic Engineering, Fudan
University (SKLGE-1206).
References
|
1
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu L and Griffin JD: Modulation of Notch
signaling by mastermind-like (MAML) transcriptional co-activators
and their involvement in tumorigenesis. Semin Cancer Biol.
14:348–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weijzen S, Zlobin A, Braid M, Miele L and
Kast WM: HPV16 E6 and E7 oncoproteins regulate Notch-1 expression
and cooperate to induce transformation. J Cell Physiol.
194:356–362. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schwarz JK, Payton JE, Rashmi R, Xiang T,
Jia Y, Huettner P, Rogers BE, Yang Q, Watson M, Rader JS, et al:
Pathway-specific analysis of gene expression data identifies the
PI3K/Akt pathway as a novel therapeutic target in cervical cancer.
Clin Cancer Res. 18:1464–1471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Perez-Plasencia C, Duenas-Gonzalez A and
Alatorre-Tavera B: Second hit in cervical carcinogenesis process:
Involvement of wnt/beta catenin pathway. Int Arch Med. 1:102008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yousif NG, Sadiq AM, Yousif MG,
Al-Mudhafar RH, Al-Baghdadi JJ and Hadi N: Notch1 ligand signaling
pathway activated in cervical cancer: Poor prognosis with
high-level JAG1/Notch1. Arch Gynecol Obstet. 292:899–904. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maliekal TT, Bajaj J, Giri V, Subramanyam
D and Krishna S: The role of Notch signaling in human cervical
cancer: Implications for solid tumors. Oncogene. 27:5110–5114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu L, Aster JC, Blacklow SC, Lake R,
Artavanis-Tsakonas S and Griffin JD: MAML1, a human homologue of
Drosophila mastermind, is a transcriptional co-activator for NOTCH
receptors. Nat Genet. 26:484–489. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wael H, Yoshida R, Kudoh S, Hasegawa K,
Niimori-Kita K and Ito T: Notch1 signaling controls cell
proliferation, apoptosis and differentiation in lung carcinoma.
Lung Cancer. 85:131–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brakenhoff RH: Cancer. Another NOTCH for
cancer. Science. 333:1102–1103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
del Campo JM, Prat A, Gil-Moreno A, Pérez
J and Parera M: Update on novel therapeutic agents for cervical
cancer. Gynecol Oncol. 110(Suppl 2): S72–S76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pant S, Jones SF, Kurkjian CD, Infante JR,
Moore KN, Burris HA, McMeekin DS, Benhadji KA, Patel BK, Frenzel
MJ, et al: A first-in-human phase I study of the oral Notch
inhibitor, LY900009, in patients with advanced cancer. Eur J
Cancer. 56:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kong R, Feng J, Ma Y, Zhou B, Li S, Zhang
W, Jiang J, Zhang J, Qiao Z, Zhang T, et al: Silencing NACK by
siRNA inhibits tumorigenesis in non-small cell lung cancer via
targeting Notch1 signaling pathway. Oncol Rep. 35:2306–2314.
2016.PubMed/NCBI
|
|
14
|
Duenas-Gonzalez A, Candelaria M,
Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E and
Herrera LA: Valproic acid as epigenetic cancer drug: Preclinical,
clinical and transcriptional effects on solid tumors. Cancer Treat
Rev. 34:206–222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ranganathan P, Weaver KL and Capobianco
AJ: Notch signalling in solid tumours: A little bit of everything
but not all the time. Nat Rev Cancer. 11:338–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zagouras P, Stifani S, Blaumueller CM,
Carcangiu ML and Artavanis-Tsakonas S: Alterations in Notch
signaling in neoplastic lesions of the human cervix. Proc Natl Acad
Sci USA. 92:6414–6418. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tewari KS, Taylor JA, Liao SY, DiSaia PJ,
Burger RA, Monk BJ, Hughes CC and Villarreal LP: Development and
assessment of a general theory of cervical carcinogenesis utilizing
a severe combined immunodeficiency murine-human xenograft model.
Gynecol Oncol. 77:137–148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Talora C, Sgroi DC, Crum CP and Dotto GP:
Specific down-modulation of Notch1 signaling in cervical cancer
cells is required for sustained HPV-E6/E7 expression and late steps
of malignant transformation. Genes Dev. 16:2252–2263. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Talora C, Cialfi S, Segatto O, Morrone S,
Kim Choi J, Frati L, Paolo Dotto G, Gulino A and Screpanti I:
Constitutively active Notch1 induces growth arrest of HPV-positive
cervical cancer cells via separate signaling pathways. Exp Cell
Res. 305:343–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu H, Zhao X, Huang S, Jian L, Qian G and
Ge S: Blocking Notch1 signaling by RNA interference can induce
growth inhibition in HeLa cells. Int J Gynecol Cancer. 17:511–516.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: Emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar
|
|
22
|
Huang BH, Laban M, Leung CH, Lee L, Lee
CK, Salto-Tellez M, Raju GC and Hooi SC: Inhibition of histone
deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression,
independent of histone deacetylase 1. Cell Death Differ.
12:395–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Teng HF, Li PN, Hou DR, Liu SW, Lin CT,
Loo MR, Kao CH, Lin KH and Chen SL: Valproic acid enhances Oct4
promoter activity through PI3K/Akt/mTOR pathway activated nuclear
receptors. Mol Cell Endocrinol. 383:147–158. 2014. View Article : Google Scholar
|
|
24
|
Shan Z, Feng-Nian R, Jie G and Ting Z:
Effects of valproic acid on proliferation, apoptosis, angiogenesis
and metastasis of ovarian cancer in vitro and in vivo. Asian Pac J
Cancer Prev. 13:3977–3982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dotto GP: Notch tumor suppressor function.
Oncogene. 27:5115–5123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Franko-Tobin LG, Mackey LV, Huang W, Song
X, Jin B, Luo J, Morris LM, Liu M, Fuselier JA, Coy DH, et al:
Notch1-mediated tumor suppression in cervical cancer with the
involvement of SST signaling and its application in enhanced
SSTR-targeted therapeutics. Oncologist. 17:220–232. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsai C, Leslie JS, Franko-Tobin LG,
Prasnal MC, Yang T, Vienna Mackey L, Fuselier JA, Coy DH, Liu M, Yu
C, et al: Valproic acid suppresses cervical cancer tumor
progression possibly via activating Notch1 signaling and enhances
receptor-targeted cancer chemotherapeutic via activating
somatostatin receptor type II. Arch Gynecol Obstet. 288:393–400.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greenblatt DY, Cayo MA, Adler JT, Ning L,
Haymart MR, Kunnimalaiyaan M and Chen H: Valproic acid activates
Notch1 signaling and induces apoptosis in medullary thyroid cancer
cells. Ann Surg. 247:1036–1040. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Greenblatt DY, Vaccaro AM, Jaskula-Sztul
R, Ning L, Haymart M, Kunnimalaiyaan M and Chen H: Valproic acid
activates notch-1 signaling and regulates the neuroendocrine
phenotype in carcinoid cancer cells. Oncologist. 12:942–951. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Platta CS, Greenblatt DY, Kunnimalaiyaan M
and Chen H: Valproic acid induces Notch1 signaling in small cell
lung cancer cells. J Surg Res. 148:31–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuncharin Y, Sangphech N, Kueanjinda P,
Bhattarakosol P and Palaga T: MAML1 regulates cell viability via
the NF-κB pathway in cervical cancer cell lines. Exp Cell Res.
317:1830–1840. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng J, Cen J, Li J, Zhao R, Zhu C, Wang
Z, Xie J and Tang W: Histone deacetylase inhibitor valproic acid
(VPA) promotes the epithelial mesenchymal transition of colorectal
cancer cells via up regulation of Snail. Cell Adhes Migr.
9:495–501. 2015. View Article : Google Scholar
|
|
35
|
Mawatari T, Ninomiya I, Inokuchi M, Harada
S, Hayashi H, Oyama K, Makino I, Nakagawara H, Miyashita T, Tajima
H, et al: Valproic acid inhibits proliferation of HER2-expressing
breast cancer cells by inducing cell cycle arrest and apoptosis
through Hsp70 acetylation. Int J Oncol. 47:2073–2081.
2015.PubMed/NCBI
|
|
36
|
Kostrouchová M, Kostrouch Z and
Kostrouchová M: Valproic acid, a molecular lead to multiple
regulatory pathways. Folia Biol (Praha). 53:37–49. 2007.
|
|
37
|
Qing H, He G, Ly PT, Fox CJ, Staufenbiel
M, Cai F, Zhang Z, Wei S, Sun X, Chen CH, et al: Valproic acid
inhibits Abeta production, neuritic plaque formation, and
behavioral deficits in Alzheimer's disease mouse models. J Exp Med.
205:2781–2789. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dueñas-González A, Lizano M, Candelaria M,
Cetina L, Arce C and Cervera E: Epigenetics of cervical cancer. An
overview and therapeutic perspectives. Mol Cancer. 4:382005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Qin H, Chen B, Xin X, Li J and Han
H: Overexpressed active Notch1 induces cell growth arrest of HeLa
cervical carcinoma cells. Int J Gynecol Cancer. 17:1283–1292. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yuan CH, Filippova M, Krstenansky JL and
Duerksen-Hughes PJ: Flavonol and imidazole derivatives block HPV16
E6 activities and reactivate apoptotic pathways in HPV(+) cells.
Cell Death Dis. 7:20602016. View Article : Google Scholar
|
|
41
|
Cai Q, Lv L, Shao Q, Li X and Dian A:
Human papillomavirus early proteins and apoptosis. Arch Gynecol
Obstet. 287:541–548. 2013. View Article : Google Scholar
|