Introduction
Hepatocellular carcinoma (HCC) remains a major
health concern worldwide (1).
Hepatitis B virus (HBV) is the dominant risk factor for HCC
(2), accounting for more than half
of all cases (3). HBV X protein
(HBx) is the only expressed HBV protein and plays critical roles in
hepatocarcinogenesis (4). Previous
reports have shown that HBx interferes with many signal
transduction pathways including Hippo, nuclear factor-κB,
WNT/β-catenin, and p53 pathways (2,4-7).
However, the molecular mechanisms underlying the hepatocellular
carcinogenesis induced by HBx remain unclear.
The Notch pathway plays crucial roles in
organogenesis and morphogenesis, and influences various biological
processes including apoptosis, proliferation, and differentiation
(8). Emerging evidence
demonstrates that dysregulation of the Notch pathway is associated
with various types of malignancies (9,10).
Persistent activation of the Notch pathway leads to liver
malignancies (11). A link between
HBx and the Notch pathway has been reported previously (12,13).
Studies have demonstrated that HBx activates the Notch1 pathway
that further upregulates ERK and AKT pathways to promote cell
proliferation (14,15). However, the detailed molecular
mechanisms whereby Notch1 activates ERK and AKT pathways in HCC are
unresolved.
In this study, we further explored the link between
HBx and Notch1 in HCC and elucidated the molecular mechanisms
underlying Notch1/ERK and Notch1/AKT activations by HBx.
Materials and methods
Patients and clinical specimens
A total of 121 human liver tissue samples were
collected from patients who underwent surgical resections at the
Hepatic Surgery Centre, Tongji Hospital of Huazhong University of
Science and Technology (Wuhan, China). Detailed clinicopathological
parameters are listed in Table I.
The procedure of human specimen collection was approved by the
Ethics Committee of Tongji Hospital, Huazhong University of Science
and Technology, and the study was conducted according to the
Declaration of Helsinki.
 | Table ICorrelation between the factors and
clinicopathological parameters in HCC patients (n=121). |
Table I
Correlation between the factors and
clinicopathological parameters in HCC patients (n=121).
| Clinicopathological
variables | Relative Notch1
expression
| P-value |
|---|
| Low | High |
|---|
| Sex | | | 0.322 |
| Male (n=91) | 16 | 75 | |
| Female (n=30) | 3 | 27 | |
| Age (years) | | | 0.506 |
| ≤50 (n=49) | 9 | 40 | |
| >50 (n=72) | 10 | 62 | |
| HBV | | | <0.001 |
| Negative
(n=13) | 7 | 6 | |
| Positive
(n=108) | 12 | 96 | |
| AFP
(µg/l) | | | 0.410 |
| ≤20 (n=41) | 8 | 33 | |
| >20 (n=80) | 11 | 69 | |
| Cirrhosis | | | 0.044 |
| No (n=29) | 8 | 21 | |
| Yes (n=92) | 11 | 81 | |
| Tumor size
(cm) | | | 0.023 |
| ≤5 (n=54) | 13 | 41 | |
| >5 (n=67) | 6 | 61 | |
Cell lines, cell culture, and
reagents
The human hepatoma cell line HepG2 and HBV
genome-transfected HepG2 (HepG2.2.15) cells were obtained from the
China Center for Type Culture Collection (Wuhan, China). Cells were
cultured in high glucose Dulbecco's modified Eagle's medium (Gibco,
Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco).
Dimethyl sulfoxide (DMSO) and γ-secretase inhibitor
N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl
ester (DAPT) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). DAPT was dissolved in DMSO.
Immunohistochemical analysis
Immunohistochemistry (IHC) was performed as
described previously (16).
Primary antibodies against HBx and Notch1 were purchased from
Merck-Millipore (Billerica, MA, USA) and Santa Cruz Biotechnology
(Santa Cruz, CA, USA), respectively.
Western blotting
Western blotting was performed as described
previously (17). The primary
antibodies and their sources were as follows: anti-Notch1,
anti-Hes1, anti-NICD, anti-pERK, anti-ERK, and anti-pAKT (Cell
Signaling Technology, Beverly, MA, USA); anti-Jagged1,
anti-β-actin, anti-AKT, and anti-DUSP1 (Santa Cruz Biotechnology);
anti-HBx (Merck-Millipore); anti-PTEN (Proteintech Group, Chicago,
IL, USA). Horseradish peroxidase-conjugated secondary antibodies
were purchased from Jackson Immuno Research Laboratories (West
Grove, PA, USA).
Double immunofluorescence analysis
Double immunofluorescence immunostaining was
performed as described previously (18,19).
Double-labeling immunofluorescence was used to detect HBx and
Notch1 simultaneously. All sections were analyzed by confocal
laser-scanning microscopy using a Nikon Digital Eclipse C1 system
(Nikon Corp., Japan).
Reverse transcription and real-time
quantitative PCR (qPCR) analysis
PCR was performed as described previously (20). The primer sequences for HBx were as
follows: sense, 5′-GGCTGCTAGGCTGTGCTGCC-3′; antisense,
5′-GTTCCTGTGGGCGTTCACGG-3′. Images of electrophoresed PCR products
were acquired using the Alpha Innotech Fluorochem Imaging system.
Real-time PCR primers are listed in Table II. Cycle threshold values were
reported relative to β-actin mRNA. Expression values were obtained
in triplicate and normalized to β-actin expression. Results were
calculated as fold induction relative to controls.
| Table IIPrimer sequences for real-time
PCR. |
Table II
Primer sequences for real-time
PCR.
| Gene | Primer
sequences |
|---|
| HBx | F:5′-CAC CTC TCT
TTA CGC GGA CT-3′ |
| R: 5′-GGT CGT TGA
CAT TGC AGA GA-3′ |
| Hes1 | F: 5′-AAG AAA GAT
AGC TCG CGG CAT-3′ |
| R: 5′-CCA GCA CAC
TTG GGT CTG T-3′ |
| DUSP1 | F: 5′-CCA GTA CAA
GAG CAT CCC TGT-3′ |
| R: 5′-AGT GGA CAA
ACA CCC TTC CTC-3′ |
| PTEN | F:5′-AGC GTG CAG
ATA ATG ACA AGG-3′ |
| R: 5′-TGG ATC AGA
GTC AGT GGT GTC-3′ |
| β-actin | F: 5′-CAA GGC CAA
CCG CGA GAA GAT-3′ |
| R: 5′-CCA GAG GCG
TAC AGG GAT AGC AC-3′ |
Transient RNA interference
Small interfering RNA (siRNA) duplexes targeting
human HBx, Hes1, DUSP1, and PTEN sequences and a scrambled siRNA
were designed as described previously (5,21-23).
All siRNAs were synthesized by Ribobio (Guangzhou, China).
Transfection of the siRNA duplexes was performed by jetPRIME
(Polyplus-transfection SA, Illkirch, France) according to the
manufacturer's instructions.
Cell viability assay
The cell viability assay was performed as described
previously (24). HepG2.2.15 cells
(2×103 per well) were seeded and cultured in 96-well
plates for the indicated times. Cell Counting Kit-8 (CCK-8,
Dojindo, Japan) was added to the cells, and the optical density
value at 450 nm was measured after 2 h.
Transcriptional response assay
Luciferase assays were performed as described
previously (25). Cell lysates was
subjected to luciferase assays using the Dual-luciferase Reporter
assay system (Promega) according to the manufacturer's
instructions. DUSP1-luc reporter, PTEN-luc reporter, and Hes1
expression vectors were transfected as reported previously
(26-28). Relative luciferase activities were
determined by a Glomar 20/20 Illuminometer (Promega) and normalized
against Renilla luciferase as an internal control.
Chromatin immunoprecipitation (ChIP)
Quantitative ChIP analysis was performed as
described previously (29).
Briefly, cells were crosslinked, sonicated, and immunoprecipitated
with anti-Hes1 antibodies or IgG (negative control). A mixture of
two anti-Hes1 antibodies (H140, Santa Cruz Biotechnology; 4H1,
Novus Biologicals) was used for immunoprecipitation. Then, DNA was
eluted and crosslinking was reversed, followed by purification and
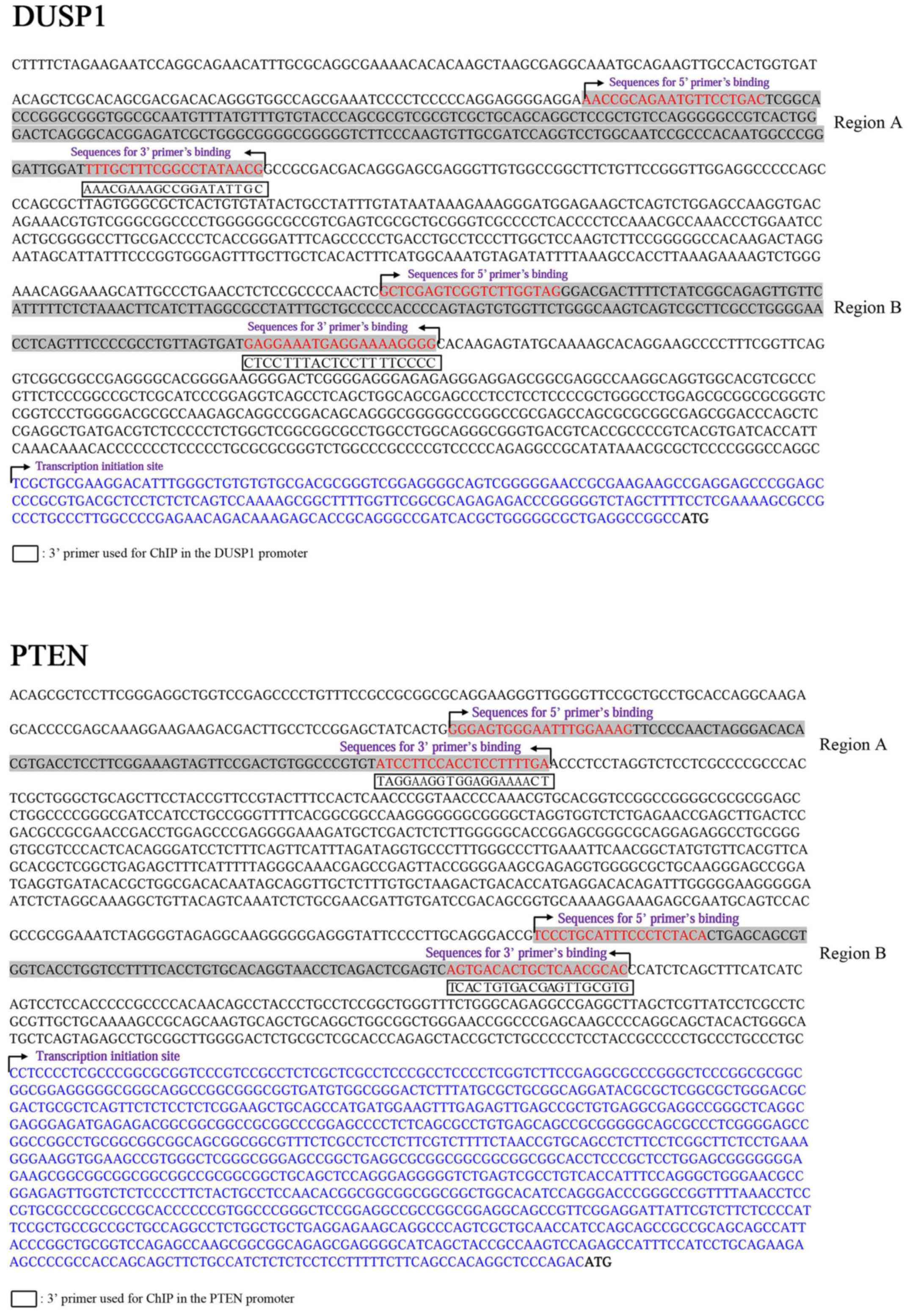
amplification for PCR analysis. The primers used in qPCR are listed
in Table III.
| Table IIIPrimers used for ChIP. |
Table III
Primers used for ChIP.
| ChIP primers | Primer
sequences |
|---|
| DUSP1 | F: 5′-AAC CGC AGA
ATG TTC CTG AC-3′ |
| promoter A | R: 5′-CGT TAT AGG
CCG AAA GCA AA-3′ |
| DUSP1 | F: 5′-GCT CGA GTC
GGT CTT GGT AG-3′ |
| promoter B | R: 5′-CCC CTT TTC
CTC ATT TCC TC-3′ |
| PTEN | F: 5′-GGG AGT GGG
AAT TTG GAA AG-3′ |
| promoter A | R: 5′-TCA AAA GGA
GGT GGA AGG AT-3′ |
| PTEN | F: 5′-TCC CTG CAT
TTC CCT CTA CA-3′ |
| promoter B | R: 5′-GTG CGT TGA
GCA GTG TCA CT-3″ |
Statistical analysis
Data were analyzed using GraphPad Prism 5.0 (La
Jolla, CA, USA) or SPSS 13.0 (Chicago, IL, USA). All experiments
were independently performed at least three times. The results are
presented as the mean ± standard error of the mean. Comparisons
between different groups were evaluated by one-way analysis of
variance. P<0.05 was considered as statistically
significant.
Results
Expression of Notch1 in human liver and
HCC tissues
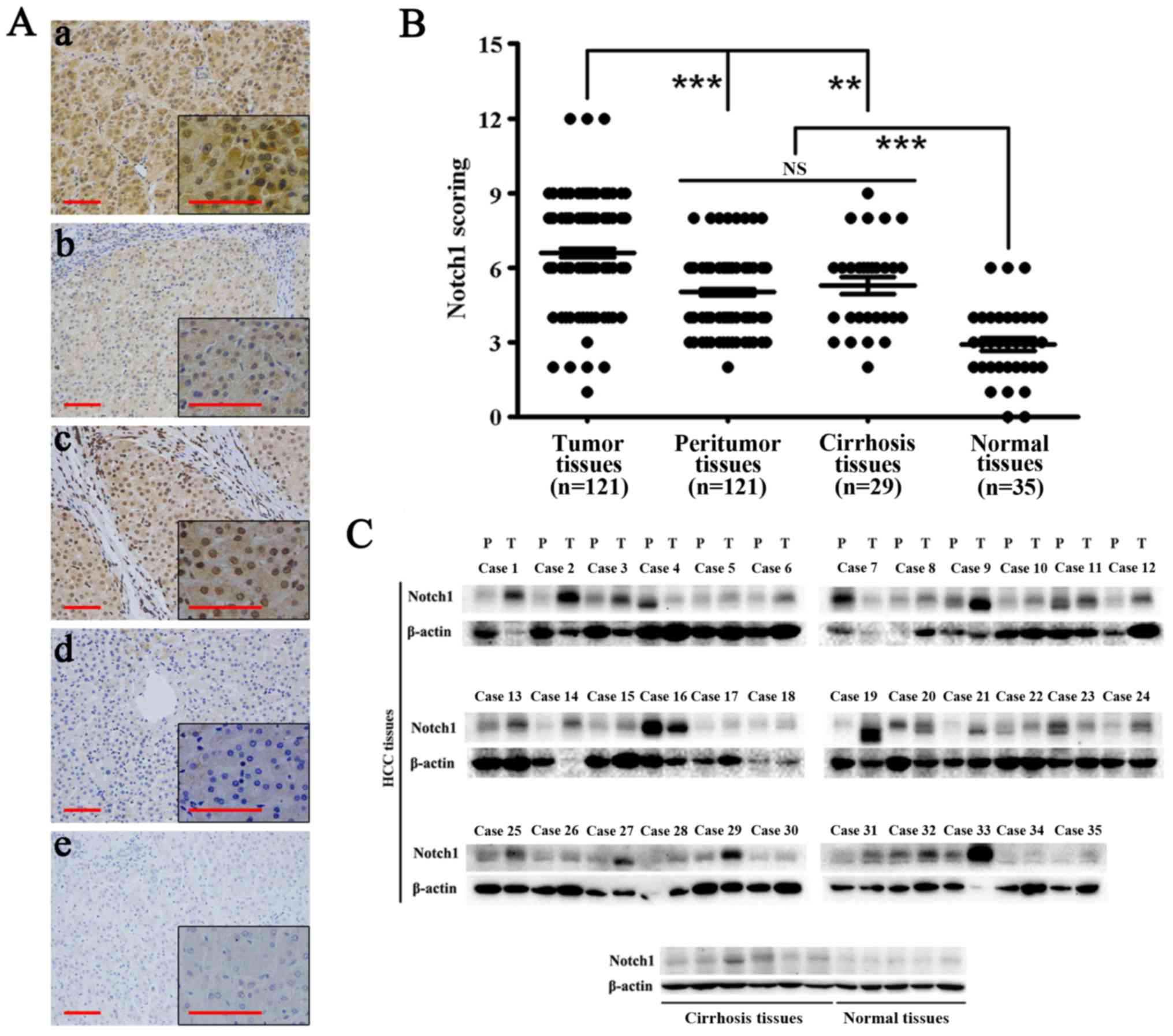
Immunohistochemical analysis showed that Notch1 was
highly expressed in HCC tissues. Notch1 also exhibited expression
in adjacent non-tumor and cirrhosis tissues. However, its
expression was significantly lower than that in HCC tissues
(P<0.01). Moreover, the expression of Notch1 in normal liver
tissues was significantly lower than that in the above-mentioned
tissues (P<0.001) (Fig. 1A and
B). Western blotting showed similar results (Fig. 1C). In most HCC tissues, the
expression of Notch1 was highly elevated compared with that in the
other tissues. Notch1 was weakly expressed in all normal liver
tissues.
Specifically, among 121 HCC tissues, 19 of them had
weak expression and 102 of them had high expression. In the 121
corresponding peritumoral tissues, 59 of them had weak expression
and 62 of them had high expression (P<0.001) (Table IV).
| Table IVExpression of Notch1 in HCC and
peritumor liver tissues. |
Table IV
Expression of Notch1 in HCC and
peritumor liver tissues.
| Cases tested | Relative Notch1
expression
| P-value |
|---|
| Low | High |
|---|
| Tumor tissues | 121 | 19 | 102 | <0.001 |
| Peritumor
tissues | 121 | 59 | 62 | |
Table I shows the
correlation between the factors and clinicopathological parameters
in the 121 HCC patients. As shown in the table, sex, age, and the
AFP level were unrelated to Notch1 expression. The level of Notch1
was significantly more elevated in HBV-positive patients
(P<0.001), cirrhosis patients, (P<0.05), and patients with
large HCCs (P<0.05).
Co-localization and relationship of
Notch1 with HBx in HCC tissues and the HepG2.2.15 cell line
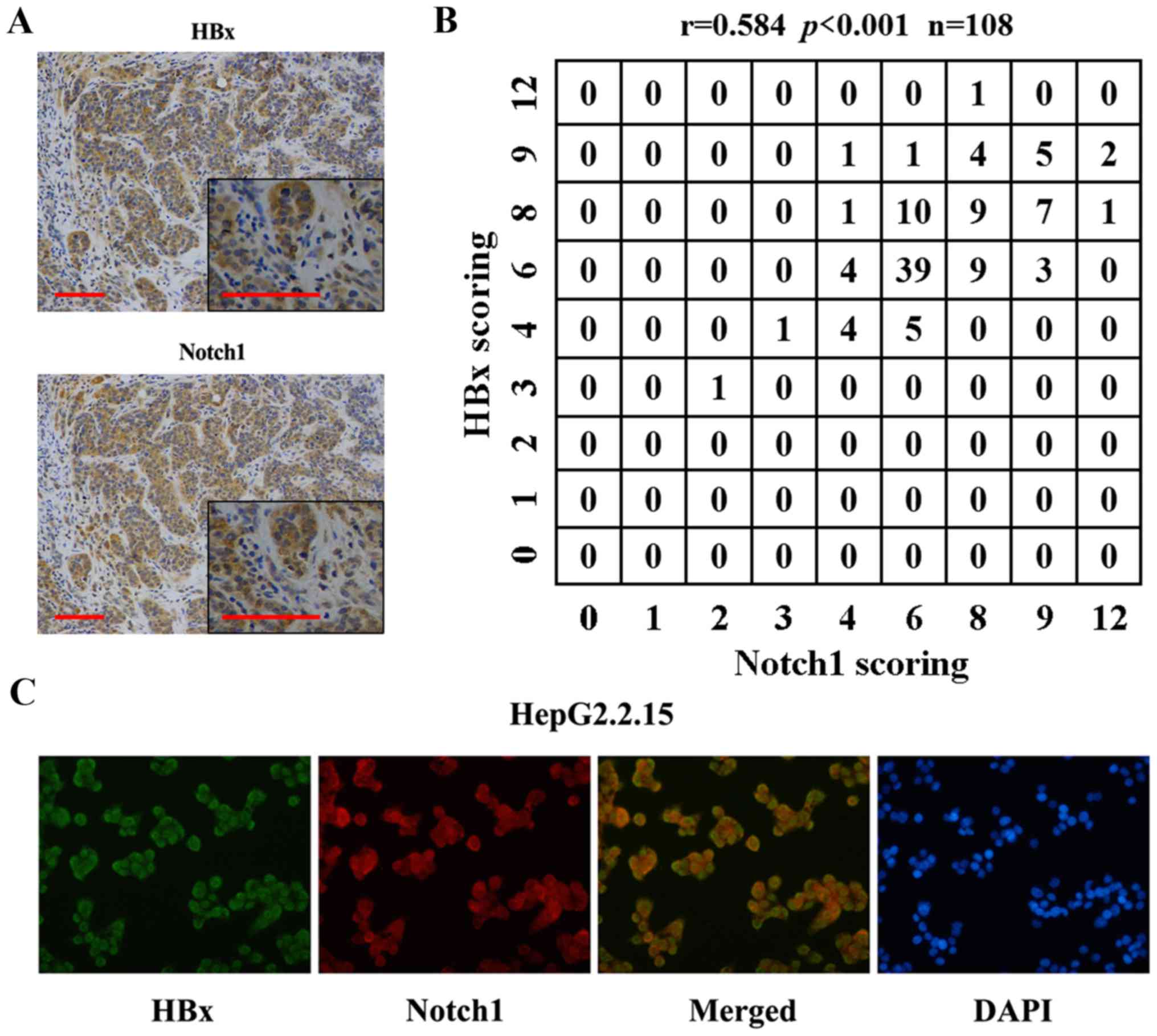
Previous reports have shown the relationship of
Notch1 with HBx. Here, we found co-localization of the two
proteins. IHC results showed that Notch1 and HBx were co-expressed
in HCC tissues (Fig. 2A). Fig. 2B shows the correlation of Notch1
and HBx expression levels. Spearman's rho was 0.584, indicating
that Notch1 levels were positively correlated with HBx expression
in the 108 HBV-positive HCC tissues. Fig. 2C shows HBx (green) and Notch1 (red)
staining in HepG2.2.15 cells. The yellow staining in dual labeling
experiments indicated overlapping areas of HBx and Notch1 staining,
suggesting co-expression of Notch1 with HBx in HepG2.2.15
cells.
Regulation of Notch1 by HBx in HepG2.2.15
cells
Because a relationship of HBx with Notch1 was found
in HCC tissues and cells, we determined whether HBx could regulate
the expression of Notch1 in HepG2.2.15 cells in vitro. To
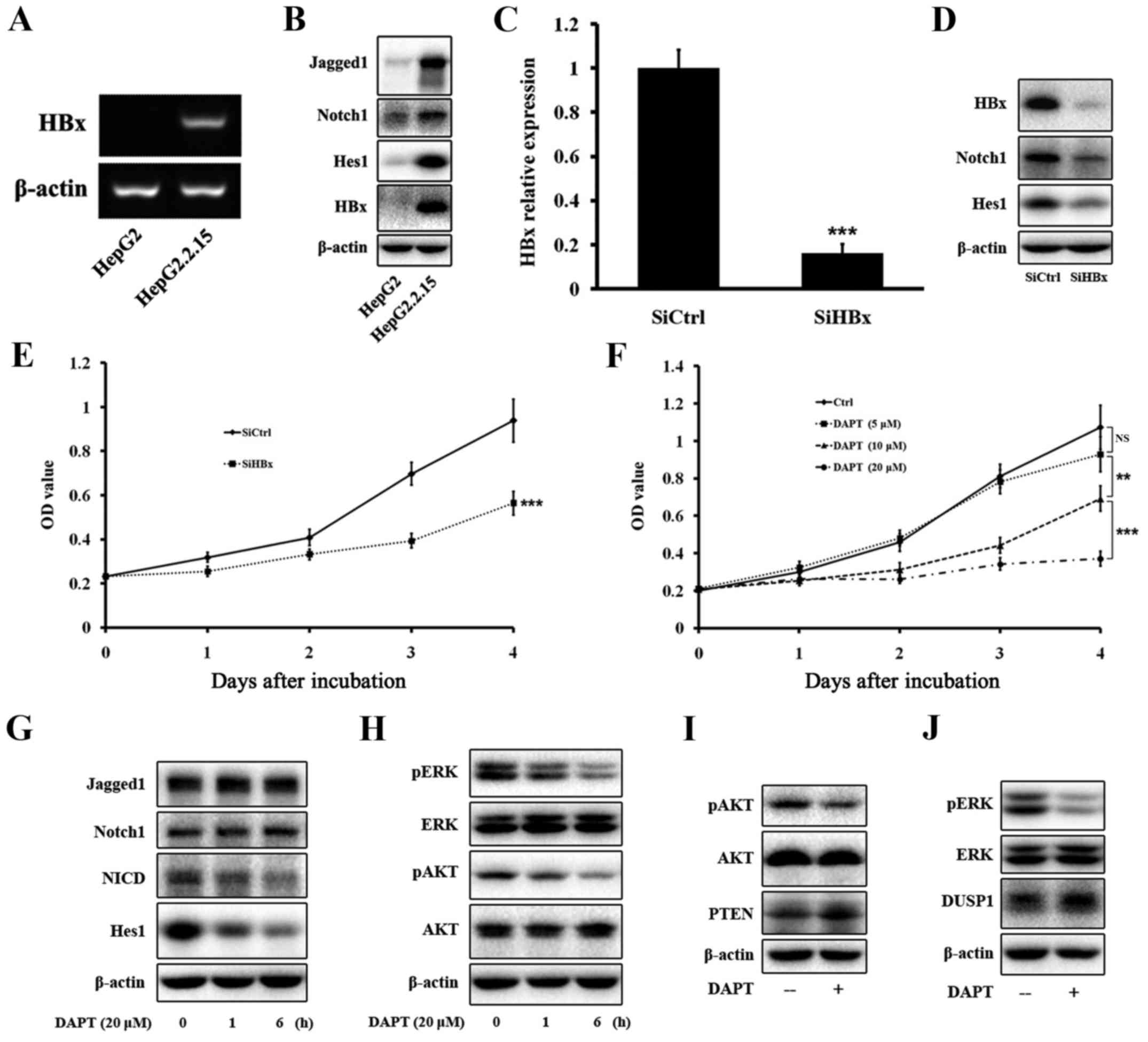
test this hypothesis, we compared the expression of HBx and some
members of the Notch1 pathway between HepG2 and HepG2.2.15 cells,
and between HepG2.2.15-SiCtrl and HepG2.2.15-SiHBx cells. Western
blotting and PCR confirmed that HepG2 cells did not express HBx,
whereas the HBx gene and protein were expressed in HepG2.2.15 cells
(Fig. 3A and B). Next, we tested
the expression of some members of the Notch1 pathway, such as
Jagged1, Notch1, and Hes1. Western blotting showed that the protein
levels of the three proteins were much more elevated in HepG2.2.15
cells than in HepG2 cells (Fig.
3B). Next, we knocked down HBx expression in HepG2.2.15 cells
by HBx-specific siRNA. Western blotting and PCR confirmed that HBx
expression was significantly decreased by the siRNA (Fig. 3C and D). Western blotting showed
that the decrease of HBx expression led to a decrease of Notch1 and
Hes1 expression in HepG2.2.15 cells (Fig. 3D), verifying that HBx regulated the
expression of Notch1 in vitro.
Suppression of HBx and the Notch1 pathway
inhibits the growth of HepG2.2.15 cells
Our results indicated that Notch1 was highly
expressed in HCC tissues, especially in large HCC tissues, and the
Notch1 pathway was regulated by HBx, we therefore determined
whether inhibition of the Notch1 pathway attenuated cell growth. To
inhibit the Notch1 pathway, we used the γ-secretase inhibitor DAPT.
HepG2.2.15 cells were treated with increasing concentrations of
DAPT for 4 days, and then cell viability was evaluated by CCK-8
assays. As shown in Fig. 3E,
inhibition of HBx attenuated the growth of HepG2.2.15 cells.
Increasing concentrations and treatment times of DAPT resulted in
progressive inhibition of HepG2.2.15 cell viability (Fig. 3F). At day 1, DAPT treatment at
various concentrations did not reduce cell viability. However, DAPT
treatment for more than 2 days triggered a significant time- and
dose-dependent decrease in the viability of HepG2.2.15 cells. We
ultimately selected a DAPT treatment concentration of 20 µM
for further experiments.
Inhibition of Notch1, ERK, and AKT
pathways after DAPT treatment in HepG2.2.15 cells
HepG2.2.15 cells treated with 20 µM DAPT for
1 and 6 h were assessed for inhibition levels of the Notch1 pathway
by western blot analyses of Jagged1, Notch1, NICD, and Hes1
expression. DAPT treatment significantly decreased the amount of
NICD and Hes1 in a time-dependent manner, but did not have any
effect on Jagged1 or Notch1 (Fig.
3G). Our data showed that DAPT treatment greatly inhibited the
growth of HepG2.2.15 cells. Therefore, we tested expression of ERK
and AKT pathways that are closely related to cell proliferation.
Western blotting indicated that DAPT greatly reduced the amount of
pERK and pAKT in a time-dependent manner, but did not have any
effect on total ERK or AKT (Fig.
3H).
Upregulation of DUSP1 and PTEN after DAPT
treatment in HepG2.2.15 cells
Previous reports have revealed the regulatory
circuit linking the pathway with DUSP1 expression and ERK activity
(21). A link has also been found
between the Notch1 pathway and PTEN and AKT activities (28). Our data showed that DUSP1 and PTEN
were upregulated after DAPT treatment in HepG2.2.15 cells by
western blotting (Fig. 3I and
J).
Upregulation of DUSP1 and PTEN after
SiHes1 treatment of HepG2.2.15 cells
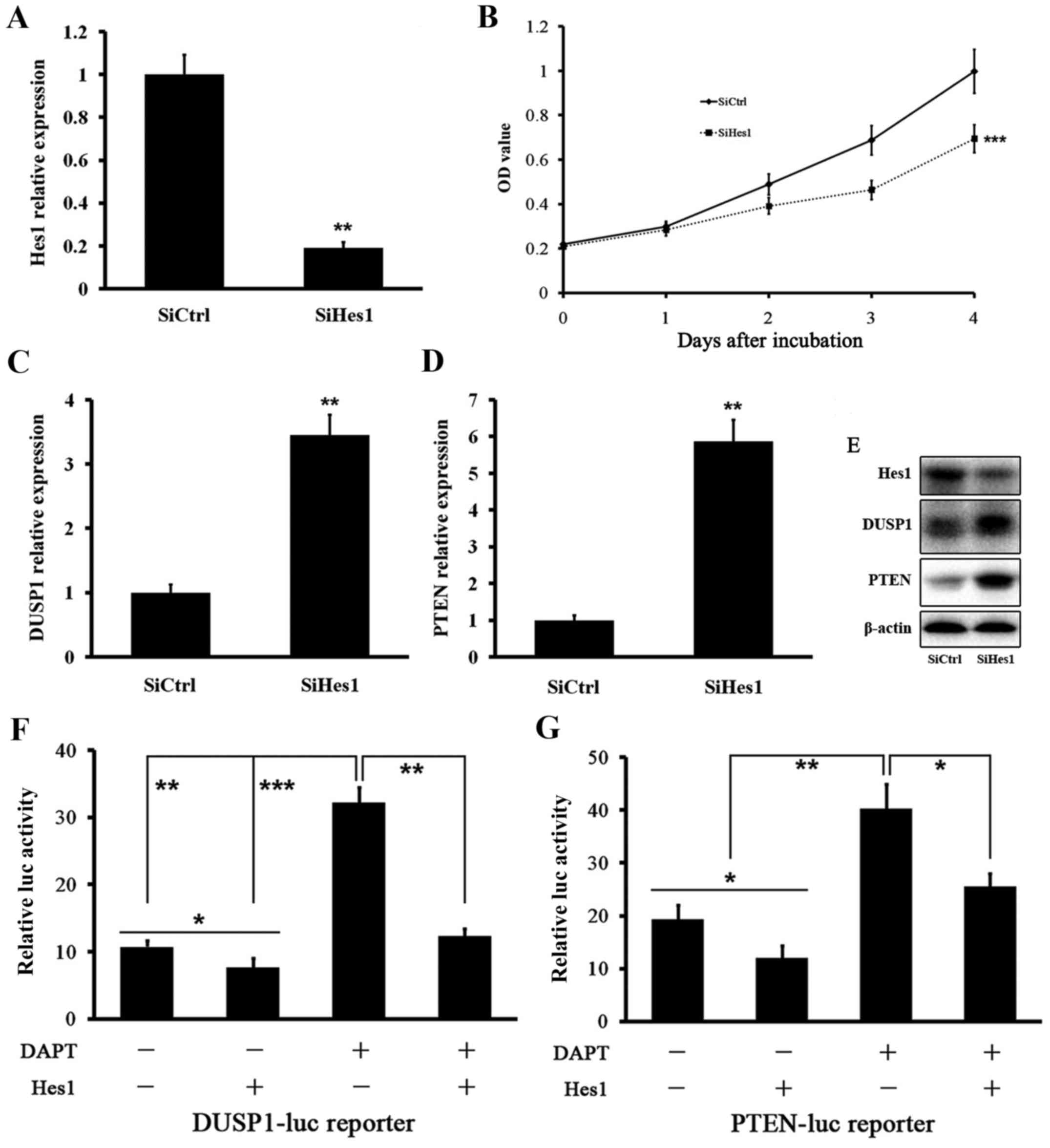
We tested the effect of Hes1 inhibition on DUSP1 and
PTEN levels. Treatment of HepG2.2.15 cells with siRNA targeting
Hes1 mRNA (SiHes1) effectively reduced the Hes1 mRNA level
(Fig. 4A). Growth of HepG2.2.15
cells was inhibited after SiHes1 treatment (Fig. 4B). Next, we found that the mRNA
levels of DUSP1 and PTEN were greatly increased after SiHes1
treatment (Fig. 4C and D). Western
blot analysis showed the same results. The Hes1 protein level was
signifi-cantly reduced and the protein levels of DUSP1 and PTEN
were elevated significantly (Fig.
4E).
Hes1 suppresses and directly binds to the
promoters of DUSP1 and PTEN genes in HepG2.2.15 cells
Based on the above data, we investigated whether
Hes1 suppressed DUSP1 and PTEN. We performed DUSP1 and PTEN
promoter assays using a luciferase reporter. The basal activities
of DUSP1 and PTEN gene promoters were reduced by Hes1. Treatment of
HepG2.2.15 cells with DAPT induced DUSP1 and PTEN gene promoters,
which was reversed by cotransfection of Hes1 (Fig. 4F and G). We next performed ChIP
assays to determine whether Hes1 directly bound to the promoters of
DUSP1 and PTEN genes using two mixed antibodies against Hes1. We
designed several primers in the promoter regions of DUSP1 and PTEN
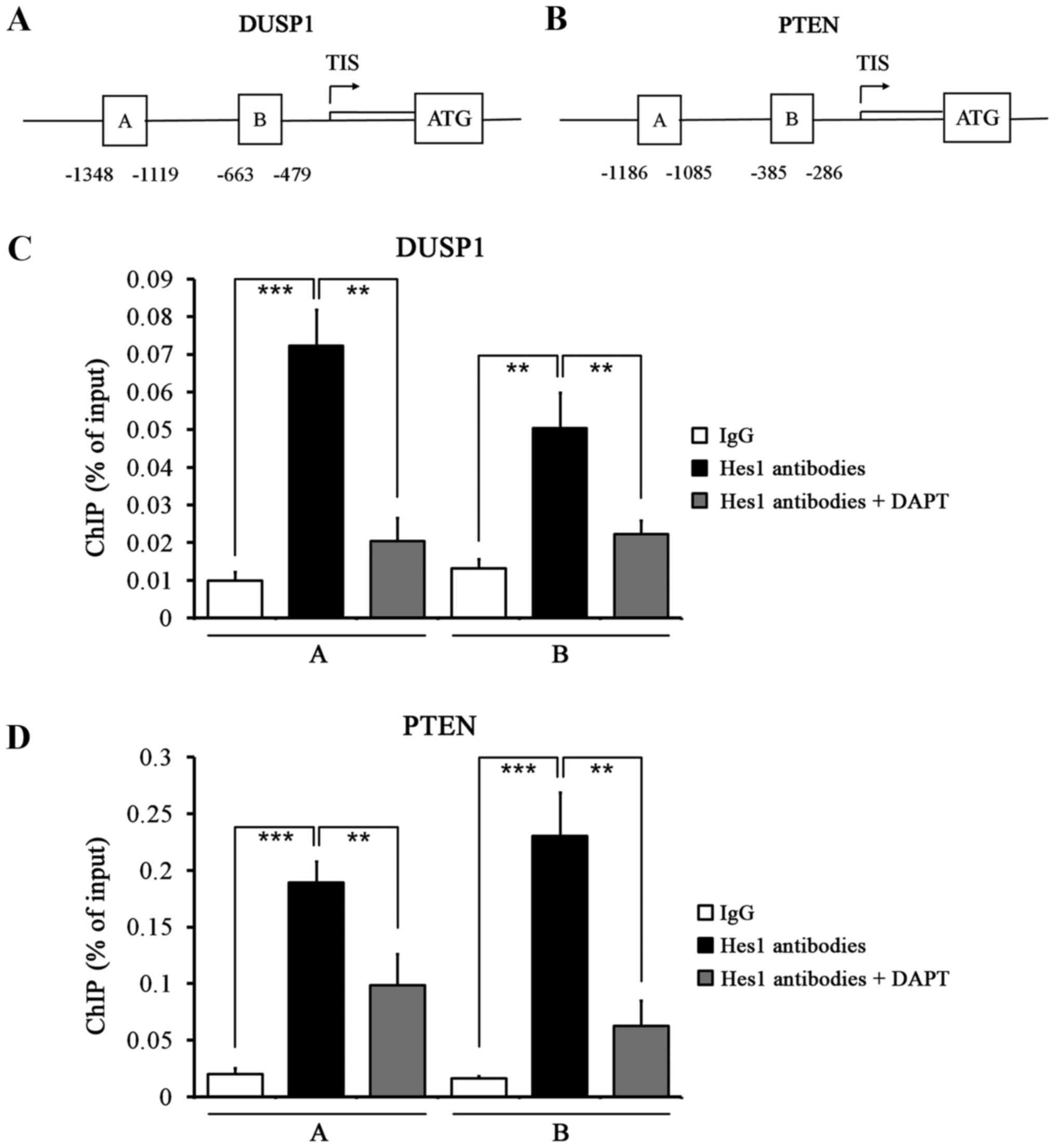
genes (Fig. 5). ChIP assays showed
that Hes1 bound to the regulatory sequences in the promoters of
DUSP1 and PTEN genes in HepG2.2.15 cells (Fig. 6A and B). Quantitative ChIP assays
revealed significant enrichment of two regions of the DUSP1 and
PTEN gene promoters compared with control IgG immunoprecipitation,
which was reversed by treatment with DAPT (Fig. 6C and D). These results further
reinforce our previous findings that DAPT upregulated DUSP1 and
PTEN expression by downregulating Hes1.
SiDUSP1/SiPTEN treatment of HepG2.2.15
cells elevates pERK/pAKT
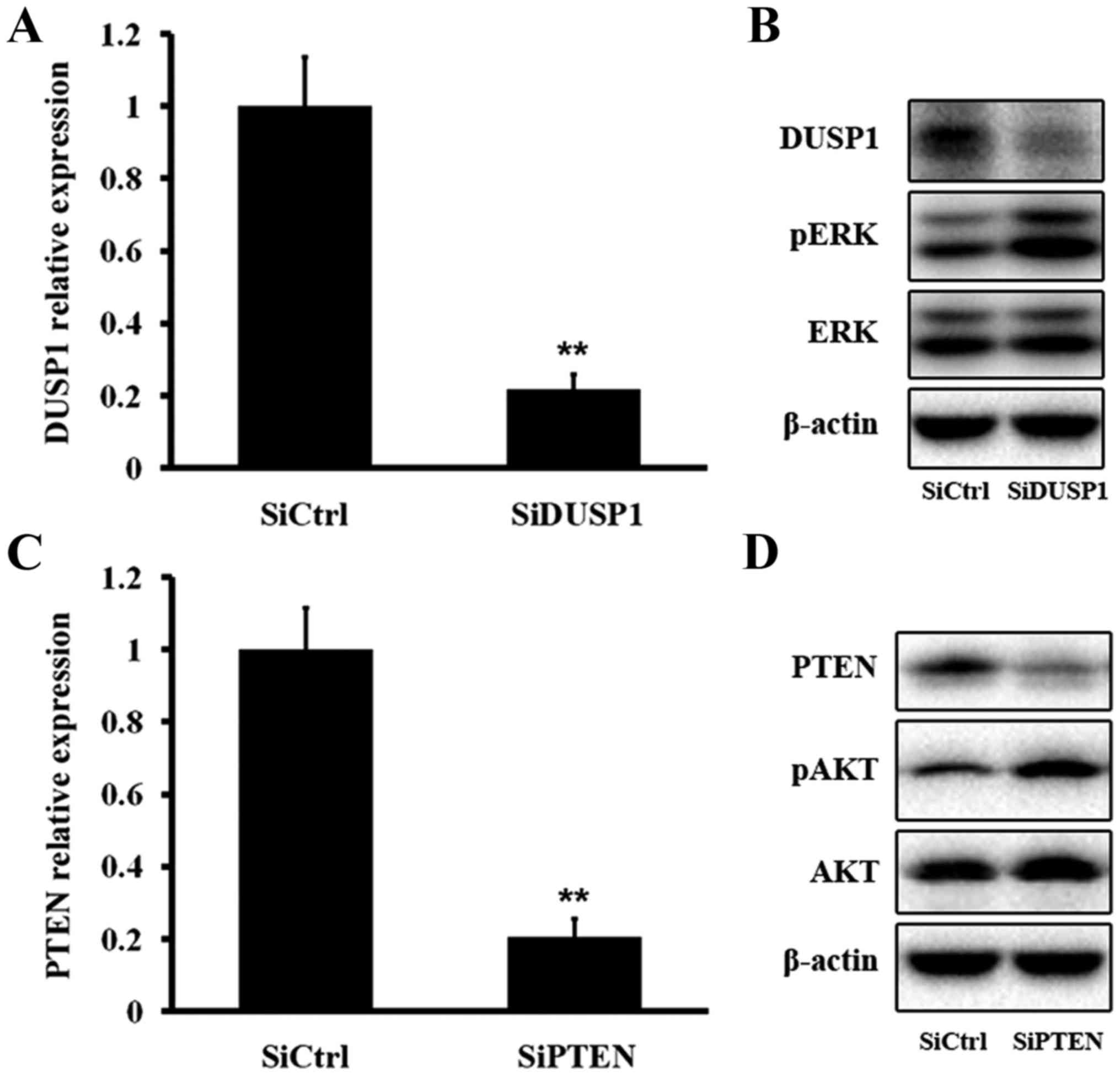
We next tested the effect of DUSP1 inhibition on ERK
phosphorylation. Treatment of HepG2.2.15 cells with siRNA targeting
DUSP1 mRNA (SiDUSP1) effectively reduced DUSP1 mRNA and protein
levels, resulting in an elevated pERK level (Fig. 7A and B). We then evaluated the
effect of PTEN inhibition on AKT phosphorylation. Treatment with
SiPTEN effectively decreased PTEN mRNA and protein levels, which
induced an increase of the pAKT level (Fig. 7C and D). The above findings
demonstrated that DUSP1 dephosphorylated pERK and PTEN
dephosphorylated pAKT.
Discussion
Notch1 is overexpressed in HCC (14). Consistent with the results of
previous studies, we found that Notch1 expression in tumor tissues
was much more elevated than that in peri-tumoral, cirrhosis, and
normal tissues by IHC and western blot analyses. Previous reports
have shown co-localization of Notch1 with HBx (14). In accordance with these findings,
IHC and confocal analyses demonstrated that Notch1 morphologically
co-localized with HBx in HCC tissues and HepG2.2.15 cells.
Furthermore, there was a positive correlation between Notch1 and
HBx expression. It has been demonstrated that HBx activates the
Notch1 pathway. We also found that the Notch1 pathway was
upregulated by HBx. The growth of HepG2.2.15 cells was inhibited by
treatment with SiHBx or DAPT, indicating that HBx stimulated cell
proliferation via the Notch1 pathway. A previous report has shown
that HBx inhibition attenuates the phosphorylation of ERK and AKT
in HepG2.2.15 cells (15). In our
study, we found that inhibition of the Notch1 pathway downregulated
the phosphorylated levels of ERK and AKT.
Previous studies have shown that Hes1, the key
member of the Notch1 pathway, represses expression of DUSP1 that is
active against pERK in non-small cell lung carcinoma (21). Another study has shown that Hes1
decreases expression of PTEN, a negative regulator of the AKT
pathway in T-cell leukemia (28).
Thus, we hypothesized that the pathway influenced expression of
DUSP1 and PTEN, and further affected ERK and AKT pathways in
HepG2.2.15 cells. To test this hypothesis, we performed PCR,
western blotting, luciferase assays, and ChIP. We found that mRNA
and protein levels of DUSP1 and PTEN were increased after Hes1
inhibition. To investigate the mechanisms that link inhibition of
the Notch1 pathway with dephosphorylation of ERK and AKT, we
analyzed the transcriptional changes induced by DAPT. Promoter
induction of DUSP1 and PTEN genes after DAPT treatment were
confirmed in HepG2.2.15 cells. Previous data have shown that ERK
and AKT pathways are important to regulate the Notch1 pathway
downstream of HBx. However, a direct link between Notch1 and
ERK/AKT pathways has not been revealed (15).
Hes1 is a well-known transcriptional regulator of
multiple genes (30,31). Based on our finding that the Hes1
level was reduced upon DAPT treatment of HepG2.2.15 cells, we
hypothesized that dephosphorylation of ERK or AKT induced by DAPT
could be regulated by Hes1-mediated decreases of DUSP1 or PTEN. Our
luciferase reporter assays showed that Hes1 was a negative
regulator of DUSP1 and PTEN. It directly repressed DUSP1 and PTEN
gene promoters, which was reversed by DAPT treatment. Next, we
found that increased levels of pERK and pAKT were induced by
inhibition of DUSP1 and PTEN, respectively. This result could
explain the decrease in ERK and AKT phosphorylations upon DAPT
treatment.
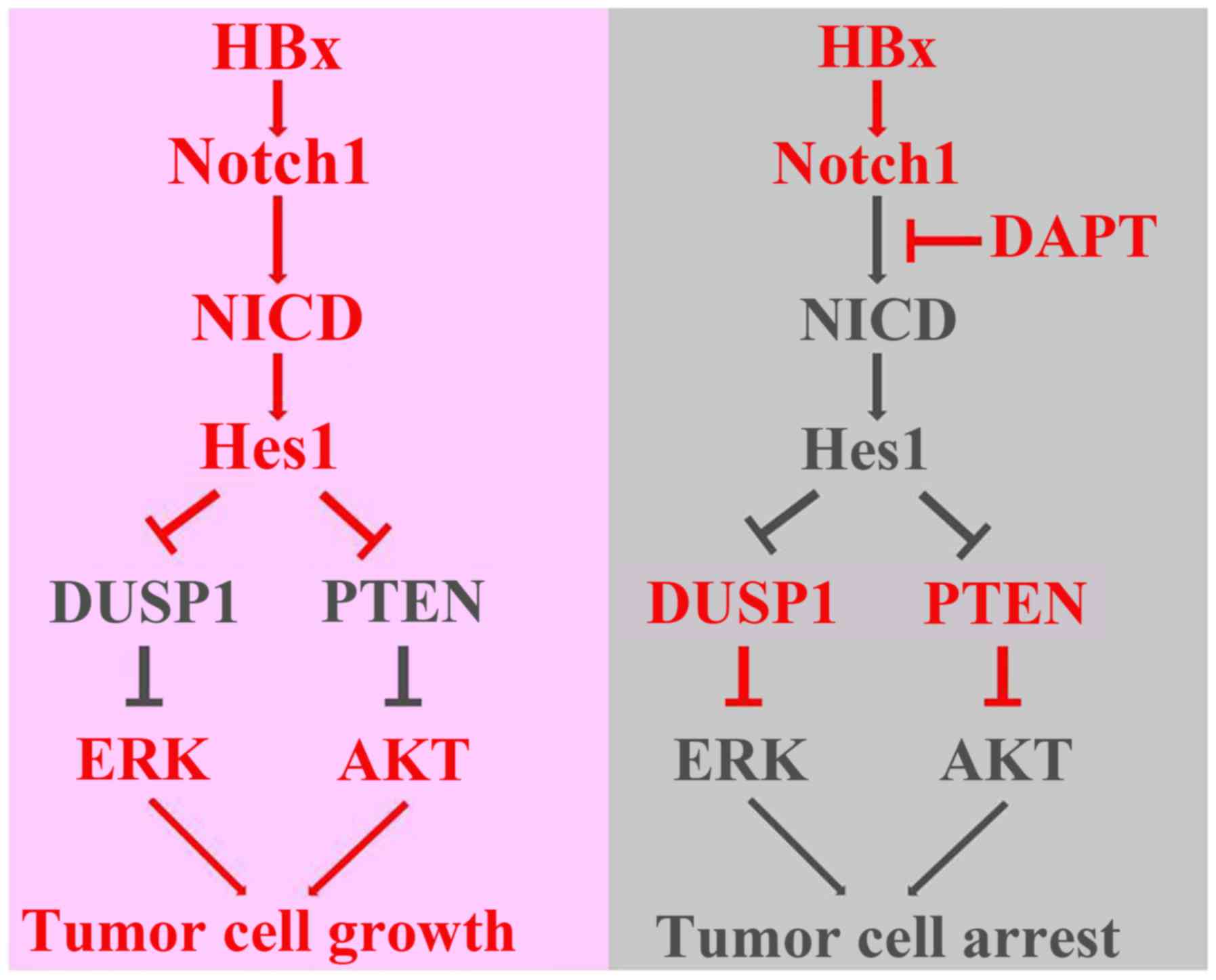
In conclusion, this study explored a direct link
among HBx, the Notch1 pathway, DUSP1/PTEN, and ERK/AKT pathways
(Fig. 8). We found that HBx
activated the Notch1 pathway to promote cell growth, which was
correlated with the capacity of Hes1 to increase ERK/AKT activities
through decrease of DUSP1/PTEN expression. Nevertheless, we cannot
exclude that other molecular mechanisms might take part in
mediating the effects of DAPT. Therefore, the underlying mechanisms
need to be elucidated further.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81272421 and 81202300) and the
Project of Hubei Natural Science Foundation of China (no.
2015CFB462).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sherman M: Hepatocellular carcinoma:
Epidemiology, surveillance, and diagnosis. Semin Liver Dis.
30:3–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geng M, Xin X, Bi LQ, Zhou LT and Liu XH:
Molecular mechanism of hepatitis B virus X protein function in
hepatocar-cinogenesis. World J Gastroenterol. 21:10732–10738. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang T, Zhang J, You X, Liu Q, Du Y, Gao
Y, Shan C, Kong G, Wang Y, Yang X, et al: Hepatitis B virus X
protein modulates oncogene Yes-associated protein by CREB to
promote growth of hepatoma cells. Hepatology. 56:2051–2059. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu C, Zhou W, Wang Y and Qiao L: Hepatitis
B virus-induced hepatocellular carcinoma. Cancer Lett. 345:216–222.
2014. View Article : Google Scholar
|
|
7
|
Levrero M and Zucman-Rossi J: Mechanisms
of HBV-induced hepatocellular carcinoma. J Hepatol. 64(Suppl):
S84–S101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miele L, Golde T and Osborne B: Notch
signaling in cancer. Curr Mol Med. 6:905–918. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rizzo P, Osipo C, Foreman K, Golde T,
Osborne B and Miele L: Rational targeting of Notch signaling in
cancer. Oncogene. 27:5124–5131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Geisler F and Strazzabosco M: Emerging
roles of Notch signaling in liver disease. Hepatology. 61:382–392.
2015. View Article : Google Scholar
|
|
12
|
Wang F, Zhou H, Xia X, Sun Q, Wang Y and
Cheng B: Activated Notch signaling is required for hepatitis B
virus X protein to promote proliferation and survival of human
hepatic cells. Cancer Lett. 298:64–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang F, Zhou H, Yang Y, Xia X, Sun Q, Luo
J and Cheng B: Hepatitis B virus X protein promotes the growth of
hepatocel-lular carcinoma by modulation of the Notch signaling
pathway. Oncol Rep. 27:1170–1176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao J, Xiong Y, Wang Y, Wang Y, Zheng G
and Xu H: Hepatitis B virus X protein activates Notch signaling by
its effects on Notch1 and Notch4 in human hepatocellular carcinoma.
Int J Oncol. 48:329–337. 2016. View Article : Google Scholar
|
|
15
|
Kongkavitoon P, Tangkijvanich P, Hirankarn
N and Palaga T: Hepatitis B virus HBx activates Notch signaling via
delta-like 4/Notch1 in hepatocellular carcinoma. PLoS One.
11:e01466962016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ding ZY, Jin GN, Wang W, Chen WX, Wu YH,
Ai X, Chen L, Zhang WG, Liang HF, Laurence A, et al: Reduced
expression of transcriptional intermediary factor 1 gamma promotes
metastasis and indicates poor prognosis of hepatocellular
carcinoma. Hepatology. 60:1620–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen L, Zhang W, Zhou QD, Yang HQ, Liang
HF, Zhang BX, Long X and Chen XP: HSCs play a distinct role in
different phases of oval cell-mediated liver regeneration. Cell
Biochem Funct. 30:588–596. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiang S, Dong HH, Liang HF, He SQ, Zhang
W, Li CH, Zhang BX, Zhang BH, Jing K, Tomlinson S, et al: Oval cell
response is attenuated by depletion of liver resident macrophages
in the 2-AAF/partial hepatectomy rat. PLoS One. 7:e351802012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong HH, Xiang S, Chen XP, Liang HF, Zhang
W, Jing K, Zhang W, Zhang WG and Chen L: The epithelial-mesenchymal
transition promotes transdifferentiation of subcutaneously
implanted hepatic oval cells into mesenchymal tumor tissue. Stem
Cells Dev. 18:1293–1298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen L, Zhang W, Liang HF, Zhou QF, Ding
ZY, Yang HQ, Liu WB, Wu YH, Man Q, Zhang BX, et al: Activin A
induces growth arrest through a SMAD-dependent pathway in hepatic
progenitor cells. Cell Commun Signal. 12:182014. View Article : Google Scholar
|
|
21
|
Maraver A, Fernandez-Marcos PJ, Herranz D,
Cañamero M, Muñoz-Martin M, Gómez-López G, Mulero F, Megías D,
Sanchez-Carbayo M, Shen J, et al: Therapeutic effect of γ-secretase
inhibition in KrasG12V-driven non-small cell lung carcinoma by
derepression of DUSP1 and inhibition of ERK. Cancer Cell.
22:222–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Calvisi DF, Pinna F, Meloni F, Ladu S,
Pellegrino R, Sini M, Daino L, Simile MM, De Miglio MR, Virdis P,
et al: Dual-specificity phosphatase 1 ubiquitination in
extracellular signal-regulated kinase-mediated control of growth in
human hepatocellular carcinoma. Cancer Res. 68:4192–4200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Song M, Chen W,
Dimitrova-Shumkovska J, Zhao Y, Cao Y, Song Y, Yang W, Wang F,
Xiang Y, et al: MicroRNA-21 Contributes to Liver Regeneration by
Targeting PTEN. Med Sci Monit. 22:83–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liao B, Liang H, Chen J, Liu Q, Zhang B
and Chen X: Suberoylanilide hydroxamic acid enhances
chemosensitivity to 5-fluorouracil in hepatocellular carcinoma via
inhibition of thymidylate synthase. Tumour Biol. 36:9347–9356.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ding ZY, Jin GN, Liang HF, Wang W, Chen
WX, Datta PK, Zhang MZ, Zhang B and Chen XP: Transforming growth
factor β induces expression of connective tissue growth factor in
hepatic progenitor cells through Smad independent signaling. Cell
Signal. 25:1981–1992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tchen CR, Martins JR, Paktiawal N, Perelli
R, Saklatvala J and Clark AR: Glucocorticoid regulation of mouse
and human dual specificity phosphatase 1 (DUSP1) genes: Unusual
cis-acting elements and unexpected evolutionary divergence. J Biol
Chem. 285:2642–2652. 2010. View Article : Google Scholar :
|
|
27
|
Real PJ, Tosello V, Palomero T, Castillo
M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C,
Homminga I, et al: Gamma-secretase inhibitors reverse
glucocorticoid resistance in T cell acute lymphoblastic leukemia.
Nat Med. 15:50–58. 2009. View
Article : Google Scholar
|
|
28
|
Palomero T, Sulis ML, Cortina M, Real PJ,
Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, et
al: Mutational loss of PTEN induces resistance to NOTCH1 inhibition
in T-cell leukemia. Nat Med. 13:1203–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding ZY, Liang HF, Jin GN, Chen WX, Wang
W, Datta PK, Zhang MZ, Zhang B and Chen XP: Smad6 suppresses the
growth and self-renewal of hepatic progenitor cells. J Cell
Physiol. 229:651–660. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iso T, Kedes L and Hamamori Y: HES and
HERP families: Multiple effectors of the Notch signaling pathway. J
Cell Physiol. 194:237–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sang L, Roberts JM and Coller HA:
Hijacking HES1: How tumors co-opt the anti-differentiation
strategies of quiescent cells. Trends Mol Med. 16:17–26. 2010.
View Article : Google Scholar
|