Introduction
Cis-diamminedichloroplatinum (II), best known
as cisplatin, is a classical chemotherapy drug that is widely used
for treating advanced cancers, including ovarian cancer. The
antitumor activity of cisplatin is due to its interaction with
nuclear DNA, which results in the formation of DNA adducts and
subsequent DNA damage-mediated apoptotic signaling (1,2).
Cisplatin also has unrelated effects and accumulates in different
organelles, including the endoplasmic reticulum (ER), lysosomes,
and mitochondria, thus resulting in activation of apoptotic
signaling pathways (3). The effect
of cross-talk between different organelles on the antitumor
efficacy of cisplatin has received substantial attention in recent
years. The regulatory networks between the ER and mitochondria
involve mitochondrial energy metabolism, lipid metabolism,
Ca2+ signaling transmission and cell apoptosis (4). ER stress not only triggers a cascade
of cellular events that lead to apoptosis but also amplifies the
apoptotic signal. ER stress enhances the mitochondrial apoptosis
pathway via highly efficient transportation of Ca2+ from
the ER to the mitochondria (5).
Hence, studying the cellular mechanisms underlying the signaling
between the ER and mitochondria should provide new insights into
cisplatin resistance in ovarian cancer.
The ER is a central membrane-bound organelle that is
the main Ca2+ storage compartment in most cell types and
has an extremely important role in maintaining Ca2+
homeostasis. The large release of Ca2+ from the ER, an
early event in cell apoptosis, results in cytosolic and
mitochondrial Ca2+ overload (6). Calpains are a complex family of
Ca2+-dependent cysteine proteases that cleave various
protein substrates. The ubiquitous calpain isoforms can be divided
into calpain-1 (μ-calpain) and calpain-2 (m-calpain) according to
the cytosolic Ca2+ concentration (μM or mM range)
required for their activation (7).
Recent research has shown that calpain plays a key role in ER
stress-mediated apoptosis and mitochondria-mediated apoptosis
(8–10). Huang et al have reported
that the ER stress inducer thapsigargin (TG) increases
intracellular Ca2+ levels and subsequently activates
calpains; this activation is followed by the subsequent activation
of caspase-3 and caspase-9 and the induction of CHOP. As expected,
EGTA and BAPTA-AM, intracellular Ca2+ chelators, inhibit
the TG-induced activation of the calpains, caspase-3, and caspase-9
and suppress the TG-induced CHOP protein in hepatic stellate cells
(8). Interestingly, after
silencing calpain-1 and calpain-2, researchers have demonstrated
that calpain-1 (but not calpain-2) triggers ER stress in an in
vitro model of hypoxia/reoxygen-ation (10). However, the relationship between
calpains and ER-mitochondria interactions remains unclear.
Ca2+ can also be taken up by the mitochondria through
the mitochondrial calcium channel uniporter (MCU), which causes
mitochondrial depolarization, thereby contributing to the opening
of the permeability transition pore (PTP) and altering the
permeability of the inner membrane of the mitochondria (IMM). The
PTP can further lead to mitochondrial swelling and mitochondrial
outer membrane permeabilization (MOMP), with a consequent release
of Cyto C and caspase-activating factors and the induction of cell
apoptosis (11). In our previous
study, using the cytoplasmic Ca2+-indicating fluorophore
Fluo-4/AM and the mitochondrial Ca2+-indicating
fluorophore Rhod-2/AM, we found that cisplatin causes pro-apoptotic
Ca2+ release from the ER to the cytosol and
mitochondria, thus leading to cytosolic and mitochondrial
Ca2+ overload, which contributes to ER-mediated
apoptosis and mitochondria-mediated apoptosis in
cisplatin-sensitive SKOV3 ovarian cancer cells (12). These results have confirmed that a
large release of Ca2+ from the ER to the cytosol and
mitochondria is very important for ER-mediated apoptosis and
mitochondrial apoptosis. However, the exact mechanism by which the
cisplatin-induced ER Ca2+ release enters the
mitochondria is not known.
In eukaryotic cells, intracellular organelles
determine cell fate and maintain cellular homeostasis through their
physical interactions with one another; the functional or physical
interaction between mitochondria and the ER (referred to as
mitochondria-associated ER membranes, MAM) is a prime example
(13,14). MAM serves to establish close
communication between the mitochondria and ER, including efficient
transportation of Ca2+ from the ER to the mitochondria.
The subcellular compartment and function of MAM is under intense
investigation because it is increasingly recognized as an important
region controlling cell physiology. In the MAM, Ca2+
release has been proposed to occur through the ER Ca2+
channel inositol 1,4,5-trisphosphate receptor (IP3R, the major ER
Ca2+ release channel) to voltage-dependent anion channel
1 (VDAC1, the main mitochondrial Ca2+ uptake pathway) on
the outer mitochondrial membrane (OMM) (13). Glucose-regulated protein 75 (Grp75)
and mitofusin 2 (Mfn2), known as molecular bridges, help to
increase ER-mitochondria contact sites (15). Electron microscopy and fluorescence
microscopy, two important technologies, have been widely used to
observe the regions of close contacts between the ER and
mitochondria (16). Moreover,
Wieckowski et al have provided a detailed introduction on
the optimized protocols to isolate MAM fraction from tissues and
cells (17). By adapting the
protocol, Paillard at the Université de Montpellier and his
colleagues have found that lethal hypoxia-reoxygenation contributes
to H9c2 cell injury accompanied by a significant increase in Grp75
in the MAM, which indicates increased ER and mitochondria physical
interactions. Using Rhod-2/AM loading, they have also shown that
increased ER and mitochondrial interactions lead to lethal
reperfusion injury through mitochondrial Ca2+ overload
(18). Guo et al have shown
that overexpression of Mfn2 enhances
H2O2-induced vascular smooth muscle cell
apoptosis, perhaps through excessive ER-mitochondria tethering and
Ca2+ transfer (19).
These results suggest that there is a positive correlation between
MAM and cell apoptosis. However, it is unclear whether there is a
link between MAM and cisplatin resistance in ovarian cancer.
Although a number of indicators suggest that MAM may be useful in
cancer treatment, the potential of MAM in cancer treatment has not
been studied systematically. Hence, it is very important to explore
the effects of MAM on cisplatin-induced cell apoptosis to clarify
the mechanism of cisplatin resistance.
A systematic survey of the transcriptional profiles
of various cancer cell types has indicated that the dysregulation
of Bcl-2 is a key distinguishing factor between normal and cancer
cells. Additionally, empirical evidence has shown a positive
correlation between Bcl-2 expression and cisplatin resistance in
cancer cells (20). In our
previous studies, we have demonstrated that Bcl-2 expression in
cisplatin-resistant SKOV3/DDP cells is significantly higher than
that in cisplatin-sensitive SKOV3 cells (21). During ER stress, by interacting
with the IP3R, Bcl-2 and Bcl-xL have been shown to decrease the ER
Ca2+ load, thereby preventing excessive pro-apoptotic
Ca2+ signals and mitochondrial Ca2+ overload,
and finally to inhibit cell apoptosis (22). High expression of Bcl-2 also
results in partial VDAC closure, which is accompanied by a marked
decrease in mitochondrial Ca2+ levels (23). Interestingly, by analyzing MAM
composition in Chinese hamster ovary cells, Meunier and Hayashi
have demonstrated that Bcl-2 is enriched at the MAM (24). However, the exact role of Bcl-2 in
cisplatin-induced SKOV3/DDP cell apoptosis remains unknown.
ABT-737, a potent small-molecule inhibitor of Bcl-xL and Bcl-2, can
bind to the hydrophobic groove in Bcl-2 and Bcl-xL and consequently
prevent them from sequestering proapoptotic BH3-only protein.
ABT737 exhibits synergistic cytotoxicity and induced significant
apoptosis in various cancer types, including ovarian, lung and
bladder cancers (25). Moreover,
ABT737 has been reported to enhance the cisplatin-induced apoptosis
in cancer cells (25). Hence, we
propose that ABT737 reverses cisplatin resistance by regulating
ER-mitochondria Ca2+ signal transduction in human
ovarian cancer cells.
Materials and methods
Antibodies and drugs
Anti-caspase-3 (sc-7272), anti-caspase-4 (sc-56056),
anti-cleaved caspase-4 (sc-22173-R), and anti-caspase-9 (sc-56073)
antibodies (Abs) were purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Anti-cleaved caspase-3 (ab2302),
anti-cleaved caspase-9 (ab2324), anti-PDI (ab2792), anti-VDAC1
(ab14734), anti-CHOP (ab11419), anti-IP3R (ab5804) Abs were
purchased from Abcam Ltd. (Hong Kong, China). Anti-β-actin
(60008-1-Ig), anti-Bax (50599-2-Ig), anti-Bcl-2 (12789-1-AP),
anti-cytochrome c (Cyto C) (10993-1-AP), anti-Grp78/BIP
(11587-1-AP), anti-Grp75 (14887-1-AP), anti-mitofusin 2 (Mfn2)
(12186-1-AP), anti-β-tubulin (10068-1-AP), anti-calreticulin
(10292-1-AP), peroxidase-conjugated AffiniPure goat anti-mouse IgG
(H+L) (SA00001-1), peroxidase-conjugated AffiniPure goat
anti-rabbit IgG (H+L) (SA00001-2) and anti-IgG control (30000-0-AP)
Abs were purchased from Proteintech Group, Inc. (Chicago, IL, USA).
The anti-calpain-1 catalytic subunit (#31038-1) Ab was purchased
from SAB (College Park, MD, USA). Cisplatin was purchased from
Sigma-Aldrich (St. Louis, MO, USA) and dissolved in normal saline
(NS) for in vitro use and animal studies. ABT-737 was
provided by Abbott Laboratories (Abbott Park, IL, USA) and
dissolved in dimethyl sulfoxide (DMSO) for in vitro use and
animal studies. 2-aminoethyl diphenylborinate (2-APB) (ab120124)
was purchased from Abcam Ltd.
Cell culture
The cisplatin-resistant clone SKOV3/DDP was obtained
from the Peking Union Medical College (Beijing, China). COC1/DDP
and A2780/DDP cells were purchased from the American Type Culture
Collection (Manassas, VA, USA). SKOV3/DDP, COC1/DDP and A2780/DDP
cells were maintained in IMDM (Gibco; Thermo Fisher Scientific,
Inc., Carlsbad, CA, USA), supplemented with 10% (v/v) fetal calf
serum (Gibco; Thermo Fisher Scientific, Inc.), 100 mg/ml
streptomycin and 100 U/ml penicillin (each from Genview, Galveston,
TX, USA). The cells were incubated at 37°C in an atmosphere
containing 5% CO2.
Cell viability assay
Cell viability was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Beyotime Institute of Biotechnology, Haimen, China). The
cisplatin-resistant ovarian cancer cells, during the exponential
growth phase, were seeded into 96-well culture plates in 100 μl of
IMDM at a density of 1.0×104 cells/well. After a 24-h
incubation, the indicated drugs were added for another 24 h in four
parallel wells. The MTT assays were performed as follows: 20 μl of
MTT solution [5 mg/ml in phosphate-buffered saline (PBS)] was added
to each well, and the cells were incubated at 37°C for 4 h. At the
end of the incubation, 150 μl of DMSO (Beijing Chemical Industry
Co., Ltd., Beijing, China) was added to each well. The cells were
agitated for 10 min prior to measuring the absorbance at 570 nm
using a model 680 microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The growth inhibition rate was calculated as
follows: Inhibition (%) = [1 − (absorbance of experimental
group/absorbance of control group)] × 100. The mean value of four
replicate wells was calculated for each treatment group.
Annexin V and cell death assay
The Muse™ Annexin V Dead Cell kit (Ref. MCH 100105,
Merck Millipore, Darmstadt, Germany) was used to monitor cell
death. Exponentially growing cisplatin-resistant ovarian cancer
cells were seeded into 6-well culture plates at a density of
2×105 cells/well. After exposure to different
experimental conditions, the cells were trypsinized and resuspended
in IMDM with 10% FBS at a concentration of 1×106
cells/ml. Cells were incubated with Annexin V and Dead Cell reagent
in a dark room at room temperature for 20 min. Finally, the samples
were measured by flow cytometry (Muse Cell Analyzer, Merck
Millipore).
Mitochondrial membrane potential
(ΔΨm)
Changes in the mitochondrial membrane potential
(ΔΨm) during the early stages of apoptosis were assayed using the
Muse MitoPotential assay (Ref. MCH 100110, Merck Millipore) in
SKOV3/DDP cells treated with cisplatin alone or in combination with
ABT737. Briefly, cells were harvested, and the cell pellet was
suspended in assay buffer (1×105 cells/100 μl).
MitoPotential dye working solution was added, and the cell
suspension was incubated at 37°C for 20 min. After the addition of
Muse MitoPotential 7-AAD dye (propidium iodide) and incubation for
5 min, changes in ΔΨm and in cellular plasma membrane
permeabilization were assessed on the basis of the fluorescence
intensities of both the dyes, which were analyzed by flow cytometry
(Muse Cell Analyzer, Merck Millipore).
Immunoblotting
Whole cell protein extracts from SKOV3/DDP cells
were prepared with cell lysis buffer (50 mM Tris-HCl, pH 7.5, 150
mM NaCl, 1 mM Na2EDTA, 1 mM EDTA, 1% Triton, 2.5 mM
sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 mM NaF, 1 μg/ml leupeptin, and 1 mM
PMSF) for western blotting. The protein extracts were quantified
using a Bio-Rad kit (Pierce). Then, the protein extracts were
resolved by 10% SDS-PAGE and transferred to a PVDF membrane
(GS0914, Millipore). After being blocked in 5% non-fat milk in
buffer [10 mM Tris-HCl (pH 7.6), 100 mM NaCl and 0.1% Tween-20],
the membranes were probed with primary antibodies at 37°C for 2 h.
The membranes were then incubated with HRP-conjugated secondary
antibodies at room temperature for 1.5 h. Immunodetection was
performed using enhanced chemiluminescence reagents (Thermo
Scientific, Rockford, IL, USA), and images were captured using a
Syngene Bio Imaging System (Synoptics, Cambridge, UK). Specific
proteins were quantified by densitometry using Quantity One
software (Bio-Rad Laboratories), normalized to actin, and presented
as the mean ± SD of three independent experiments.
Calcium concentration analysis
The cytoplasmic Ca2+-sensitive
fluorescent dye Fluo-4/AM (Molecular Probes) and the mitochondrial
Ca2+-sensitive fluorescent dye Rhod-2/AM (AAT Bioquest,
Sunnyvale, CA, USA) were used to measure the Ca2+
concentration according to the manufacturer’s instructions. Before
exposure to different experimental conditions, the cells were
incubated with Fluo-4/AM or Rhod-2/AM for 30 min at 37°C. Cell
samples were then analyzed by confocal laser microscopy. All
experiments were performed in triplicate.
Immunofluorescence staining and confocal
laser microscopy
Cells were seeded onto coverslips in 24-well plates
at a density of 5×104 cells/well and allowed to recover
overnight. After treatment with the indicated drugs, cells were
washed three times with cold 0.1 M PBS and fixed in 4% (w/v)
paraformaldehyde/PBS for 20–30 min, stained with the nuclear stain
Hoechst 33342/H2O (2 μg/ml, Sigma) for 30 min, washed
with 0.01 M PBS, and examined using Olympus FV1000 confocal laser
microscopy to reveal chromatin condensation. The co-localization of
IP3R and VDAC1 and the expression of PDI were examined by the
indirect immunofluorescence method. Cells were cultured on
coverslips overnight, then treated with the indicated drugs, and
rinsed with 0.1 M PBS three times. After incubation, the cells were
fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1%
Triton X-100 (Sigma-Aldrich) for 5 min, washed three times with
0.01 M PBS, and then blocked for 30 min in 5% (w/v) non-immune
animal serum (goat) (Beyotime Biotechnology, Shanghai, China) PBS,
and incubated with primary antibody (IP3R, VDAC1, PDI) overnight at
4°C. The next day, the slides were incubated with the Alexa
Fluor-488/546-conjugated secondary antibody (1:400 dilution;
Invitrogen) for 1 h, then stained with Hoechst 33342 (2 μg/ml) for
2 min, and washed three times with PBS. After mounting, the cells
were examined by Olympus FV1000 confocal laser microscopy.
Subcellular fractionation
The purification of the cytoplasmic fraction, ER
fraction, pure mitochondrial fraction, crude mitochondrial fraction
and the MAM fraction were performed as previously described
(17). The above fractions were
lysed in RIPA buffer [1% sodium deoxycholate, 0.1% SDS, 1% Triton
X-100, 10 mM Tris (pH 8.0), 0.14 M NaCl] for immunoblotting with
antibodies against IP3R, VDAC1, Bcl-2, calreticulin, Cyto C, Mfn2,
Grp75 and tubulin. For co-immunoprecipitation analysis, the MAM
lysates were pre-cleared with protein G agarose beads (Proteintech
Group, Inc.) for 1 h at 4°C. Then, equal amounts of sample lysates
were incubated with either 2 μg of IgG or anti-IP3R antibody for 20
h at 4°C, and this was followed by precipitation with protein G
agarose beads. Immunoprecipitated proteins from MAM lysates and
total MAM lysates were subjected to immunoblot analysis with the
anti-IP3R antibody and anti-VDAC1 antibody. Total MAM lysates were
used as the input control. The results are representative of three
independent experiments.
Electron microscopy
Electron microscopy and morphometric analysis were
performed as described previously (26). Cells were fixed for 30 min with
ice-cold 2.5% glutaraldehyde in 0.1 M cacodylate buffer, embedded
in Epon, and processed for transmission electron microscopy by
standard procedures. Representative areas were chosen for
ultra-thin sectioning and examined on a transmission electron
microscope at a magnification of ×20,000.
Bcl-2 knockdown by small interfering
RNA
Small interfering RNA (siRNA) sequences targeting
human Bcl-2 and a non-target sequence were constructed by Genechem
(Shanghai, China). The Bcl-2 siRNA sequence was
CCG-CAT-TTA-ATT-CAT-GGT-ATT and that of the non-target siRNA
(Scramble) was TTC-TCC-GAA-CGT-GTC-ACG-T. Transfections with the
siRNAs were performed using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s protocol.
Briefly, cisplatin-resistant SKOV3/DDP cells were placed into
6-well plates and transfected the next day with 4 μg of Bcl-2 siRNA
or siScramble, using 10 μl of Lipofectamine 2000 (at 1 μg/μl).
Cells were harvested 2 days after transfection; whole cell lysates
were isolated for western blots. For MTT assays, transfected cells
were treated with cisplatin for 24 h, and then subjected to the MTT
assay to determine cell viability.
Human tumor xenografts
BALB/c nude mice (4–6 weeks old) were purchased from
Beijing Vital River Laboratory Animal Technology Co. Ltd., China.
Animals were maintained in specific pathogen-free conditions and a
controlled light and humidity environment. Animal experiments were
conducted in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory animals. SKOV3/DDP cells
(5×106) were subcutaneously injected into the right
flank of each mouse. Tumor volume (mm3) was measured
every 4 days using a Vernier caliper and calculated as 0.4 × (short
length)2 × long length. Treatment was initiated when
tumors reached a volume of 45–55 mm3 (day 16). The mice
were randomly divided into 4 groups (5 animals/group) and received
NS (IP, daily), 4 mg/kg cisplatin (IP, every other day), 50 mg/kg
ABT737 (IP, every other day), or a combination treatment for 20
days. The mice were sacrificed, and the experiment was terminated
at the end of 36 days. Tumors were isolated, weighed, and
imaged.
Immunohistochemistry
Immunohistochemical staining for cleaved caspase-3
was performed on 5-μm thick sections embedded in paraffin after
formalin fixation. The sections were de-paraffinized in xylene and
rehydrated in graded ethanol solutions (reducing concentration from
95 to 70%). The sections were incubated in
H2O2 solution (3% H2O2
in PBS buffer) for 30 min to block endogenous peroxidase activity.
Antigen retrieval was performed in retrieval buffer (pH 9.0, 20 mM
Tris, 0.05 mM EDTA, 0.05% Tween-20 buffer) in a 99°C bath for 20
min. The sections were subsequently incubated at 4°C overnight with
anti-cleaved caspase-3. After rinsing with PBS buffer, the
secondary antibody (MaxVision™ HRP-Polymer Anti-Rabbit IHC kit,
Maixin Bio, China) was applied for 15 min at RT. The DAB (Maixin
Bio, China) solution was used as the chromogen. Finally, the
sections were counterstained with hematoxylin (Sigma) to identify
the nuclei. The images were observed and analyzed using a
microscope (Leica DM 4000B) with Image-Pro Plus 6.0 software.
Terminal deoxynucleotidyl transferase
dUTP nick-end labeling (TUNEL)
Apoptosis analysis was performed using an In Situ
Cell Death Detection kit (Roche, Indianapolis, IN, USA) to identify
DNA breaks according to the manufacturer’s instructions. Sections
were incubated with TUNEL reaction mix containing 10 U of terminal
deoxyribosyltransferase, 10 mM dUTP biotin, and 2.5 mM cobalt
chloride in 1X terminal transferase reaction buffer for 1 h at 37°C
in a humidified atmosphere. The DAB (Maixin Bio, China) solution
was used as the chromogen. Finally, the sections were
counterstained with hematoxylin (Sigma) to identify the nuclei. The
images were observed and analyzed using a microscope (Leica DM
4000B) with Image-Pro Plus 6.0 software.
Statistical analyses
All values are presented as mean ± SEM. Statistical
analysis was performed using one-way analysis of variance (ANOVA)
followed by the Bonferroni multiple comparison test with the
software GraphPad Prism version 6.00 for Mac (GraphPad Software, La
Jolla, CA, USA). A value of P<0.05 was considered statistically
significant.
Results
ABT737 increases cisplatin-induced growth
inhibition and apoptosis in cisplatin-resistant ovarian cancer
cells
We treated cisplatin-resistant ovarian cancer cells
(SKOV3/DDP, COC1/DDP and A2780/DDP cells) with ABT737 in
combination with cisplatin to investigate whether ABT737 would
enhance the antitumor effects of cisplatin. On the basis of our
previous studies (unpublished data), the non-toxic dose of ABT737
in SKOV3/DDP, COC1/DDP and A2780/DDP cells for 24 h is 5 μM. Thus,
we treated SKOV3/DDP, COC1/DDP and A2780/DDP cells with 15 μg/ml
cisplatin and/or 5 μM ABT737 for 24 h. MTT assays indicated that
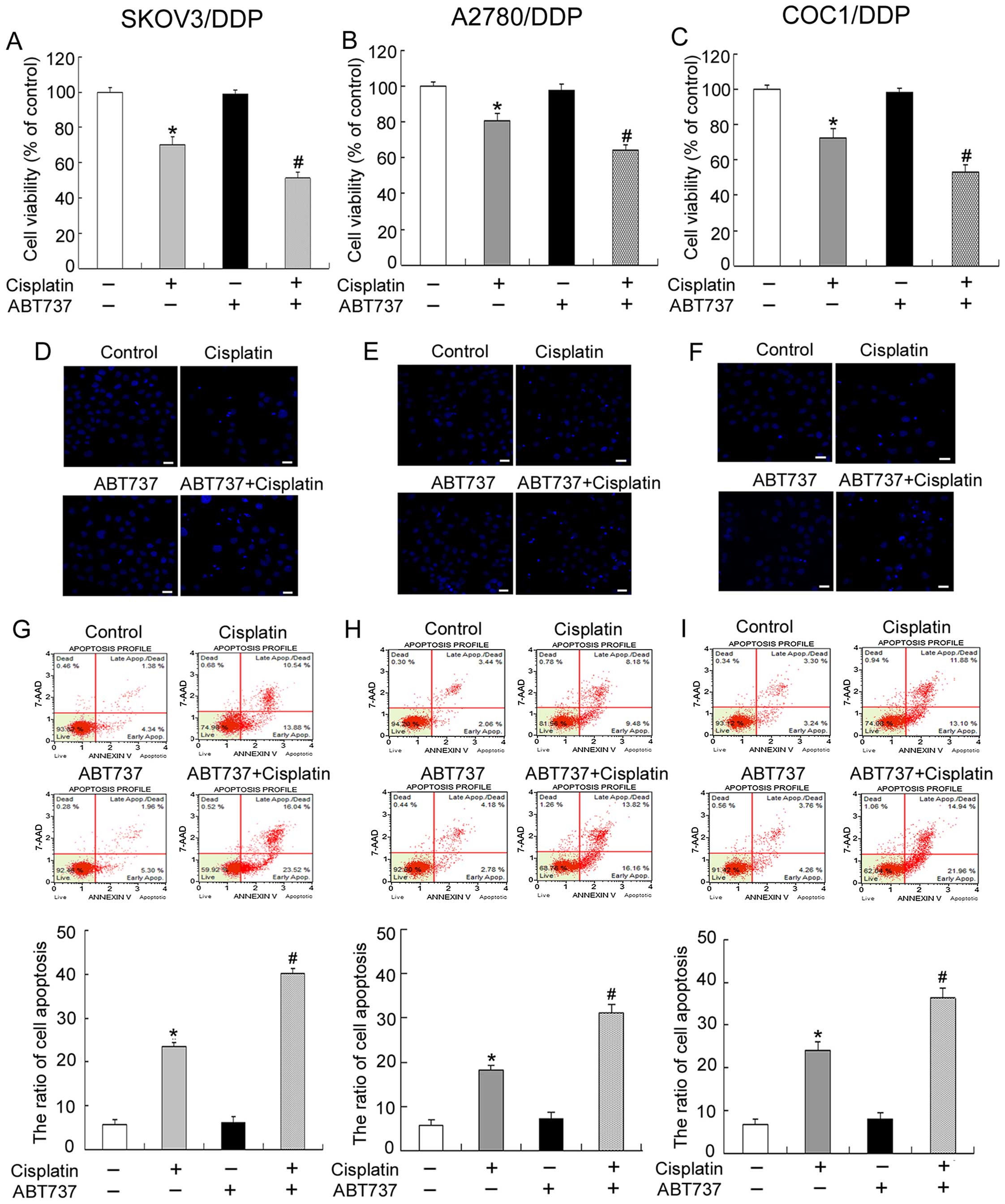
ABT737 increased cisplatin-induced growth inhibition (Fig. 1A–C).
On the basis of these results, we treated cells with
cisplatin and/or ABT737 for 24 h and then examined apoptotic
chromatin condensation with Hoechst 33342 staining. These results
showed that ABT737 increased cisplatin-induced apoptotic chromatin
condensation (Fig. 1D–F).
Additionally, flow cytometry analysis revealed that the apoptosis
rate was clearly higher in SKOV3/DDP, COC1/DDP and A2780/DDP cells
exposed to cisplatin and ABT737 for 24 h compared with cells
treated with cisplatin alone (Fig.
1G–I). These results indicate that ABT737 restores sensitivity
to cisplatin in cisplatin-resistant ovarian cancer cells.
ABT737 and cisplatin have a synergistic
effect in the treatment of human ovarian cancer xenograft mouse
models
After the in vitro experiments, in
vivo experiments were performed to determine whether ABT737
contributes to suppressing tumor growth in combination with
cisplatin. SKOV3/DDP cells were subcutaneously inoculated into
BALB/c nude mice to establish a subcutaneous transplant tumor
model. The tumor growth curve over time was recorded (Fig. 2A). As predicted, ABT737 plus
cisplatin caused marked tumor growth inhibition compared with
treatment with cisplatin-only (Fig.
2B). The final tumor weights were 1.01, 0.39, 0.97 and 0.19 g
for BALB/c nude mice injected i.p. with NS, cisplatin, ABT737 or a
combination treatment, respectively (Fig. 2C). Next, we used
immunohistochemical methods to detect the expression of the
apoptosis-related protein cleaved caspase-3 in mice exposed to
cisplatin and/or ABT737 for 20 days. As shown in Fig. 2D, ABT737 combined with cisplatin
treatment led to the upregulation of cleaved caspase-3 compared
with treatment with cisplatin only. Furthermore, TUNEL assays were
used to determine whether ABT737 had a synergistic effect with
cisplatin in inducing cell apoptosis in mouse models of ovarian
cancer. These results showed that the number of apoptotic cells in
the cisplatin plus ABT737 group was significantly greater than that
of the cisplatin group (Fig. 2E and
F). Together, these data suggest that ABT737 and cisplatin have
a synergistic effect in the treatment of human ovarian cancer
xenograft mouse models.
ABT737 enhances cisplatin-induced
mitochondrial apoptosis and ER-mediated apoptosis in SKOV3/DDP
cells
First, we investigated whether ABT737 would enhance
cisplatin-induced mitochondrial apoptosis. To determine the effect
of cisplatin and/or ABT737 on mitochondrial function, we measured
ΔΨm by using flow cytometry after 6 h. As shown in Fig. 3A, the combination treatment further
reduced the cisplatin-induced decrease of ΔΨm. Moreover, western
blot analysis showed that the combination treatment increased the
ratio of Bax/Bcl-2 and enhanced the expression of mitochondrial
apoptosis-related proteins (calpain-1, Cyto C, cleaved caspase-9
and cleaved caspase-3), as compared with the cisplatin treatment
(Fig. 3B and C).
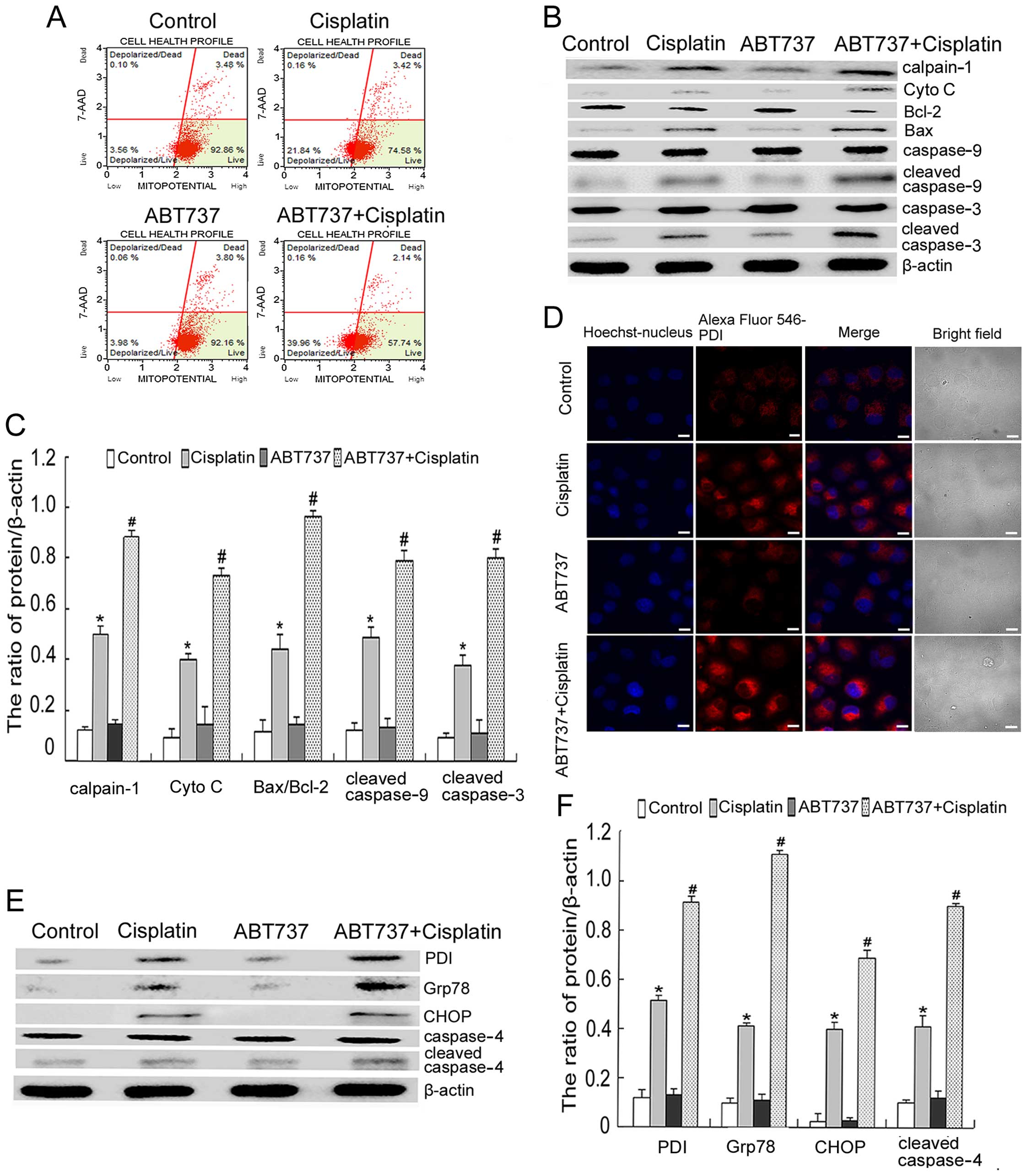 | Figure 3ABT737 enhances cisplatin-induced
mitochondrial apoptosis and ER-mediated apoptosis in SKOV3/DDP
cells. (A) SKOV3/DDP cells were treated with 15 μg/ml cisplatin
and/or 5 μM ABT737 for 6 h. ΔΨm was assessed by staining for
MitoPotential dye and 7-AAD, and analysed by Muse cell analyser. (B
and C) SKOV3/DDP cells were treated with 15 μg/ml cisplatin and/or
5 μM ABT737 for 24 h. Western blot analysis for the expression of
calpain-1, Cyto C, Bax, Bcl-2, cleaved caspase-9 and cleaved
caspase-3 after normalization to β-actin and quantification. Data
are presented as mean ± SD, n=3. *P<0.05 vs. control,
#P<0.05 vs. cisplatin. (D) SKOV3/DDP cells were
treated with 15 μg/ml cisplatin and/or 5 μM ABT737 for 24 h.
Distribution of PDI was observed by confocal microscopy (bar, 10
μm). (E and F) SKOV3/DDP cells were treated with 15 μg/ml cisplatin
and/or 5 μM ABT737 for 24 h. Western blot analysis for the
expression of PDI, Grp78, CHOP and cleaved caspase-4 after
normalization to β-actin and quantification. Data are presented as
mean ± SD, n=3. *P<0.05 vs. control,
#P<0.05 vs. cisplatin. |
A growing body of research suggests that cisplatin
triggers ER stress and induces ER stress-mediated apoptotic events
(2). Hence, we further
investigated whether ABT737 enhanced cisplatin-induced ER
stress-mediated apoptosis in SKOV3/DDP cells. Using confocal
microscopy, we found that ABT737 increased the expression of PDI
induced by cisplatin (Fig. 3D). We
assessed the expression of ER stress-related proteins (PDI, Grp78,
CHOP and cleaved caspase-4) by western blot analysis. These results
showed that the combination treatment enhanced the expression of
PDI, Grp78, CHOP and cleaved caspase-4, as compared with the
cisplatin treatment (Fig. 3E and
F). Thus, we demonstrated that the intrinsic mitochondrial
apoptotic pathway and ER-mediated apoptosis are actively involved
in the ABT737-mediated increase in the cytotoxic effects of
cisplatin on SKOV3/DDP cells.
ABT737 increases cisplatin-induced
Ca2+ transfer from the ER to the cytosol and
mitochondria
Ca2+ is not only a key regulator of cell
survival but also triggers cell apoptosis in response to various
physiological and pathological conditions. A growing body of
literature shows that cytoplasmic and mitochondrial Ca2+
overload can contribute to ER-mediated apoptosis and mitochondrial
apoptosis (22,27). As described in Introduction, the ER
is the most important Ca2+ storage compartment in
eukaryotic cells. Therefore, we determined whether mitochondrial
and cytoplasmic Ca2+ overload is a result of
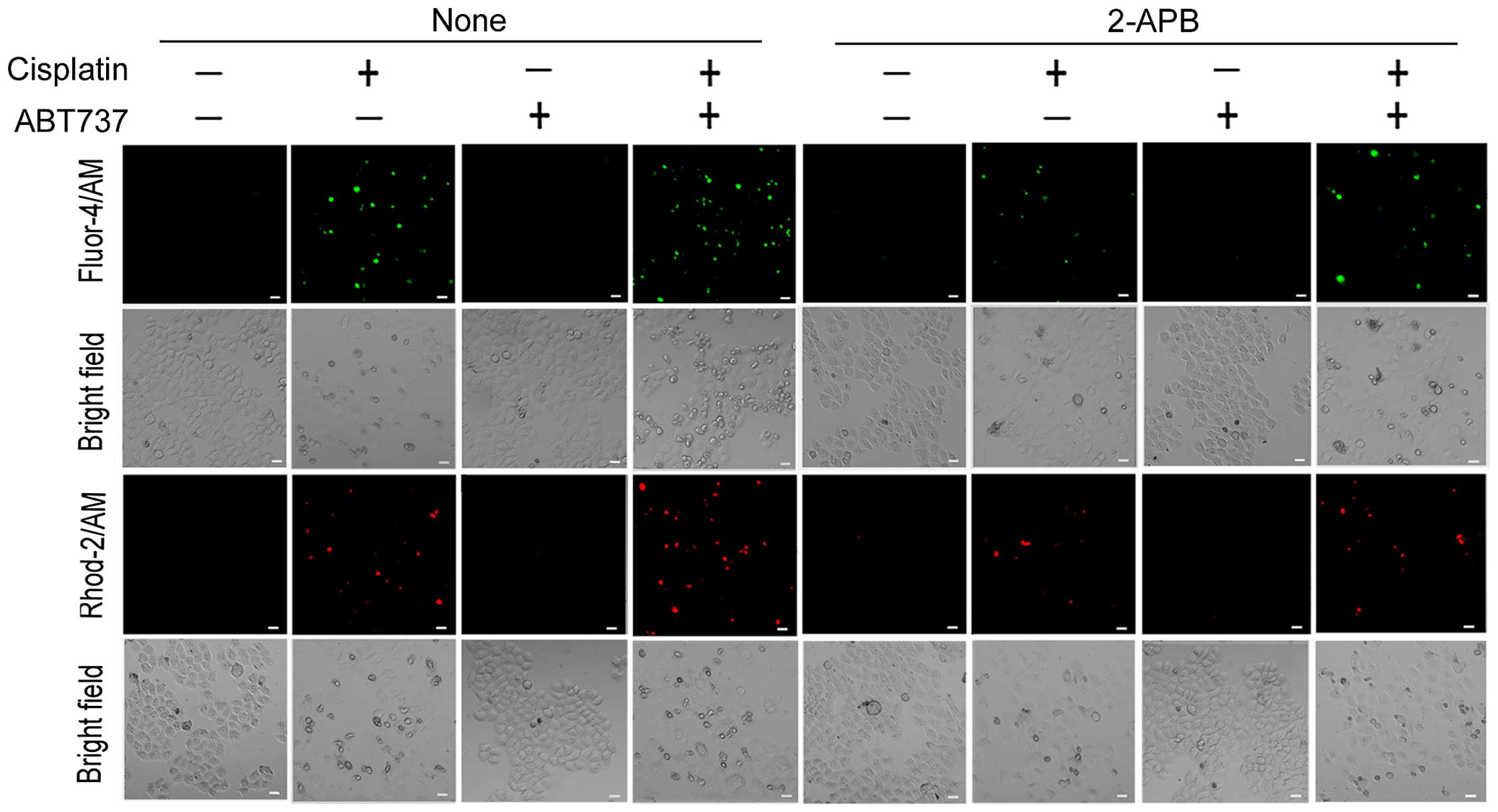
Ca2+ release from the ER. We used 2-APB, an IP3R
antagonist that inhibits ER Ca2+ efflux (27). The fluorescent dyes Fluo-4/AM and
Rhod-2/AM were used to detect cytoplasmic and mitochondrial
Ca2+ levels in SKOV3/DDP cells, respectively. As shown
in Fig. 4, the cytoplasmic and
mitochondrial Ca2+ elevation induced by cisplatin or/and
ABT737 in SKOV3/DDP cells was markedly reduced in the presence of
2-APB. These results strongly suggest that ABT737 increases
cisplatin-induced Ca2+ transfer from the ER to the
cytosol and mitochondria.
ABT737 enhances cisplatin-induced
ER-mitochondria contacts in SKOV3/DDP cells
We showed that ABT737 enhanced cisplatin-increased
mitochondrial Ca2+ levels in the previous experiments
(Fig. 4). Therefore, we next
explored whether the increased mitochondrial Ca2+ levels
were mediated by increasing the ER-mitochondria contact sites. IP3R
and VDAC1 localize to the mitochondria and ER, respectively
(13–15,28).
We used confocal microscopy to analyze the co-localization of IP3R
and VDAC1. The yellow areas represent co-localization between IP3R
and VDAC1, and these results demonstrated that ABT737 enhanced
cisplatin-induced ER-mitochondria contact sites (Fig. 5A).
 | Figure 5ABT737 enhances cisplatin-induced
ER-mitochondria contacts in SKOV3/DDP cells. (A) Colocalization of
IP3R and VDAC1 in SKOV3/DDP cells treated with 15 μg/ml cisplatin
and/or 5 μM ABT737 for 24 h (bar, 5 μm). (B) Protein components of
subcellular fractions prepared from SKOV3/DDP cells revealed by
immunoblot analysis. H, homogenate; Mc, crude mitochondrial
fraction; Mp, pure mitochondrial fraction; MAM,
mitochondria-associated membrane. C, cytosol. (C and E) Analysis of
Grp75 and Mfn2 at the MAM interface in SKOV3/DDP cells treated with
cisplatin and/or ABT737 for 24 h. Data are presented as mean ± SD,
n=3. *P<0.05 vs. control, #P<0.05 vs.
cisplatin. (D and F) MAM fraction from SKOV3/DDP cells was
subjected to immunoprecipitation with anti-IP3R antibody or normal
IgG and then immunoblotted with anti-VDAC1 antibody. Lysate
indicates the MAM fraction that was not subjected to
immunoprecipitation. Data are presented as mean ± SD, n=3.
*P<0.05 vs. control, #P<0.05 vs.
cisplatin. (G) Representative transmission electron microscopy
photomicrographs of SKOV3/DDP cells treated with cisplatin and/or
ABT737 for 24 h (bar, 200 nm). White arrowheads indicate
ER-mitochondria contacts. (H) Quantification of ER-mitochondria
contacts of electron microscopy images. Data are presented as mean
± SD, n=3. *P<0.05 vs. control, #P<0.05
vs. cisplatin. |
Next, we detected the changes in the ER-mitochondria
contact sites by western blotting and immunoprecipitation. Before
studying the changes in ER-mitochondria interactions, we assessed
the quality of each fraction by immunoblotting for subcellular
organelle marker proteins: IP3R and calreticulin for the ER and
MAM; VDAC1 for the mitochondria and MAM; Bcl-2 which enriches
mitochondria and ER and is also present in MAM; Cyto C for the
mitochondria; tubulin for the cytoplasm (17,24,28).
As expected, the data showed that we obtained highly pure
fractions, and there was no cross-contamination (Fig. 5B). By analyzing MAM composition, we
found that ABT737 enhanced the cisplatin-induced interactions of
VDAC1 with Grp75 and Mfn2 (Fig. 5C and
E), thus suggesting that ER-mitochondria contact sites induced
by cisplatin were enhanced in the combination treatment. Then, we
treated cells with cisplatin and/or ABT737 for 24 h. The MAM
fraction was immunoprecipitated using the anti-IP3R antibody
followed by western blotting using the anti-VDAC1 antibody. These
results showed that ABT737 enhanced the cisplatin-induced
IP3R-VDAC1 interactions (Fig. 5D and
F).
Finally, we used electron microscopy to examine the
ultrastructural changes in MAM in the cisplatin and/or ABT737 group
for 24 h. White arrowheads indicate the ER-mitochondria
interactions. The contact points between the two organelles were
significantly increased in the combination-treatment group compared
with the control group (Fig. 5G and
H). In summary, we concluded that ABT737 enhances
cisplatin-induced mitochondrial Ca2+ levels by
increasing ER-mitochondria contact sites.
Bcl-2 siRNA increases sensitivity to
cisplatin by increasing cytoplasmic and mitochondrial
Ca2+ levels
To better clarify the role of Bcl-2 in cisplatin
resistance in SKOV3/DDP cells, we depleted the endogenous Bcl-2
using a specific siRNA in SKOV3/DDP cells and observed that the
Bcl-2 protein expression was clearly downregulated (Fig. 6A). MTT assays showed that Bcl-2
siRNA combined with cisplatin for 24 h markedly decreased cell
viability compared with that in the cisplatin plus siScramble group
(Fig. 6B). Additionally, flow
cytometry analysis revealed that the apoptosis rate was
substantially higher in SKOV3/DDP cells exposed to cisplatin
combined with Bcl-2 siRNA for 24 h compared with cells treated with
cisplatin plus siScramble (Fig. 6C and
D). Consistently with the results in Fig. 4, Bcl-2 siRNA also enhanced the
cisplatin-induced cytoplasmic and mitochondrial Ca2+
elevations (Fig. 6E). Finally, we
used electron microscopy to examine the ultrastructural changes of
MAM after treatment with cisplatin and/or Bcl-2 siRNA. White
arrowheads indicate the ER-mitochondria interactions. The contact
points between the two organelles were significantly increased
after the cisplatin plus Bcl-2 siRNA treatment for 24 h compared
with the cisplatin plus siScramble treatment (Fig. 6F and G). Together, these findings
suggest that Bcl-2 siRNA improves the sensitivity of ovarian cancer
cells to cisplatin by increasing cytoplasmic and mitochondrial
Ca2+ levels.
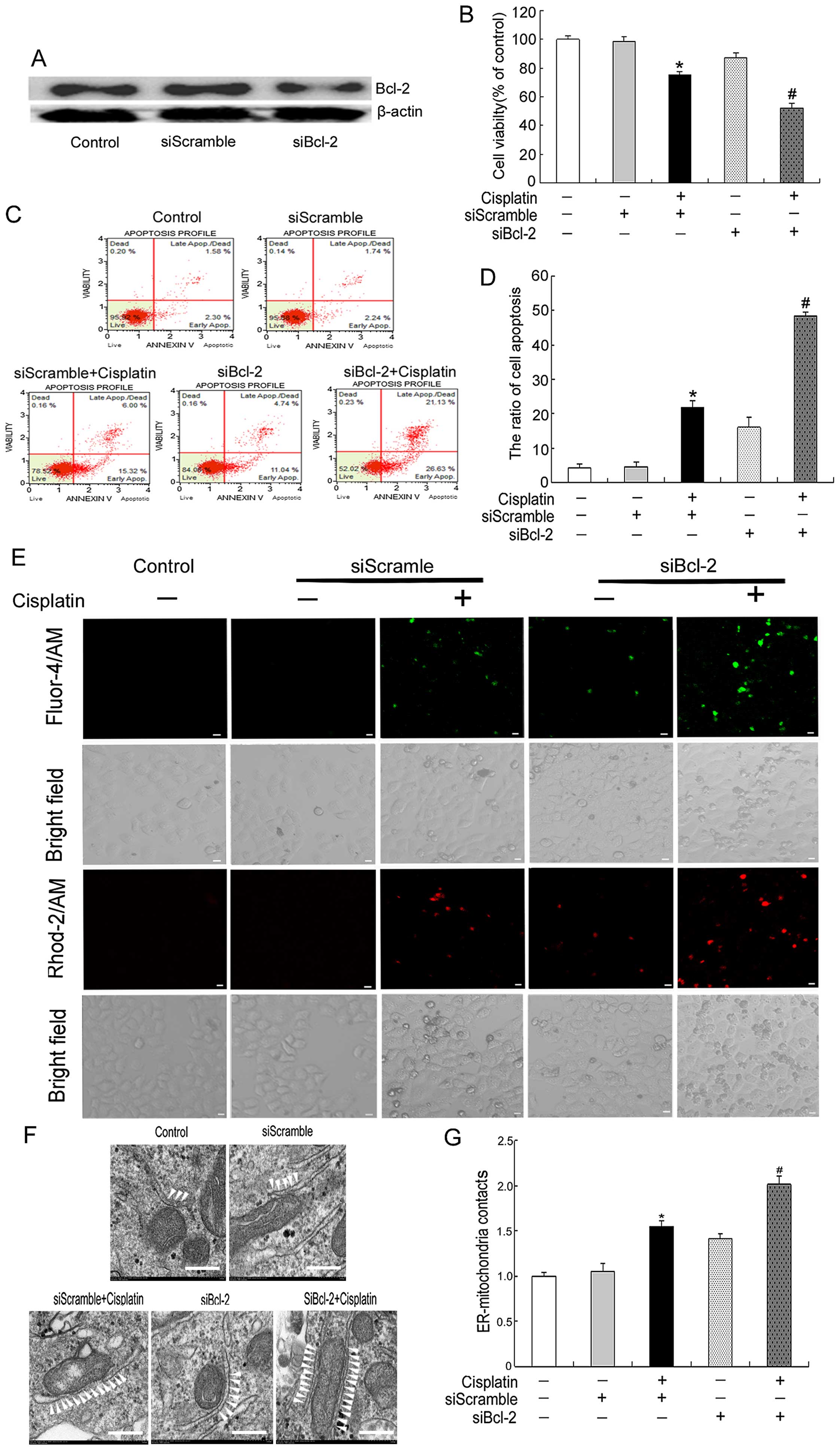 | Figure 6Bcl-2 siRNA increases sensitivity to
cisplatin by increasing cytoplasmic and mitochondrial
Ca2+ levels. SiBcl-2 increases sensitivity to cisplatin
by decreasing Bcl-2 expression at ER and mitochondria. (A) Western
blotting analysed the knockdown efficiency of Bcl-2, siScramle was
used as negative control. (B) Cell viability was determined by MTT
assay. Data are presented as mean ± SD, n=3. *P<0.05
vs. control, #P<0.05 vs. cisplatin plus siScramle
group. (C) Apoptosis was assessed by staining for Annexin V and
7-AAD, and analysed by Muse cell analyser. (D) The quantification
of apoptosis in SKOV3/DDP cells exposed to different treatment for
24 h. Data are presented as mean ± SD, n=3. *P<0.05
vs. control, #P<0.05 vs. cisplatin plus siScramle
group. (E) Confocal microscopy was used to detect cytoplasmic and
mitochondrial Ca2+ in cells exposed to different
treatment for 24 h (scale bar, 10 μm). (F) Representative
transmission electron microscopy photomicrographs of SKOV3/DDP
cells exposed to different treatment for 24 h (bar, 200 nm). White
arrowheads indicate ER-mitochondria contacts. (G) Quantification of
ER-mitochondria contacts of electron microscopy images. Data are
presented as mean ± SD, n= 3. *P<0.05 vs. control,
#P<0.05 vs. cisplatin plus siScramle group. |
Discussion
Bcl-2 overexpression is recognized as one of the key
mechanisms underlying tumor cell apoptosis escape and intrinsic or
acquired cisplatin resistance (20,29,30).
Hence, Bcl-2 has attracted considerable attention as a possible
target for the treatment of cancer, including ovarian cancer.
ABT-737, a small molecule compound, inhibits Bcl-2 by mimicking the
function of the BH3-only protein (31–33).
Using a xenograft model of hepatoblastoma (HB), Lieber et al
have reported that ABT737 restores the cisplatin sensitivity of
cisplatin-resistant HB cells and significantly decreases tumor
growth, as compared with that observed after cisplatin monotherapy
(34). Additionally, our previous
results have shown that ABT737 enhances cisplatin-induced Bcl-2
downregulation and cisplatin-induced apoptosis through the
regulation of mitochondrial dynamics in cholangiocarcinoma cells
(25). In this study, ABT737
enhanced cisplatin-induced apoptotic chromatin condensation and
cell apoptosis in cisplatin-resistant ovarian cancer cells
(Fig. 1). To evaluate the effect
of cisplatin combined with ABT737 on antitumor activity in
vivo, a BALB/c nude mouse subcutaneous transplant tumor model
was established using SKOV3/DDP cells. The results indicated that
cisplatin combined with ABT737 had an additive inhibitory effect on
tumorigenesis (Fig. 2).
An increasing number of studies have found that
cisplatin is actively involved in mitochondria-mediated and
ER-mediated apoptosis (35,36).
Interestingly, Bcl-2 is predominantly located in the mitochondria
and ER (30). Therefore, this
finding suggests that ABT737 may increase the cytotoxic effects of
cisplatin via the above two pathways. In this study, we found that
ABT737 enhanced cisplatin-induced ΔΨm collapse and Cyto C release
and increased the cisplatin-induced elevated Bax/Bcl-2 ratio and
calpain-1, cleaved caspase-9, cleaved caspase-3 expression.
Additionally, ABT737 increased Grp78, CHOP and cleaved caspase-4
induced by cisplatin (Fig. 3). All
of our results demonstrate that ABT737 improved the response of
SKOV3/DDP cells to cisplatin by mitochondria-mediated and
ER-mediated apoptosis.
Ca2+, a ubiquitous intracellular
messenger, is a key determinant of cell function and survival.
Altered Ca2+ homeostasis has an adverse effect on
proliferating cells (37,38). A membrane-permeant inhibitor of
IP3R, 2-APB, specifically inhibits IP3R-mediated Ca2+
release (39,40). Using 2-APB, Splettstoesser et
al revealed that cisplatin-induced ER Ca2+ release
results in programmed cell death due to activation of calpain
(41). Calpain-1, a
Ca2+-dependent cysteine endopeptidase, can be activated
by a small increase in cytoplasmic Ca2+ levels (10,42).
Studies by Zheng et al and Altznauer et al and have
shown that Ca2+-induced calpain-1 triggers
mitochondria-mediated and ER-mediated apoptosis (10,43).
Recently, we showed that cisplatin increases cytosolic and
mitochondrial Ca2+ levels, activates calpain-1, and
eventually leads to mitochondria- and ER-mediated apoptosis
(27). However, the effects of
cisplatin plus ABT737 on intracellular Ca2+ signals have
not been reported in the literature. In this study, to investigate
the effect of cisplatin plus ABT737 on cytosolic and mitochondrial
Ca2+ levels, we used the fluorescent dyes Fluo-4/AM and
Rhod-2/AM to detect cytoplasmic and mitochondrial Ca2+
levels in SKOV3/DDP cells. Our experimental results showed that
ABT737 increased cytoplasmic and mitochondrial Ca2+
levels induced by cisplatin and enhanced cisplatin-mediated
calpain-1 activation, eventually leading to apoptosis (Figs. 3B and 4). These results indicate that
cytoplasmic and mitochondrial Ca2+ overload increased
SKOV3/DDP cell sensitivity to cisplatin-induced cytotoxicity, thus
possibly providing a new approach for treating cisplatin-resistant
ovarian cancer.
The signaling mechanisms linking MAM and cancer cell
apoptosis have attracted substantial interest (44–47)
The phosphofurin acidic cluster sorting protein-2 (PACS-2), a
cytosolic sorting protein, is required for the integrity of MAM
(48). Using siRNA depletion of
PACS-2, Simmen et al have demonstrated that low expression
of PACS-2 blocks the apoptosis of STS-mediated human melanoma A7
cells, possibly through decreased transfer of Ca2+ from
the ER to the mitochondria (48).
Similarly, using fluorescence labeling of PDI (the marker protein
of ER) and VDAC1 (the marker protein of mitochondria), our previous
studies showned that cisplatin increases ER-mitochondria contacts,
and this is followed by highly efficient transportation of
Ca2+ from the ER to the mitochondria, ultimately
resulting in mitochondrial Ca2+ overload and apoptosis
in SKOV3 cells (12). Next, we
determined whether the increase in mitochondrial Ca2+
levels induced by cisplatin and ABT737 is achieved through the
increase in ER-mitochondrial cross-talk in SKOV3/DDP cells. In our
study, through the fluorescence labeling of IP3R and VDAC1, we
showed that ABT737 increased cisplatin-induced ER-mitochondria
contacts. We analyzed MAM composition and found that ABT737
increased cisplatin-induced Grp75 and Mfn2 expression at the MAM
interface. These results indicate that ABT737 increases
cisplatin-induced ER-mitochondria interactions, a finding
consistent with the results of electron microscopy (Fig. 5). All of these results suggest that
enhanced ER-mitochondria coupling leads to mitochondrial
Ca2+ overload and subsequent apoptosis by favoring
Ca2+ transfer from the ER to the mitochondria.
Interestingly, it has been reported that ER close to the
mitochondria may sense local cytoplasmic Ca2+ increases,
and the mitochondrial Ca2+ increase is closely coupled
to the increase in cytoplasmic Ca2+ levels (49). Cytoplasmic
Ca2+-dependent calpain-1 activation is an important
element of mitochondria-mediated and ER-mediated apoptosis. We
suggest that increased ER-mitochondria contact sites allow for
calpain-1-mediated apoptotic effects. However, further
investigations are required to test this possibility.
To further explore whether the downregulation of
Bcl-2 can increase the cytotoxic effects of cisplatin, we inhibited
Bcl-2 expression by using siRNA transfection in SKOV3/DDP cells.
Our study revealed that Bcl-2 siRNA enhanced the inhibitory effect
of cisplatin on SKOV3/DDP cell proliferation and also enhanced
cisplatin-induced apoptosis (Fig.
6B–D). Moreover, Bcl-2 knockdown significantly enhanced
cisplatin-induced cytoplasmic and mitochondrial Ca2+
elevation and cisplatin-induced ER-mitochondria cross-talk in
SKOV3/DDP cells (Fig. 6E–G). These
results further indicated that increasing MAM formation plays a
fundamental role in cell apoptosis by driving higher
Ca2+ accumulation in the mitochondria.
In conclusion, in a departure from conventional
therapeutic strategies for overcoming cisplatin resistance, we
explored an alternative therapeutic strategy linking cisplatin
resistance to Bcl-2-regulated Ca2+ signals in ovarian
cancer cells. The pharmacological inhibition of Bcl-2 or Bcl-2
siRNA reversed the cisplatin resistance of SKOV3/DDP cells by
increasing cisplatin-induced cytoplasmic and mitochondrial
Ca2+ levels. MAM not only promotes higher calcium
transfer from the ER to the mitochondria, thus leading to
mitochondrial Ca2+ overload, but also may allow for
calpain-1-mediated apoptosis. Our experimental results elucidate
the antitumor mechanism of Bcl-2 via MAM, suggesting a possibility
for individualized treatment of ovarian cancer patients.
Acknowledgements
This study was supported by the National Nature and
Science Foundation of China (NSFC 81372793, 81472419, 81541148 and
81202552) and the Department of Education of Jilin Province Project
(no. 2016237). We thank Nature Publishing Group for editing the
English in this manuscript.
References
|
1
|
Ma L, Xu Y, Su J, Yu H, Kang J, Li H, Li
X, Xie Q, Yu C, Sun L, et al: Autophagic flux promotes cisplatin
resistance in human ovarian carcinoma cells through ATP-mediated
lysosomal function. Int J Oncol. 47:1890–1900. 2015.PubMed/NCBI
|
|
2
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2012. View Article : Google Scholar
|
|
3
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Vliet AR, Verfaillie T and Agostinis
P: New functions of mitochondria associated membranes in cellular
signaling. Biochim Biophys Acta. 1843:2253–2262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farooqi AA, Li KT, Fayyaz S, Chang YT,
Ismail M, Liaw CC, Yuan SS, Tang JY and Chang HW: Anticancer drugs
for the modulation of endoplasmic reticulum stress and oxidative
stress. Tumour Biol. 36:5743–5752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krebs J, Agellon LB and Michalak M: Ca(2+)
homeostasis and endoplasmic reticulum (ER) stress: An integrated
view of calcium signaling. Biochem Biophys Res Commun. 460:114–121.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moretti D, Del Bello B, Allavena G and
Maellaro E: Calpains and cancer: Friends or enemies? Arch Biochem
Biophys. 564:26–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang Y, Li X, Wang Y, Wang H, Huang C and
Li J: Endoplasmic reticulum stress-induced hepatic stellate cell
apoptosis through calcium-mediated JNK/P38 MAPK and
Calpain/Caspase-12 pathways. Mol Cell Biochem. 394:1–12. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martinez JA, Zhang Z, Svetlov SI, Hayes
RL, Wang KK and Larner SF: Calpain and caspase processing of
caspase-12 contribute to the ER stress-induced cell death pathway
in differentiated PC12 cells. Apoptosis. 15:1480–1493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng D, Wang G, Li S, Fan GC and Peng T:
Calpain-1 induces endoplasmic reticulum stress in promoting
cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim
Biophys Acta. 1852:882–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fonteriz RI, de la Fuente S, Moreno A,
Lobatón CD, Montero M and Alvarez J: Monitoring mitochondrial
[Ca(2+)] dynamics with rhod-2, ratiometric pericam and aequorin.
Cell Calcium. 48:61–69. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Wang C, Su J, Xie Q, Ma L, Zeng L,
Yu Y, Liu S, Li S, Li Z, et al: Tolerance to endoplasmic reticulum
stress mediates cisplatin resistance in human ovarian cancer cells
by maintaining endoplasmic reticulum and mitochondrial homeostasis.
Oncol Rep. 34:3051–3060. 2015.PubMed/NCBI
|
|
13
|
Anelli T, Bergamelli L, Margittai E,
Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R
and Sitia R: Ero1α regulates Ca(2+) fluxes at the endoplasmic
reticulum-mitochondria interface (MAM). Antioxid Redox Signal.
16:1077–1087. 2012. View Article : Google Scholar
|
|
14
|
Hayashi T, Rizzuto R, Hajnoczky G and Su
TP: MAM: More than just a housekeeper. Trends Cell Biol. 19:81–88.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ouyang YB and Giffard RG: ER-mitochondria
crosstalk during cerebral ischemia: Molecular chaperones and
ER-mitochondrial calcium transfer. Int J Cell Biol.
2012:4939342012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rowland AA and Voeltz GK: Endoplasmic
reticulum-mitochondria contacts: Function of the junction. Nat Rev
Mol Cell Biol. 13:607–625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wieckowski MR, Giorgi C, Lebiedzinska M,
Duszynski J and Pinton P: Isolation of mitochondria-associated
membranes and mitochondria from animal tissues and cells. Nat
Protoc. 4:1582–1590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paillard M, Tubbs E, Thiebaut PA, Gomez L,
Fauconnier J, Da Silva CC, Teixeira G, Mewton N, Belaidi E, Durand
A, et al: Depressing mitochondria-reticulum interactions protects
cardiomyocytes from lethal hypoxia-reoxygenation injury.
Circulation. 128:1555–1565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo X, Chen KH, Guo Y, Liao H, Tang J and
Xiao RP: Mitofusin 2 triggers vascular smooth muscle cell apoptosis
via mitochondrial death pathway. Circ Res. 101:1113–1122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leisching G, Loos B, Botha M and
Engelbrecht AM: Bcl-2 confers survival in cisplatin treated
cervical cancer cells: Circumventing cisplatin dose-dependent
toxicity and resistance. J Transl Med. 13:3282015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu N, Xu Y, Sun JT, Su J, Xiang XY, Yi
HW, Zhang ZC and Sun LK: The BH3 mimetic S1 induces endoplasmic
reticulum stress-associated apoptosis in cisplatin-resistant human
ovarian cancer cells although it activates autophagy. Oncol Rep.
30:2677–2684. 2013.PubMed/NCBI
|
|
22
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and BAK
regulation of endoplasmic reticulum Ca2+: A control
point for apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giorgi C, Missiroli S, Patergnani S,
Duszynski J, Wieckowski MR and Pinton P: Mitochondria-associated
membranes: Composition, molecular mechanisms, and
physiopathological implications. Antioxid Redox Signal.
22:995–1019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meunier J and Hayashi T: Sigma-1 receptors
regulate Bcl-2 expression by reactive oxygen species-dependent
transcriptional regulation of nuclear factor kappaB. J Pharmacol
Exp Ther. 332:388–397. 2010. View Article : Google Scholar :
|
|
25
|
Fan Z, Yu H, Cui N, Kong X, Liu X, Chang
Y, Wu Y, Sun L and Wang G: ABT737 enhances cholangiocarcinoma
sensitivity to cisplatin through regulation of mitochondrial
dynamics. Exp Cell Res. 335:68–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo
J and Hu X: Shikonin circumvents cancer drug resistance by
induction of a necroptotic death. Mol Cancer Ther. 6:1641–1649.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen L, Wen N, Xia M, Zhang YU, Liu W, Xu
YE and Sun L: Calcium efflux from the endoplasmic reticulum
regulates cisplatin-induced apoptosis in human cervical cancer HeLa
cells. Oncol Lett. 11:2411–2419. 2016.PubMed/NCBI
|
|
28
|
Lee GH, Lee HY, Li B, Kim HR and Chae HJ:
Bax inhibitor-1-mediated inhibition of mitochondrial
Ca2+ intake regulates mitochondrial permeability
transition pore opening and cell death. Sci Rep. 4:51942014.
|
|
29
|
Tóthová E, Fricova M, Stecová N, Kafková A
and Elbertová A: High expression of Bcl-2 protein in acute myeloid
leukemia cells is associated with poor response to chemotherapy.
Neoplasma. 49:141–144. 2002.PubMed/NCBI
|
|
30
|
Akl H, Vervloessem T, Kiviluoto S,
Bittremieux M, Parys JB, De Smedt H and Bultynck G: A dual role for
the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus
endoplasmic reticulum. Biochim Biophys Acta. 1843:2240–2252. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Zhong X, Zhang X, Shang D, Zhou
YI and Zhang C: Enhanced anticancer effect of ABT-737 in
combination with naringenin on gastric cancer cells. Exp Ther Med.
11:669–673. 2016.PubMed/NCBI
|
|
32
|
Antignani A, Sarnovsky R and FitzGerald
DJ: ABT-737 promotes the dislocation of ER luminal proteins to the
cytosol, including pseudomonas exotoxin. Mol Cancer Ther.
13:1655–1663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gardner EE, Connis N, Poirier JT, Cope L,
Dobromilskaya I, Gallia GL, Rudin CM and Hann CL: Rapamycin rescues
ABT-737 efficacy in small cell lung cancer. Cancer Res.
74:2846–2856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lieber J, Dewerth A, Wenz J, Kirchner B,
Eicher C, Warmann SW, Fuchs J and Armeanu-Ebinger S: Increased
efficacy of CDDP in a xenograft model of hepatoblastoma using the
apoptosis sensitizer ABT-737. Oncol Rep. 29:646–652. 2013.
|
|
35
|
Gatti L, Cassinelli G, Zaffaroni N, Lanzi
C and Perego P: New mechanisms for old drugs: Insights into
DNA-unrelated effects of platinum compounds and drug resistance
determinants. Drug Resist Updat. 20:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cullen KJ, Yang Z, Schumaker L and Guo Z:
Mitochondria as a critical target of the chemotheraputic agent
cisplatin in head and neck cancer. J Bioenerg Biomembr. 39:43–50.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Greenberg B, Butler J, Felker GM,
Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B,
Lyon AR, et al: Calcium upregulation by percutaneous administration
of gene therapy in patients with cardiac disease (CUPID 2): A
randomised, multinational, double-blind, placebo-controlled, phase
2b trial. Lancet. 387:1178–1186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mattson MP and Chan SL: Calcium
orchestrates apoptosis. Nat Cell Biol. 5:1041–1043. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Amcheslavsky A, Safrina O and Cahalan MD:
State-dependent block of Orai3 TM1 and TM3 cysteine mutants:
Insights into 2-APB activation. J Gen Physiol. 143:621–631. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee HC, Yoon SY, Lykke-Hartmann K, Fissore
RA and Carvacho I: TRPV3 channels mediate Ca2+ influx
induced by 2-APB in mouse eggs. Cell Calcium. 59:21–31. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Splettstoesser F, Florea AM and Büsselberg
D: IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced
Ca2+-influx in HeLa-S3 cells and prevents activation of
calpain and induction of apoptosis. Br J Pharmacol. 151:1176–1186.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Villalpando Rodriguez GE and Torriglia A:
Calpain 1 induce lysosomal permeabilization by cleavage of
lysosomal associated membrane protein 2. Biochim Biophys Acta.
1833:2244–2253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Altznauer F, Conus S, Cavalli A, Folkers G
and Simon HU: Calpain-1 regulates Bax and subsequent Smac-dependent
caspase-3 activation in neutrophil apoptosis. J Biol Chem.
279:5947–5957. 2004. View Article : Google Scholar
|
|
44
|
Giorgi C, De Stefani D, Bononi A, Rizzuto
R and Pinton P: Structural and functional link between the
mitochondrial network and the endoplasmic reticulum. Int J Biochem
Cell Biol. 41:1817–1827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marchi S, Patergnani S and Pinton P: The
endoplasmic reticulum-mitochondria connection: One touch, multiple
functions. Biochim Biophys Acta. 1837:461–469. 2014. View Article : Google Scholar
|
|
46
|
López-Crisosto C, Bravo-Sagua R,
Rodriguez-Peña M, Mera C, Castro PF, Quest AF, Rothermel BA,
Cifuentes M and Lavandero S: ER-to-mitochondria miscommunication
and metabolic diseases. Biochim Biophys Acta. 1852A:2096–2105.
2015. View Article : Google Scholar
|
|
47
|
Raturi A and Simmen T: Where the
endoplasmic reticulum and the mitochondrion tie the knot: The
mitochondria-associated membrane (MAM). Biochim Biophys Acta.
1833:213–224. 2013. View Article : Google Scholar
|
|
48
|
Simmen T, Aslan JE, Blagoveshchenskaya AD,
Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM and
Thomas G: PACS-2 controls endoplasmic reticulum-mitochondria
communication and Bid-mediated apoptosis. EMBO J. 24:717–729. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hajnóczky G, Davies E and Madesh M:
Calcium signaling and apoptosis. Biochem Biophys Res Commun.
304:445–454. 2003. View Article : Google Scholar : PubMed/NCBI
|