Introduction
As one of the most lethal and aggressive cancers,
pancreatic ductal adenocarcinoma (PDAC) is associated with a very
poor prognosis (1). As such, it
remains a clinically challenging disease to treat with a 5-year
survival rate of 5%, despite that continual improvements have been
made in therapies in recent decades (1). PDAC has several defining features
that influence its aggressive biology and resistance to multiple
therapeutic modalities, which is characterized by a distinct and
exuberant stromal reaction (desmoplasia), together with the poor
vascularization, creating a highly hypoxic and nutrient-poor
microenvironment, accompanied with genomic complexity and an
altered metabolism (2,3). Moreover, the aforementioned tumor
biology of PDAC contributes to uncontrolled cancer growth, early
metastasis and recurrence, resistance to chemotherapy and
radiotherapy, and finally short survival (1,3).
Metabolic alterations have been regarded as a
hallmark of cancer cells, which is an important research area that
has gained increasing attention in recent years (4). Unlike majority of normal cells which
depend on mitochondrial oxidative phosphorylation to provide
energy, cancer cells apply aerobic glycolysis as their primary
energy resource (4). Several
molecules, including Oncogenic K-Ras, hypoxia-inducible factor 1α
and c-Myc, have been suggested to play important roles in promoting
glycolysis and thus be involved in 'metabolic reprogramming' in the
cancer cells (5–8). Oncogenic K-Ras mutation, which occurs
in typical cases, is the signature event in PDAC, serving a
critical role in tumor initiation (5). There are considerable evidences
showing that K-Ras also plays vital roles in metabolic changes in
PDAC cancer cells and promotes cancer development by inducing
mitochondrial dysfunction (5). The
HIF1α pathway enables tumor cells to survive by changing glucose
metabolism toward a glycolytic phenotype, regulating pH balance and
proliferation rate, and eventually angiogenesis (6). c-Myc induces aerobic glycolysis by
enhancing glucose uptake and lactate production as well as
providing glycolytic intermediates for nucleotide, amino acid, and
lipid biosynthesis (7,8). In this regard, an understanding of
cellular metabolic derangements in PDAC could lead to novel
therapeutic approaches.
N-myc downstream regulated gene 1 (NDRG1) (also
known as Drg1, RTP, Rit42, PROXY-1 or Cap43), a member of the NDRG
family, has been well described as a tumor/metastasis suppressor in
a number of cancers (9), including
pancreas (10). It is believed
that NDRG1 participates in suppression of tumourigenesis or
carcinogenesis (10). However, it
is unclear whether NDRG1 is involved in cancer metabolism changes,
which is a critical aspect during carcinogenesis and metastasis.
Previously, we demonstrated NDRG1 exerts inhibition of colonic and
prostate cancer cell migration by blocking Src activation (11). Of note, studies revealed that Src
regulate breast cancer cell glucose metabolism by reducing c-Myc
translation and transcription of the glucose transporter GLUT1
(12). Therefore, it is reasonable
to speculate NDRG1 may play a role in cancer cell metabolism
regulation. In this study, we examined whether NDRG1 expression
could play any role in PDAC cancer cell metabolism. Moreover, we
also investigated the potential target molecules of NDRG1 involved
in the regulation of PDAC cancer cell metabolism.
Materials and methods
Cells and reagents
The human pancreatic cancer cell lines MIA PaCa-2
and PANC-1 cells were obtained from ATCC and cultured in DMEM
supplemented with 10% FBS. All of the cell culture media contained
100 U/ml penicillin and 100 mg/ml streptomycin. iKras cell lines
expressing K-RasG12D upon doxycycline (Dox) treatment
was generously provided by Professor Paul J. Chiao from MD Anderson
Cancer Center. Doxycycline and MAPK inhibitor U0126 were obtained
from Selleck and used at a final concentration of 2 µg/ml
and 10 µM, respectively.
Plasmid and stable cell line
generation
In order to overexpress NDRG1 in MIA PaCa-2 and
PANC-1 cells, we generated NDRG1 overexpression constructs.
pCDH-CMV-MCS-EF1-Puro (System Biosciences) plamid was used to
generate NDRG1 expression constructs. NDRG1 lentiviral expression
construct was co-transfected with psPAX2 and pMD2.G into HEK293T
cells to produce lentiviral particles. Stable cell lines were
obtained by infection with lentiviral particles and followed by
puromycin selection.
siRNA transfection
In order to silence ERK1 expression in pancreatic
cancer cells, we silenced ERK1 expression by using siRNA mediated
silencing. siRNA targets against ERK1 were CCUGCUGGACCGGAUGUUA and
UCAUGGAGA CAGACCUGTA, respectively.
Cell proliferation assay and colony
formation assay
Cell proliferation was examined using water-soluble
tetrazolium salt assay using the Cell Counting Kit-8. In short,
cells (1.5×103/well) were seeded in 96-well culture
plates in triplicate and incubated for 4 days at 37°C/5%
CO2 in a humidified incubator. Viable cells were
quantified at each 24 h interval by measuring OD450
using microplate reader.
Cells were seeded in triplicate in 6-well
plates at an initial density of 500 cells/well
After 10–14 days, colonies were clearly visible, and
the cells were fixed with 4% paraformaldehyde for 15 min at room
temperature and stained with 4 mg/ml of crystal violet. The
colonies containing more than 50 cells were counted using light
microscopy. The average number of colonies was determined from
three independent experiments.
Western blotting
Western blotting was carried out as previously
described (13). Briefly,
whole-cell protein lysates were extracted, separated by SDS-PAGE
and followed by immunoblotting. Antibodies used were purchased from
designated manufacturers, NDRG1 (Abcam, ab133572), GLUT1
(Proteintech, 21829-1-AP), HK2 (Proteintech, 22029-1-AP), PDK1
(Proteintech, 10026-1-AP), LDHA (Proteintech, 19987-1-AP), HIF1α
(Proteintech, 20960-1-AP), K-Ras (Proteintech, 12063-1-AP), ERK1
(Abcam, ab119933), p44/p42 MAPK (ERK1/2) Rabbit mAb (Cell Signaling
Technology, 4695), Phspho-p44/p42 MAPK(ERK1/2) (Thr202/Tyr204)
(Cell Signaling Technology, 4370), Akt1 (Proteintech, 10176-2-AP),
Anti-Akt1 (Phospho S473) (Abcam, ab81283), Anti-S6K (Abcam,
ab32529), phosphor-p70S6 kinase (Thr389) (Cell Signaling
Technology, 9234) and β-actin (Proteintech, 60008-1-Ig).
Oxygen consumption rate and extracellular
acidification rate
Cellular mitochondrial function was measured using
the Seahorse XF Cell Mito Stress test kit and the Bioscience XF96
Extracellular Flux Analyzer, according to the manufacturer's
instructions. The glycolytic capacity was determined using the
Glycolysis Stress test kit as per the manufacturer's instructions.
Briefly, 4×104 cells were seeded onto 96-well plates and
incubated overnight. After washing the cells with Seahorse buffer
(DMEM with phenol red containing 25 mmol/l glucose, 2 mmol/l sodium
pyruvate, and 2 mmol/l glutamine), 175 ml of Seahorse buffer plus
25 ml each of 1 mmol/l oligomycin, 1 mmol/l FCCP, and 1 mmol/l
rotenone were automatically injected to measure the oxygen
consumption rate (OCR). Then, 25 ml each of 10 mmol/l glucose, 1
mmol/l oligomycin, and 100 mmol/l 2-deoxy-glucose were added to
measure the extracellular acidification rate (ECAR). The OCR and
ECAR values were calculated after normalization to the cell number
and are plotted as the mean ± SD.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen). cDNA was synthesized by reverse transcription using a
Takara PrimeScript RT reagent kit. The expression status of
candidate genes and β-actin were determined by quantitative
real-time PCR using an ABI 7900HT Real-time PCR system (Applied
Biosystems). The reactions were run in triplicate. Primer sequences
are listed in Table I.
 | Table IPrimers used in RT-PCR analysis. |
Table I
Primers used in RT-PCR analysis.
| Gene | Primer
sequence |
|---|
| GLUT1 |
5′-CTTTGTGGCCTTCTTTGAAGT-3′ |
|
5′-CCACACAGTTGCTCCACAT-3′ |
| HK2 |
5′-GATTGTCCGTAACATTCTCATCGA-3′ |
|
5′-TGTCTTGAGCCGCTCTGAGAT-3′ |
| LDHA |
5′-TGGAGATTCCAGTGTGCCTGTATGG-3′ |
|
5′-CACCTCATAAGCACTCTCAACCACC-3′ |
| PDK1 |
5′-CTGCACCAAGACCTCGTGTTGAGA-3′ |
|
5′-GGCAGCTTTGTTATACACTGGGAG-3′ |
| β-actin |
5′-CTACGTCGCCCTGGACTTCGAGC-3′ |
|
5′-GATGGAGCCGCCGATCCACACGG-3′ |
Hypoxia response element (HRE) activity
reporter assay
HRE activity was measured using a Dual-Glo
Luciferase Assay System according to the manufacturer's protocol
(Promega, E2920). HRE-Luciferase reported construct containing
three hypoxia response elements from the Pgk-1 gene was obtained
from Addgene (Addgene Plasmid 26731) (14).
Tissue specimens
The clinical tissue samples used in this study were
obtained from patients diagnosed with pancreatic cancer at Fudan
University Shanghai Cancer Center from 2010 to 2015. Prior patient
consent and approval from the institutional research ethics
committee were obtained.
Statistical analysis
Experiments were repeated at least three times. All
data are presented as the mean ± SD. Two-tailed unpaired Student's
t-tests and one-way analysis of variance were used to evaluate the
data. SPSS version 16.0 software (IBM) was used for the data
analysis. Differences were considered significant at P<0.05.
Results
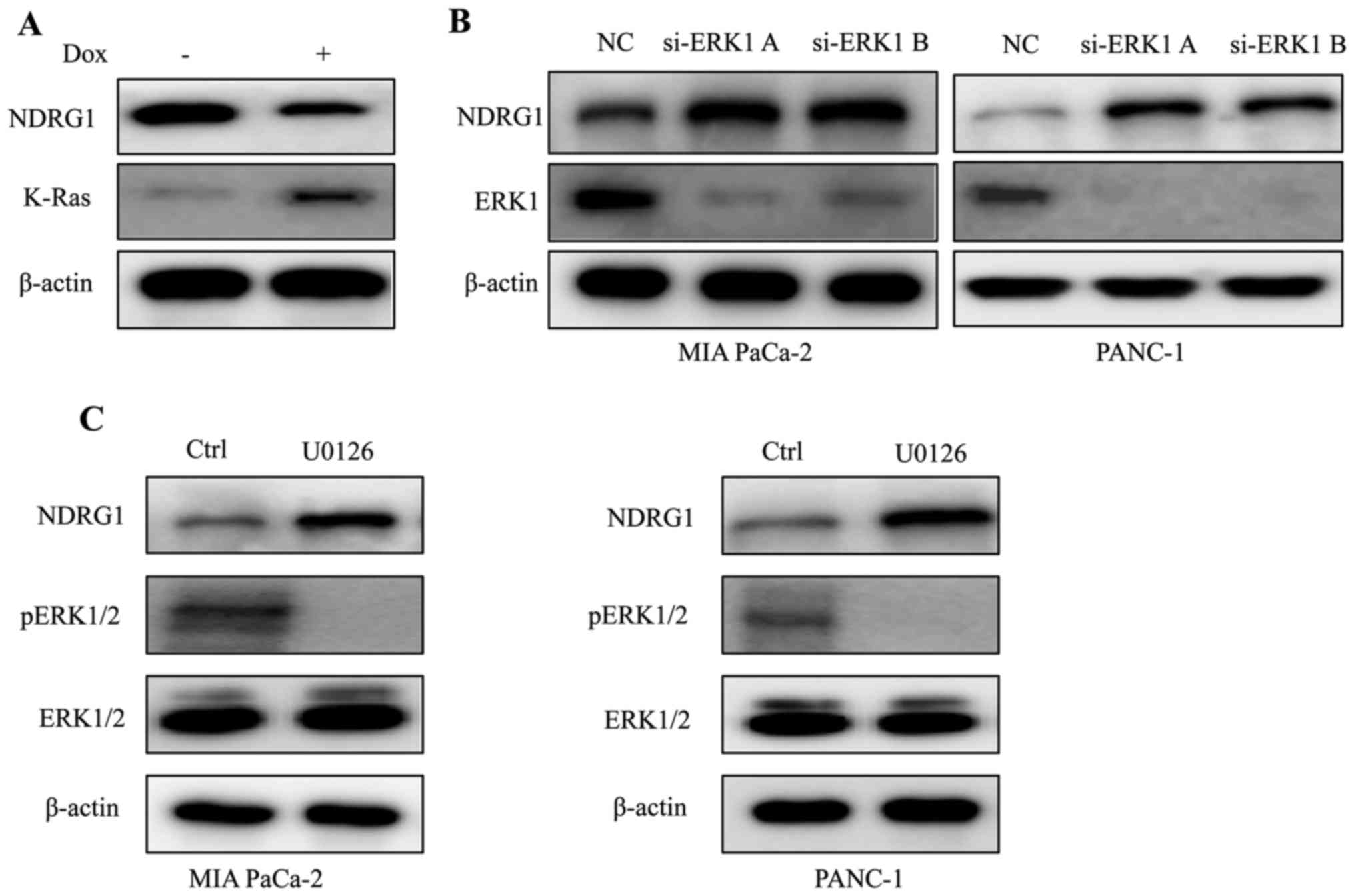
K-Ras downregulates NDRG1 expression via
ERK1 in PDAC
Considering that K-Ras mutation is the signature of
typical PDAC, we conducted experiments to elucidate the expression
of NDRG1 in K-Ras network. By inducing oncogenic K-Ras expression
in iKras cells, we observed that upon the presence of mutant K-Ras
expression, the protein level of NDRG1 decreased significantly,
indicating that NDRG1 might be a downstream effector of oncogenic
K-Ras (Fig. 1A). It is well
studied that oncogenic K-Ras mutation resulted in constitutive
activation of MAPK, therefore, we examined the effect of MAPK on
NDRG1 expression. We silenced ERK1 (MAPK3) expression in K-Ras
mutated MIA PaCa-2 and PANC-1 cells, and observed that protein
level of NDRG1 increased when ERK1 was silenced, indicating that
NDRG1 may serve as a downstream target of Ras/MAPK signaling
pathway (Fig. 1B). Next, we
treated MIA PaCa-2 and PANC-1 cells with MAPK inhibitor UO126 and
observed that NDRG1 protein level increased in UO126 treated cancer
cells (Fig. 1C). Taken together,
these results suggest that NDRG1 might be a downstream target of
oncogenic K-Ras/MAPK axis.
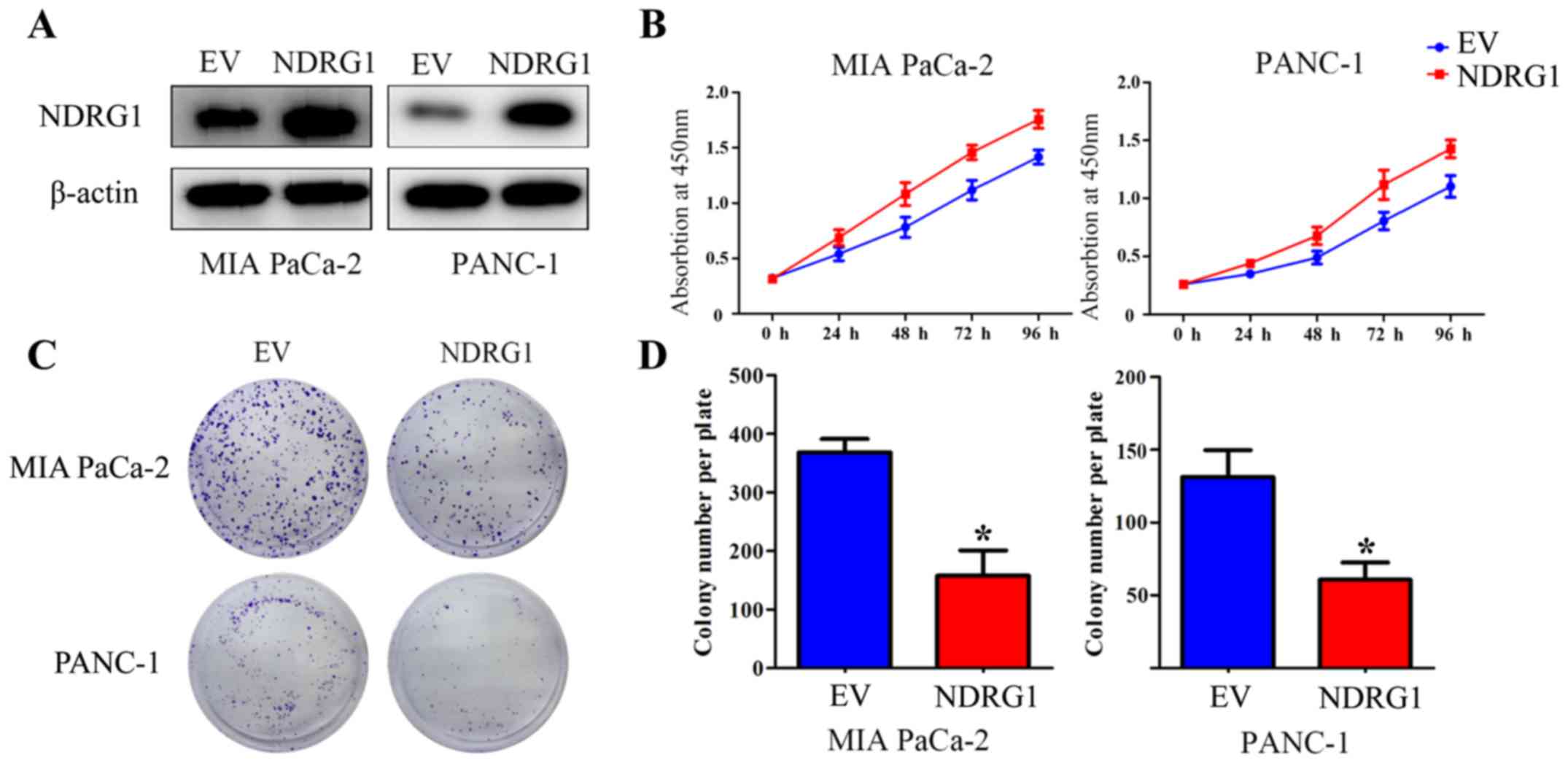
Elevated NDRG1 expression suppresses
proliferation of pancreatic cancer cells in vitro
To verify the suppressive function of NDRG1 in terms
of inhibiting proliferation in pancreatic cancer, two pancreatic
cancer cell lines, MIA PaCa-2 and PANC-1, were utilized and
infected with NDRG1 overexpressing lentiviral particles. The
transfected MIA PaCa-2 and PANC-1 cells demonstrated a significant
increase in NDRG1 expression relative to their
empty-vector-infected control cells (Fig. 2A). CCK-8 assay was then performed
to determine the growth repression role of the potent metastasis
suppressor NDRG1. As indicated, the growth of MIA PaCa-2 was
remarkably reduced with NDRG1 overexpression, relative to MIA
PaCa-2 empty-vector control cells. Similar result was observed in
the other pancreatic cancer cell line PANC-1 (Fig. 2B). To further assess the effects of
NDRG1, the role of NDRG1 expression on cell proliferation was
further investigated by a subsequent colony formation assay, in
which MIA PaCa-2 and PANC-1 cancer cell colonies were markedly
(P<0.05) decreased upon NDRG1 expression, relative to their
empty-vector-transfected control cells (Fig. 2C and D). These data confirmed that
NDRG1 inhibits proliferation and colony formation of pancreatic
cancer cells in vitro, which is in good agreement with
previous study (10).
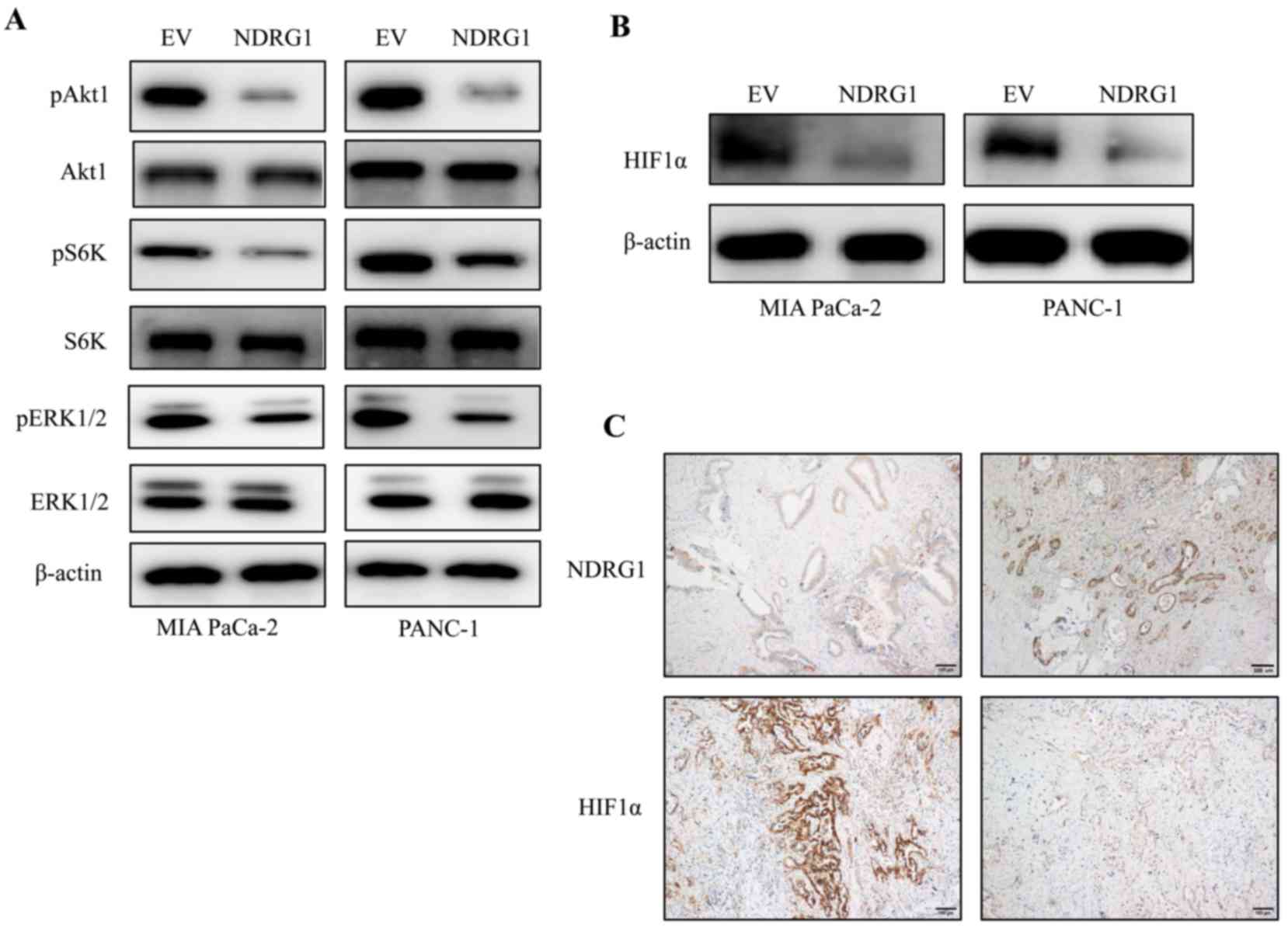
NDRG1 decreases HIF1α expression through
MAPK and Akt/mTOR signalling
The ERK1 (MAPK3) and Akt/mammalian target of
rapamycin (mTOR) signaling pathway are directly activated by Ras
activation and could be the major mediators of Ras-driven
oncogenesis (15). Regarding the
interesting phenomenon that K-Ras has an inhibitory effect on NDRG1
protein in PDAC cancer cells (Fig.
1), while NDRG1 abrogate cancer cell growth of PDAC cancer
cells (Fig. 2), we decided to
decipher whether the K-Ras downstream signaling was changed upon
NDRG1 overexpression. Herein, we performed western blotting to
observe the phosphorylated and total protein level of Akt1, the
direct mTOR target p70/p85 S6 kinase (S6K) and ERK1. Noteworthy, in
both cancer cell lines, elevated NDRG1 led to marked decrease of
phosphorylated Akt1, S6K and ERK1 protein level relative to control
cells, while there was no change on total Akt1, S6K and ERK1
protein level, indicating NDRG1 has inhibitory effect on MAPK and
Akt/mTOR signaling (Fig. 3A).
Akt/mTOR signaling has been shown to stimulate
translation of HIF1α mRNA and elevate HIF1α protein level (16), which has been implicated in
regulating many of the genes, contributing to 'Warburg effect'
providing cancer cells with a growth advantage (4,17).
These results above implied that HIF1α was a potential target of
NDRG1 in terms of inhibiting cancer cell growth. Therefore, we
conducted additional studies to investigate alterations on HIF1α in
the two pancreatic cancer cell lines and human PDAC specimens.
Importantly, HIF1α was reduced significantly by NDRG1 in the two
cancer cell lines relative to control cells (Fig. 3B). These data suggested that HIF1α
could be a downstream target of NDRG1 in current circumstance and
inhibited by NDRG1 in an Akt/mTOR signaling-mediated manner.
Considering the marked effect of NDRG1 on HIF1α
expression in pancreatic cancer cells, further investigations were
conducted to examine this effect in human PDAC specimens by
immunohistochemistry (Fig. 3C).
Similarly, there was a negative correlation between NDRG1 and HIF1α
expression in PDAC, which is consistent with our in vitro
data using MIA PaCa-2 and PANC-1 cells demonstrating the inhibitory
effect of NDRG1 on HIF1α expression in PDAC (Fig. 3C).
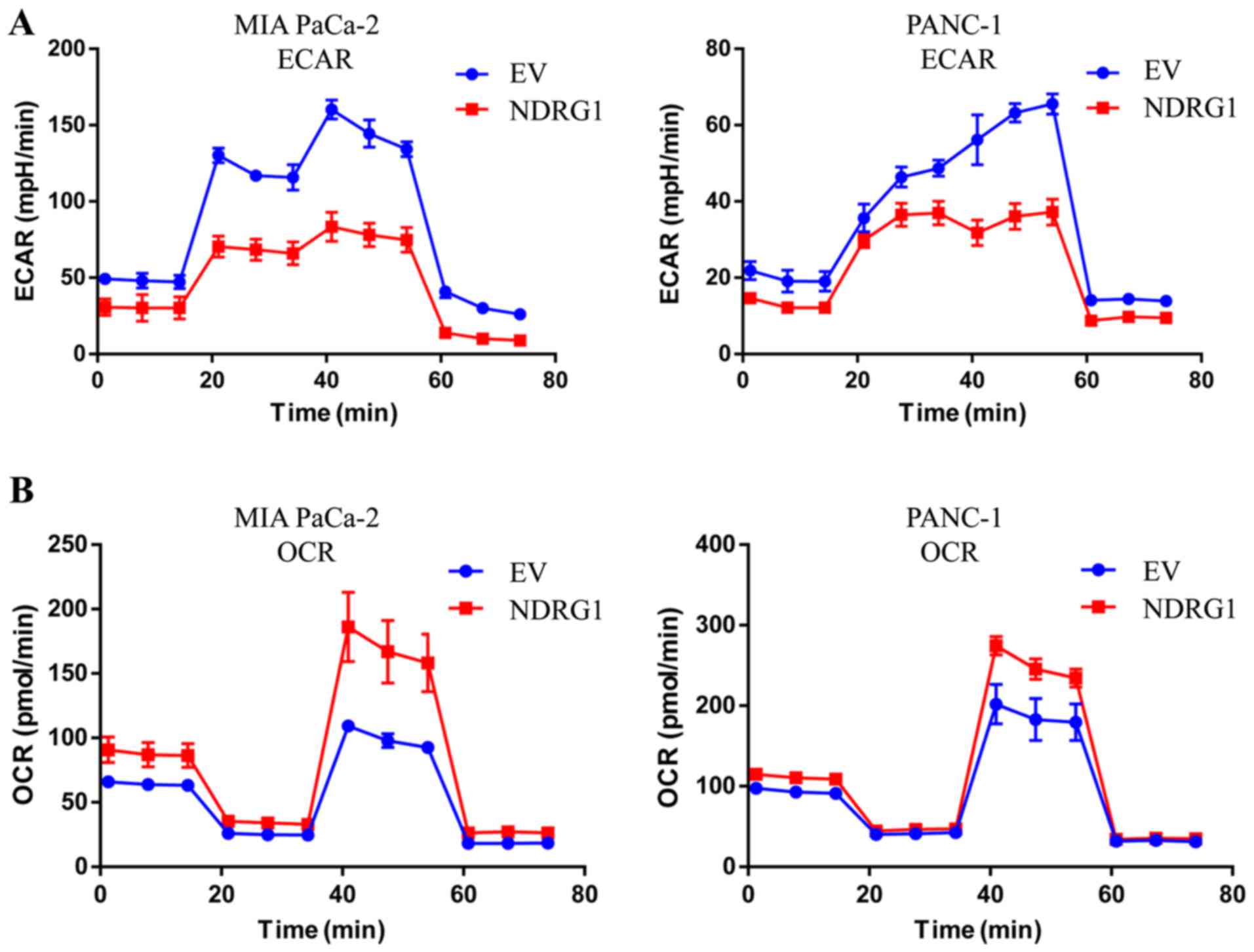
NDRG1 inhibits glucose metabolism and
promotes mitochondrial respiration in pancreatic cancer cells
The ECAR is an important measurement of glucose
metabolism and reflects the lactic acid-induced acidification of
the medium surrounding cancer cells. Considering the inhibitory
effect of NDRG1 on pancreatic cancer cell growth, further studies
were conducted to examine whether NDRG1 could modulate glycolysis
metabolism of these pancreatic cancer cells, which control the
energy feed for cell growth. Noteworthy, in both MIA PaCa-2 and
PANC-1 cells NDRG1 expression decreased the ECAR as expected
(Fig. 4A), especially from 20 min
to 60 min, which imply that it plays a suppressive role in lactic
acid formed during glycolysis. Cellular oxygen consumption reflects
mitochondrial respiration and can be measured by the OCR. To
further investigate this novel finding, the above studies were
complemented by OCR experiment to assess the effect of NDRG1 on the
'metabolism switch'. Conversely, OCR experiment demonstrated that
when MIA PaCa-2 and PANC-1 cells overexpress NDRG1, they exhibited
higher OCRs comparing to control cells (Fig. 4B), indicating that NDRG1 is a
positive regulator of basal mitochondrial respiration in pancreatic
cancer cells.
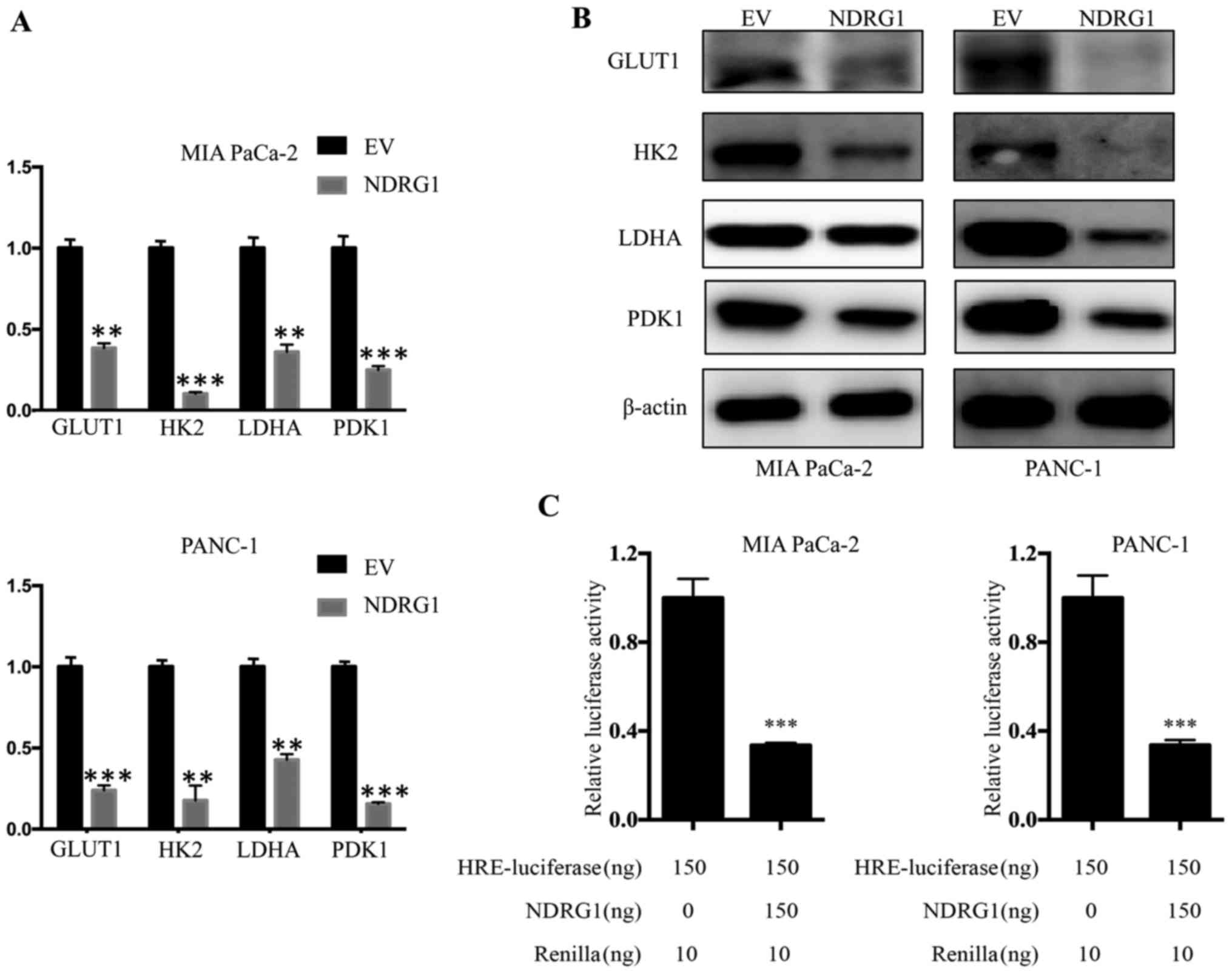
NDRG1 downregulates transcription and
expression of glucose metabolism related enzymes
The studies above indicated that NDRG1 suppress
pancreatic cancer cells metabolic level of glucose, while enhanced
the mitochondrial function. To further elucidate the underlying
mechanism of NDRG1 function, we conducted qRT-PCR to examine
whether several key enzymes of glucose metabolism, including GLUT1,
HK2, LDHA and PDK1, were affected by NDRG1 at mRNA level. For both
MIA PaCa-2 and PANC-1 cells, we observed consisted results that the
mRNA level of GLUT1, HK2, LDHA and PDK1 was significantly decreased
upon NDRG1 expression, relative to the vector control (P<0.05)
(Fig. 5A). Considering our data
above showing that NDRG1 significantly decreased GLUT1, HK2, LDHA
and PDK1 mRNA in the two cell lines, further studies were performed
to examine whether this effect was occurring at the protein level.
Therefore, we applied immunoblotting to verify protein levels of
GLUT1, HK2, LDHA and PDK1 in response to NDRG1 in both cell-types.
Similarly, in comparison to the control cells, there was a marked
reduction of GLUT1, HK2, LDHA and PDK1 protein in the presence of
NDRG1 overexpression in both cell lines (Fig. 5B). In summary, these results
suggest that the effect of NDRG1 on these key enzymes is occurring
at both transcription and post-transcription levels.
To decipher the inhibitory effect of NDRG1 on
metabolism of pancreatic cancer cells which typically grow within
hypoxic conditions, we further performed investigation using HRE
reporter assay to assess the role of NDRG1 on HIF1α activity. Both
cell lines were transfected with the reporter plasmid. For MIA
PaCa-2 cells, NDRG1 overexpression led to significant decrease of
HIF1α transcriptional activity relative to control cells
(P<0.05), as reflected by HRE-luciferase reporter assay. A
similar effect was also observed for PANC-1 cells (Fig. 5C). These results showed that NDRG1
prevented activity of HIF1α which resulted in the decrease of the
four glycolysis enzymes. Collectively, NDRG1 overexpression
remarkably reduced mRNA and protein level of GLUT1, HK2, LDHA and
PDK1, which is achieved by blocking activity of HIF1α.
Discussion
A series of evidenc has supported the role of NDRG1
in inhibiting cancer growth, development and metastasis (11,18–21).
However, complete understanding of its mechanism of action remains
an important research goal. This current investigation demonstrates
that mutant/activate K-Ras reduces NDRG1 protein via ERK-mediated
signaling (Fig. 1). Moreover, we
discovered a unique role of NDRG1 in modulating PDAC cancer cell
glycolysis, resulting in inhibited cell growth (Fig. 2), which is achieved through
NDRG1-induced downregulation of HIF1α protein levels and activity
(Figs. 3B and 5C). This effect is further confirmed by
immunohistochemical analysis using human PDAC specimens (Fig. 3C). Furthermore, many glycolysis
related-enzymes are found to be reduced at mRNA and protein levels
upon NDRG1 overexpression (Fig. 5A and
B) and, hence, ECAR as well as OCR is concurrently changed
(Fig. 4A and B). Significantly,
this is the first time that this potent metastasis suppressor has
been shown to take part in metabolic regulation of PDAC glycolysis
(Fig. 6).
An emerging theme in cancer biology is that many of
the oncogenic mutations which result in tumorigenesis drive the
altered metabolism of cancer cells (2,5,7).
Cancer cells are addicted to use glycolysis other than
mitochondrial oxidative phosphorylation for glucose-dependent ATP
production, no matter whether ample oxygen is provided or not, to
fuel mitochondrial respiration, a phenomenon known as the 'Warburg
effect' (4). Not surprisingly,
given its central role in the pathogenesis of PDAC, oncogenic K-Ras
has been recently shown to have a key role in multiple aspects of
PDAC metabolism (22,23). Of note, it seems that a number of
well-known tumor suppressors play critical roles in modulating the
'metabolic transformation' to weaken the 'Warburg effect' (24). It is believed that oncogene and
tumor suppressor networks influence the metabolic shift in cancer.
Hence, the potent tumor/metastasis suppressor NDRG1, which had been
demonstrated to negatively affect cancer growth of PDAC (10), was investigated here in K-Ras
network to understand the precise role in PDAC. Our data showed
that mutant/activated K-Ras decreased NDRG1 protein level (Fig. 1A and B). However, when ERK1, the
effector downstream of K-Ras, was abrogated by siRNA or specific
inhibitor U0126, the inhibitory effect of K-Ras on NDRG1 was
reversed indicating that K-Ras inhibits NDRG1 through ERK-mediated
signaling.
The K-Ras oncogene has been shown to promote
glycolysis so as to drive uncontrolled proliferation and enhance
survival of cancer cells (5). For
instance, the glucose transporter GLUT1 is transcriptionally
upregulated in response to K-Ras mutations, leading to elevated
glucose uptake and glycolysis (5,25).
In addition to the above mentioned case, glutamine consumption,
anomalous pentose phosphate pathway (PPP) and autophagy is also
enhanced downstream of K-Ras in different and complementary ways
(5,26), resulting in 'metabolic addiction'
of pancreatic tumor cells. Cell proliferation assay and colony
formation assay results suggested that cell growth of pancreatic
cancer cells was significantly suppressed by NDRG1 (Fig. 2), which is in good agreement with
previous studies (10,27). The ERK1 and Akt/mTOR signaling
pathway are direct downstream effectors of Ras, contributing to
Ras-driven oncogenesis (15).
However, NDRG1 abrogate cancer cell growth of PDAC cancer cells
(10,27). Noteworthy, our results demonstrated
overexpression of NDRG1 led to significant reduction of
phosphorylated Akt1, S6K and ERK1 protein, but had no change on
total Akt1, S6K and ERK1 protein level. These data prove that NDRG1
has inhibitory effect on MAPK and Akt/mTOR signaling in K-Ras
network, and the interplay of NDRG1 and ERK1 is
context-dependent.
As mentioned above, tissue within PDAC is highly
hypoxic. Not surprisingly, this hypoxic microenvironment leads to
its complex biology and in particular its cellular metabolism
(28). HIF1α is an important
regulator of cellular oxygen homeostasis, and is overexpressed in
pancreatic cancer and associated with poor prognosis (6). Moreover, it is suggested that HIF1α
promotes PDAC progression by up regulating VEGF, which is a key
inducer of angiogenesis (29).
Intriguingly, NDRG1 expression is associated with a higher
differentiation status of the cells and correlated with a better
prognosis as well as longer survival in patients with pancreatic
cancer (10). NDRG1 was
demonstrated to affect the angiogenic on or off switch of cancer
stroma in PDAC through attenuation of NF-κB signaling pathway
(27,30). Importantly, Akt/mTOR signaling has
been shown to stimulate translation of HIF1α mRNA and elevate HIF1α
protein level (16). The present
study showed that NDRG1 overexpression decreased both protein level
and activity of HIF1α (Fig. 3B).
Immunohistochemical analysis of PDAC specimens further provided
supports that high NDRG1 was correlated with reduced HIF1α staining
(Fig. 3C). Collectively, these
studies suggest that abundant NDRG1 protein acts as suppressor of
HIF1α in PDAC. However, more investigations are needed to elucidate
the underlying mechanisms.
One of the consequences of elevated HIF1α is the
metabolic rewiring allowing a cell to subsist in low-oxygen
environment. The transcription factor HIF1α has been implicated in
regulating many of the genes that are responsible for the metabolic
difference. Most part of glycolysis related enzymes are directly or
indirectly regulated by HIF1α, in the way that HIF1α coordinately
modulate the entire process (31).
In PDAC, HIF1α has been shown to mediate metabolic changes
downstream of MUC1 in vitro and in vivo (32). HIF1α stimulates glycolytic energy
production by trans-activating genes involved in GLUT1 (33), inducing LDHA and HK2 expression to
catalyze lactate conversion and phosphorylate glucose (34,35).
Using qRT-PCR and western, we found that NDRG1 reduced GLUT1, HK2,
LDHA and PDK1 mRNA and protein level in both cell lines (Fig. 5A and B), which is accompanied by
HIF1α transcriptional activity reduction (Fig. 5C).
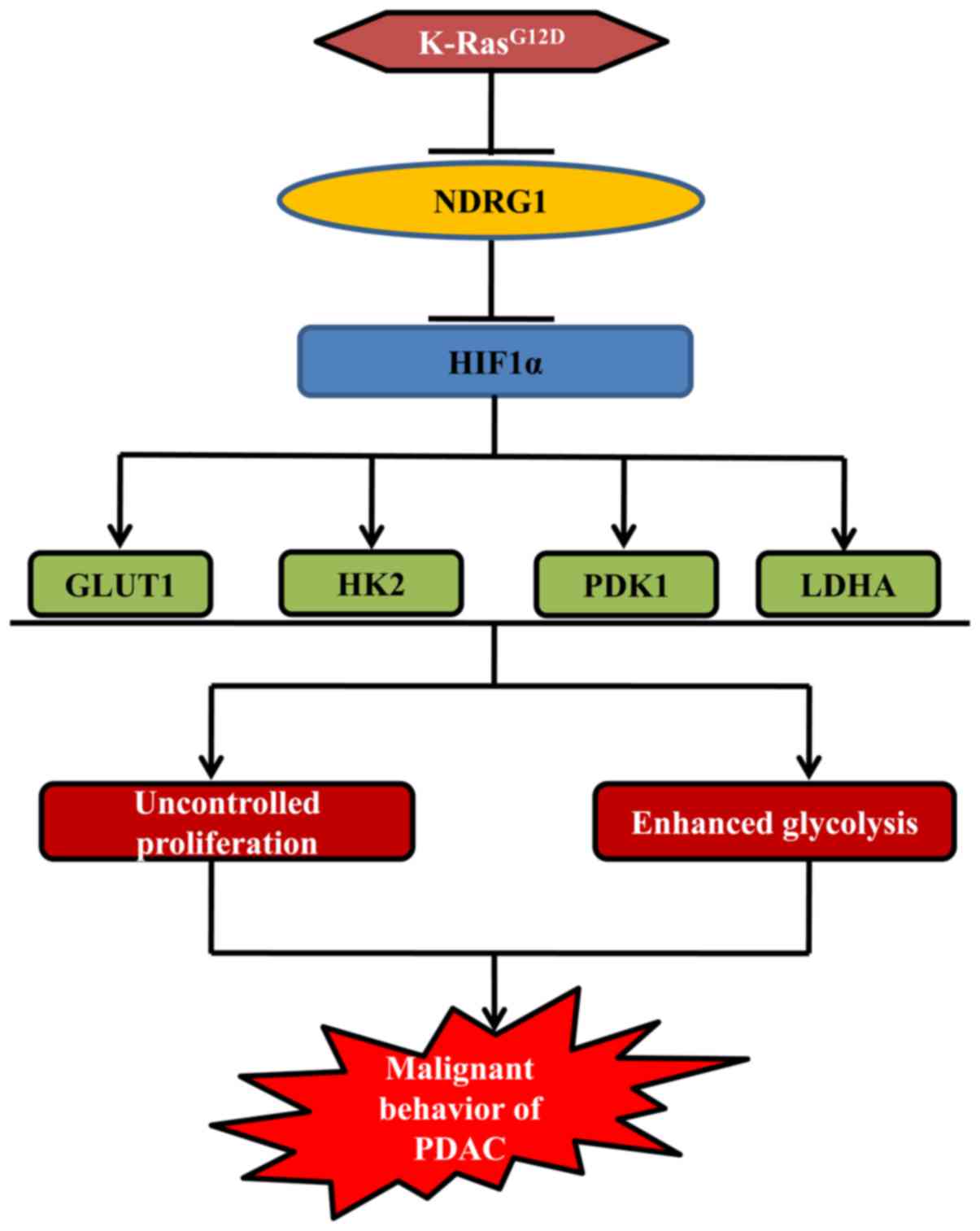
In conclusion, the investigation herein highlights a
novel facet of NDRG1 in K-Ras network (Fig. 6). NDRG1 inhibits PDAC cancer cell
growth via modulating glycolytic metabolism of PDAC (Fig. 6), which is achieved through
suppression of several glycolysis associated key enzymes (namely
GLUT1, HK2, LDHA and PDK1) at both mRNA and protein levels, leading
to decreased ECAR and improved mitochondrial respiration (Fig. 6). Moreover, NDRG1-mediated
reduction of GLUT1, HK2, LDHA and PDK1 is downstream of HIF1α which
was proved to be inhibited by NDRG1 overexpression in PDAC cancer
cell lines as well as specimens (Fig.
6). Therefore, NDRG1 inhibits HIF1α and its downstream key
enzymes of glycolysis to exert its notable anti-metabolic switch
activity in PDAC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81372651, 81502031 and 81602058),
Science and Technology Commission of Shanghai Municipality
(13DZ1942802 and 14ZR1407700) and Youth Program of Shanghai
Municipal Health Bureau (20154Y0090).
References
|
1
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Le A, Rajeshkumar NV, Maitra A and Dang
CV: Conceptual framework for cutting the pancreatic cancer fuel
supply. Clin Cancer Res. 18:4285–4290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sousa CM and Kimmelman AC: The complex
landscape of pancreatic cancer metabolism. Carcinogenesis.
35:1441–1450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cantor JR and Sabatini DM: Cancer cell
metabolism: One hallmark, many faces. Cancer Discov. 2:881–898.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meijer TW, Kaanders JH, Span PN and
Bussink J: Targeting hypoxia, HIF-1, and tumor glucose metabolism
to improve radiotherapy efficacy. Clin Cancer Res. 18:5585–5594.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li B and Simon MC: Molecular Pathways:
Targeting MYC-induced metabolic reprogramming and oncogenic stress
in cancer. Clin Cancer Res. 19:5835–5841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith SC and Theodorescu D: Learning
therapeutic lessons from metastasis suppressor proteins. Nat Rev
Cancer. 9:253–264. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Angst E, Dawson DW, Stroka D, Gloor B,
Park J, Candinas D, Reber HA, Hines OJ and Eibl G: N-myc downstream
regulated gene-1 expression correlates with reduced pancreatic
cancer growth and increased apoptosis in vitro and in vivo.
Surgery. 149:614–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu W, Yue F, Zheng M, Merlot A, Bae DH,
Huang M, Lane D, Jansson P, Lui GY, Richardson V, et al: The
proto-oncogene c-Src and its downstream signaling pathways are
inhibited by the metastasis suppressor, NDRG1. Oncotarget.
6:8851–8874. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain S, Wang X, Chang CC, Ibarra-Drendall
C, Wang H, Zhang Q, Brady SW, Li P, Zhao H, Dobbs J, et al: Src
inhibition blocks c-Myc translation and glucose metabolism to
prevent the development of breast cancer. Cancer Res. 75:4863–4875.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji S, Qin Y, Liang C, Huang R, Shi S, Liu
J, Jin K, Liang D, Xu W, Zhang B, et al: FBW7 (F-box and WD repeat
domain-containing 7) negatively regulates glucose metabolism by
targeting the c-Myc/TXNIP (thioredoxin-binding protein) axis in
pancreatic cancer. Clin Cancer Res. 22:3950–3960. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Emerling BM, Weinberg F, Liu JL, Mak TW
and Chandel NS: PTEN regulates p300-dependent hypoxia-inducible
factor 1 transcriptional activity through Forkhead transcription
factor 3a (FOXO3a). Proc Natl Acad Sci USA. 105:2622–2627. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kennedy AL, Adams PD and Morton JP: Ras,
PI3K/Akt and senescence: Paradoxes provide clues for pancreatic
cancer therapy. Small GTPases. 2:264–267. 2011. View Article : Google Scholar
|
|
16
|
Harada H, Itasaka S, Kizaka-Kondoh S,
Shibuya K, Morinibu A, Shinomiya K and Hiraoka M: The Akt/mTOR
pathway assures the synthesis of HIF-1alpha protein in a glucose-
and reoxygenation-dependent manner in irradiated tumors. J Biol
Chem. 284:5332–5342. 2009. View Article : Google Scholar
|
|
17
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar
|
|
18
|
Kurdistani SK, Arizti P, Reimer CL, Sugrue
MM, Aaronson SA and Lee SW: Inhibition of tumor cell growth by
RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res.
58:4439–4444. 1998.PubMed/NCBI
|
|
19
|
Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM and
Pardee AB: Drg-1 as a differentiation-related, putative metastatic
suppressor gene in human colon cancer. Cancer Res. 60:749–755.
2000.PubMed/NCBI
|
|
20
|
Bandyopadhyay S, Pai SK, Gross SC, Hirota
S, Hosobe S, Miura K, Saito K, Commes T, Hayashi S, Watabe M, et
al: The Drg-1 gene suppresses tumor metastasis in prostate cancer.
Cancer Res. 63:1731–1736. 2003.PubMed/NCBI
|
|
21
|
Liu W, Xing F, Iiizumi-Gairani M, Okuda H,
Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, et al:
N-myc downstream regulated gene 1 modulates Wnt-β-catenin
signalling and pleiotropically suppresses metastasis. EMBO Mol Med.
4:93–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mann KM, Ying H, Juan J, Jenkins NA and
Copeland NG: KRAS-related proteins in pancreatic cancer. Pharmacol
Ther. 168:29–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Logsdon CD and Lu W: The significance of
Ras activity in pancreatic cancer initiation. Int J Biol Sci.
12:338–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones RG and Thompson CB: Tumor
suppressors and cell metabolism: A recipe for cancer growth. Genes
Dev. 23:537–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yun J, Rago C, Cheong I, Pagliarini R,
Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S,
Zhou S, et al: Glucose deprivation contributes to the development
of KRAS pathway mutations in tumor cells. Science. 325:1555–1559.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hosoi F, Izumi H, Kawahara A, Murakami Y,
Kinoshita H, Kage M, Nishio K, Kohno K, Kuwano M and Ono M: N-myc
downstream regulated gene 1/Cap43 suppresses tumor growth and
angiogenesis of pancreatic cancer through attenuation of inhibitor
of kappaB kinase beta expression. Cancer Res. 69:4983–4991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guillaumond F, Leca J, Olivares O, Lavaut
MN, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini
O, et al: Strengthened glycolysis under hypoxia supports tumor
symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma.
Proc Natl Acad Sci USA. 110:3919–3924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun HC, Qiu ZJ, Liu J, Sun J, Jiang T,
Huang KJ, Yao M and Huang C: Expression of hypoxia-inducible
factor-1α and associated proteins in pancreatic ductal
adenocarcinoma and their impact on prognosis. Int J Oncol.
30:1359–1367. 2007.PubMed/NCBI
|
|
30
|
Maruyama Y, Ono M, Kawahara A, Yokoyama T,
Basaki Y, Kage M, Aoyagi S, Kinoshita H and Kuwano M: Tumor growth
suppression in pancreatic cancer by a putative metastasis
suppressor gene Cap43/NDRG1/Drg-1 through modulation of
angiogenesis. Cancer Res. 66:6233–6242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY,
et al: Cellular and developmental control of O2 homeostasis by
hypoxia-inducible factor 1 alpha. Genes Dev. 12:149–162. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chaika NV, Gebregiworgis T, Lewallen ME,
Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB,
Caffrey T, et al: MUC1 mucin stabilizes and activates
hypoxia-inducible factor 1 alpha to regulate metabolism in
pancreatic cancer. Proc Natl Acad Sci USA. 109:13787–13792. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen C, Pore N, Behrooz A, Ismail-Beigi F
and Maity A: Regulation of glut1 mRNA by hypoxia-inducible
factor-1. Interaction between H-ras and hypoxia. J Biol Chem.
276:9519–9525. 2001. View Article : Google Scholar
|
|
34
|
Firth JD, Ebert BL and Ratcliffe PJ:
Hypoxic regulation of lactate dehydrogenase A. Interaction between
hypoxia-inducible factor 1 and cAMP response elements. J Biol Chem.
270:21021–21027. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mathupala SP, Rempel A and Pedersen PL:
Glucose catabolism in cancer cells: Identification and
characterization of a marked activation response of the type II
hexokinase gene to hypoxic conditions. J Biol Chem.
276:43407–43412. 2001. View Article : Google Scholar : PubMed/NCBI
|