Introduction
Lung cancer is the most common cause of cancer
mortality, with 1.6 million deaths annually (1,2).
Non-small cell lung cancers (NSCLCs) are currently sub-classified
based on the presence of actionable driver oncogenes, and molecular
targeted therapies have been developed to specifically treat
cancers harboring these driver mutations (3). Anaplastic lymphoma kinase (ALK)
inhibitors are clinically effective for the treatment of NSCLC
patients whose tumors harbor ALK fusion genes, including the
echinoderm microtubule-associated protein-like 4 (EML4)
-ALK fusion (EML4-ALK) (4).
The first generation ALK inhibitor, crizotinib, can
elicit dramatic and durable responses in ALK-NSCLCs (5). In the last few years, alectinib and
ceritinib have been developed for the treatment of ALK-NSCLCs
(6,7). Unfortunately, the development of
resistance to ALK inhibitors is common and arises through a variety
mechanisms (8,9). One of the most important of these
mechanisms is the activation of alternative survival pathways,
including EGFR, MET, cKIT and IGFR signaling (10). Although activation of Src tyrosine
kinase signaling is linked to crizotinib resistance, this survival
pathway was not linked to the development of resistance to other
ALK inhibitors (11,12).
Heat shock protein (Hsp) 90 inhibitors are effective
for the treatment of ALK-NSCLC (13). Hsp90 is a major intracellular
molecular chaperone that interacts with a wide variety of
intracellular proteins, and promotes correct protein folding and
conformation. Hsp90 client proteins include many important
regulators of cell proliferation and differentiation, such as
protein kinases and steroid hormone receptors. Hsp90 expression is
increased in many cancers, including ALK-NSCLCs. Hsp90 inhibitors
promote ALK degradation, and are clinically effective for the
treatment of patients with tumors carrying ALK rearrangements,
regardless of resistance to ALK inhibitors (14,15).
The aims of the present study were to clarify the
mechanisms underlying resistance to ALK specific inhibitors in
NSCLC, and identify novel targets that may ultimately form the
basis for the design of more effective therapies for overcoming
acquired resistance.
Materials and methods
Cell culture and compounds
The H3122 (EML4-ALK variant 1) NSCLC cell
line was kindly provided by Dr Pasi A. Jänne at Dana-Farber Cancer
Institute (Boston, MA, USA). H2228 (EML4-ALK variant 3)
cells were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA) and maintained at 37°C in 5%
CO2 using RPMI-1640 medium (Thermo Fisher Scientific,
Waltham, MA, USA) containing 2 mM L-glutamine, 50 U/ml penicillin,
and 50 µg/ml streptomycin, supplemented with 10% fetal
bovine serum (FBS) (R-10 medium). Alectinib was provided by Chugai
Pharmaceutical Co., Ltd., (Kamakura, Japan) and AUY922 was provided
by Novartis Pharma AG (Basel, Switzerland). Ceritinib, lorlatinib,
saracatinib, dasatinib and bosutinib were purchased from Selleck
Chemicals (Houston, TX, USA). Each compound was dissolved in
dimethyl sulfoxide (DMSO; Wako Pure Chemical Industries, Ltd.,
Tokyo, Japan) for cell culture experiments.
Generation of H3122-AFR, H3122-LDKR and
H3122-PFR cell lines
H3122 cells were treated with alectinib at a
starting concentration of 1 nM and cells were maintained at ~70%
confluence. After every passage at a given concentration of drug,
the concentration of alectinib was increased until a final
concentration of 100 nM was achieved. The resulting pool of
resistant cells (designated H3122-AFR) was maintained in R-10
medium containing 100 nM alectinib. Other ALK inhibitor resistant
cell lines were established in a similar manner. The H3122
derivatives, H3122-LDKR and H3122-PFR, were maintained in R-10
medium with 1 µM ceritinib or 100 nM lorlatinib,
respectively.
Cell growth assays
For MTS assays, 3–4×103 cells were plated
into 96-well plates. After 24 h, cells were treated with drugs, and
incubated for a further 72 h. CellTiter 96®
AQueous solution (Promega, Maddison, WI, USA) was then
added and plates were incubated for an additional 4 h. Optical
density was measured at 490 nm and the data were graphically
displayed using GraphPad Prism version 6.0 (GraphPad Software,
Inc., La Jolla, CA, USA).
For colony formation assays, 5.0×104
cells were plated into 6-well plates and treated with drugs 24 h
later. After 6 days, the media containing drugs were refreshed. One
week after media replacement, plates were washed with
phosphate-buffered saline (PBS) once, and stained with 500
µl of 0.5% crystal violet with 25% methanol for 1 h and
photographed.
Immunoblot analysis
Cells grown under the previously specified
conditions were lysed in cell lysis buffer (Wako Pure Chemical
Industries) with PhosSTOP Phosphatase inhibitor cocktail (Roche,
Basel, Switzerland) and Complete EASYPack (Roche). After cell
lysis, lysates were centrifuged at 14,000 × g for 10 min at 4°C and
the supernatant was used for subsequent procedures. Western blot
analyses were conducted after protein separation by SDS-PAGE
electrophoresis and transfer to Immobilon-P membrane (Merck KGaA,
Darmstadt, Germany). Immunoblotting was performed according to the
antibody manufacturer's recommendations. Antibodies against
phospho-ALK (#4144), ALK (#3633), phospho-EGFR (#3777), EGFR
(#4267), phospho-Src (#6943), Src (#2109), phospho-AKT (#4060), AKT
(#9272), phosho-P130Cas (#4011), P130Cas (#13846), phospho-CrK II
(#3491), CrK II (#3492), phospho-CrKL (#3181), CrKL (#3182),
phospho-Paxillin (#2541), phosphor-FAK (#3283), FAK (#3285),
pan-phospho-Tyrosine (P-Tyr-100) (#9411) and β-actin (#4967) were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunofluorescence/immunocytochemistry
A total 5×104 of H3122-AFR cells were
plated in 8-well chamber slides 1 day before the drug treatment.
The cells were fixed with 2% paraformaldehyde at room temperature 8
h after drug treatment. The fixed cells were incubated with primary
P-Tyr-100 and Src antibodies diluted in PBS containing 0.5% Block
Ace (DS Pharma Biomedical, Osaka, Japan) for 1 day. After washing
with PBS, the fixed cells were incubated with appropriate secondary
antibodies conjugated to Alexa dye 488 or 594 (Thermo Fisher
Scientific) for 1 h before mounting. Nuclei were counterstained
with DAPI. The images were taken using an Olympus FluoView FV1000
confocal laser scanning microscope, and analyzed with Olympus
FluoView ver4.2a Viewer software.
Phospho-receptor tyrosine kinase (RTK)
array analysis
A PathScan RTK Signaling Antibody Array kit (Cell
Signaling Technology) was used to profile relative tyrosine
phosphorylation levels. A total of 2×102 cells was
seeded in 10-cm dishes 1 day before drug treatment with alectinib
and saracatinib, and the cells were harvested 1 day after drug
treatment. Phospho-RTK array analysis was performed according to
the manufacturer's protocol. The average pixel densities of
duplicate spots were determined using ImageJ software (http://imagej.nih.gov/ij/) and phospho-RTK percentages
were calculated relative to the average control intensity.
Next-generation sequencing
Genomic DNA from H3122-AFR cells was isolated using
the DNeasy Blood & Tissue kit (Qiagen) according to the
manufacturer's protocol. Ion AmpliSeq Comprehensive Cancer Panel
(Thermo Fisher Scientific), a previously validated panel for
targeted amplicon sequencing was utilized (Thermo Fisher
Scientific). Briefly, 10 ng of gDNA was amplified by PCR using Ion
AmpliSeq Library kit (Thermo Fisher Scientific) and the sequencing
was performed on an Ion PGM System according to the manufacturer's
protocol. Sequencing reads were multiplexed, quality-filtered and
aligned to the human reference genome (GRCh37) using the Torrent
Suite software (ver. 5.0.4; Thermo Fisher Scientific). Variants
were identified with the Variant Caller software (ver. 5.0.4.0;
Thermo Fisher Scientific). The quality of all variants called was
manually confirmed by Integrative Genomics Viewer software (IGV;
ver. 2.3.59).
Xenograft studies
All mouse xenograft experiments were performed using
age matched 8–12 week-old female Nu/Nu mice. After subcutaneous
injection of 5.0×106 H3122-AFR cells, mice were
randomized into four groups, and treated with either vehicle,
alectinib alone, saracatinib alone, or a combination of the two,
for the indicated duration of treatment. Tumor size was measured by
calipers twice a week. Tumor volume was calculated as length ×
width2 × 0.51. Treatment was initiated when tumors
exceeded 100 mm3. Nude mice bearing H3122-AFR xenografts
were treated with vehicle only (Control, n=11), alectinib (20
mg/kg, n=10), saracatinib (50 mg/kg, n=11), combination (alectinib
20 mg/kg + saracatinib 50 mg/kg, n=9) for the indicated number of
days. Each drug was prepared in the following solvents: alectinib
(0.02 N HCl, 10% DMSO, 10% CremophrEL, 15% PEG400, 15% HPCD),
saracatinib (0.5% HPMC and 0.1% Tween-80). Alectinib was
administered by oral gavage and saracatinib was administered by
intraperitoneal injection.
iTRAQ sample preparation and
analysis
Protein samples were diafiltrated, reduced,
alkylated and trypsin digested according to the iTRAQ protocol
(Thermo Fisher Scientific). The samples were then labeled using the
iTRAQ reagents. Peptides were desalted on a Strata-X 33 µm
polymeric reversed phase column (Phenomenex, Inc., Torrance, CA,
USA) and dissolved in a buffer containing 10 mM
KH2PO4 in 10% acetonitrile before separation
by strong cation exchange liquid chromatography on an Agilent 1100
HPLC system. Peptides were eluted with a linear gradient of 0–400
mM KCl. Eight fractions containing the peptides were collected and
desalted on Strata-X columns. The fractions were analyzed by
electro-spray ionization mass spectrometry using the Agilent 1260
Infinity HPLC system (Agilent Technologies, Santa Clara, CA, USA)
coupled to an Agilent 1260 Chip Cube nanospray interface (Agilent
Technologies) on an Agilent 6540 mass spectrometer (Agilent
Technologies).
Data analysis was performed according to a previous
report (16). Spectral data for
each sample were compared against the Swiss-Prot database with
taxonomy set to Homo sapiens using ProteinPilot™ 4.5
software (AB Sciex, Redwood City, CA, USA). Average ratios were
calculated by comparing the intensities of the peptide signals
before and after drug treatment. A P≤0.05 indicates statistically
significant differential expression.
Statistical analysis
The measurements are presented as means ± SEM.
Results were analyzed by the Student's t-test using GraphPad Prism
version 6.0 (GraphPad Software). P<0.05 were considered to be
statistically significant.
Results
Generation of ALK lung cancer cell line
resistance models
We generated ALK inhibitor resistant lung cancer
cell lines by exposing H3122 cells to increasing doses of the
alectinib, ceritinib and lorlatinib. The alectinib resistant H3122
cell line derivative was designated as H3122-AFR. The gDNA
extracted from H3122-AFR cells was sequenced using Next-generation
sequencing but secondary ALK mutation was not found. Ceritinib and
lorlatinib resistant H3122 cell line derivatives were designated as
H3122-LDKR and H3122-PFR, respectively.
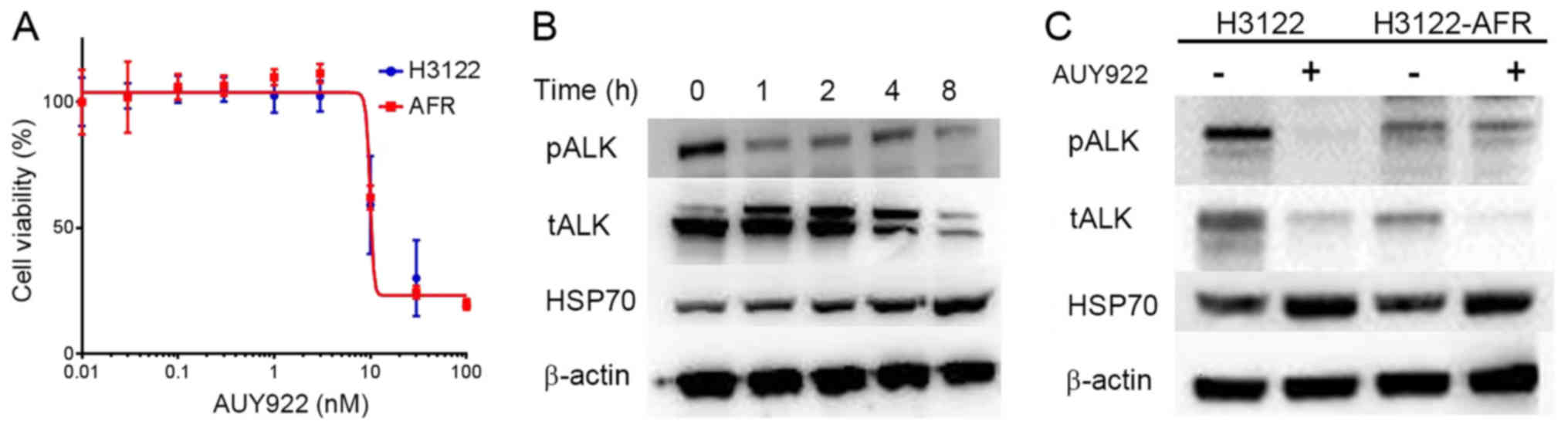
Sensitivity of HSP90 inhibitor inH3122
and H3122-AFR
H3122 and H3122-AFR cell lines had the same
sensitivity to AUY922, and ALK protein expression in H3122-AFR
cells was inhibited 4 h after the addition of AUY922 (Fig. 1A and B). Immunoblotting analysis of
H3122 and H3122-AFR before and after AUY922 treatment for 4 h
revealed that ALK proteins were degraded in both cell lines.
However, the expression of Hsp70 proteins was increased in both
H3122 and H3122-AFR after AUY922 treatment, consistent with a
compensatory activation of co-chaperones in response to Hsp90
inhibition (Fig. 1C).
Chaperone peptides/protein screening in
ALK resistant cell lines
As we previously reported (17), the treatment of H3122 cells with
Hsp90 inhibitors resulted in the degradation of ALK proteins and
the induction of apoptosis, which suggested that growth/survival
signaling in these cells was dependent on chaperone activity. To
address this hypothesis, we screened for Hsp90 client proteins in
H3122 cells before and after the addition of the Hsp90 inhibitor,
AUY922. Similarly, proteins/peptides extracted from H3122-AFR cells
were analyzed before and after treatment with AUY922 to study the
changes of client proteins after acquired resistance.
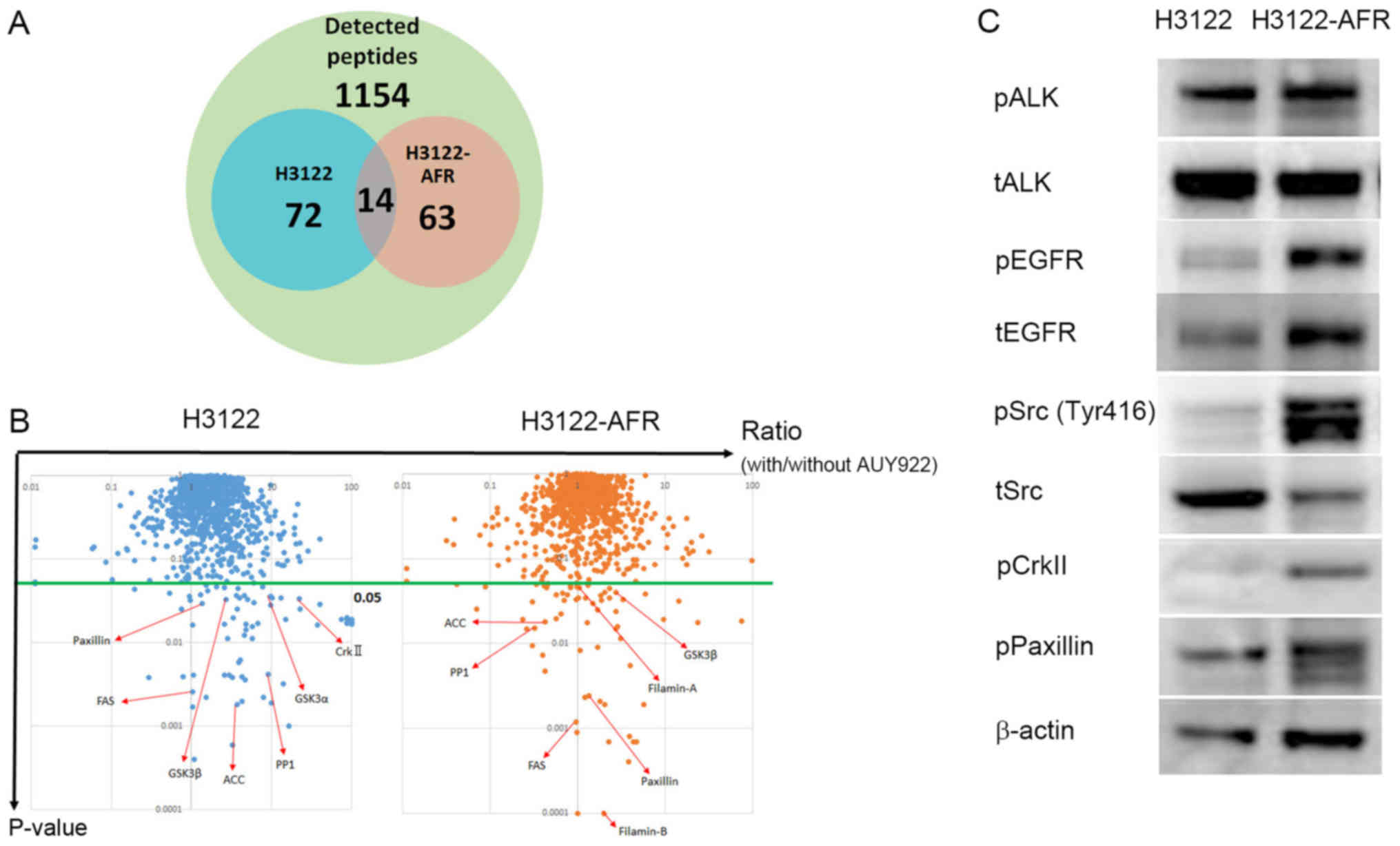
For chaperone peptides/protein screening, we
analyzed protein expression in H3122 and H3122-AFR cells in
response to AUY922 treatment by iTRAQ quantitative mass
spectrometry (16,18). iTRAQ detected 1154
proteins/peptides in each cell line, including 72 candidate
proteins in H3122, and 63 candidate proteins in H3122-AFR that had
P≤0.05. There were 14 candidate proteins that were common to both
H3122 and H3122-AFR (Fig. 2A). The
candidate proteins in the iTRAQ data were converted to gene lists
in DAVID (Database for Annotation, Visualization and Integrated
Discovery, https://david.ncifcrf.gov). KEGG
(Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) intracellular pathway
analysis of the DAVID gene lists revealed two deregulated signaling
cascades, including the focal adhesion pathway and the insulin
signaling pathway (Fig. 2B and
Table I). Activation of the focal
adhesion pathway, involving paxillin and crkII could be an
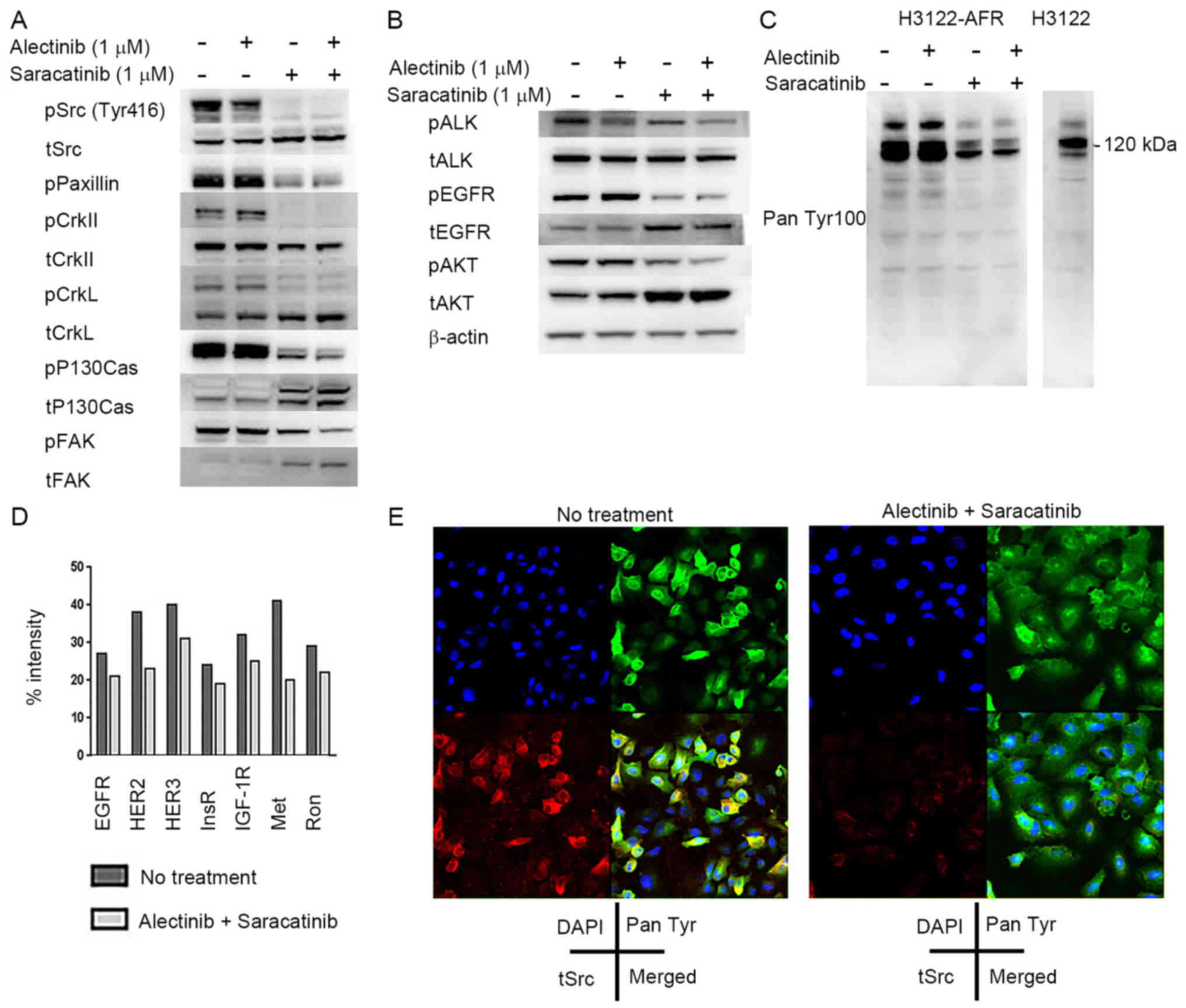
alectinib resistance mechanism. Immunoblotting revealed that
H3122-AFR cells had higher phosphorylation levels of paxillin,
crkII and Src compared with H3122 cells (Fig. 2C). Taken together, these data
suggested that these pathways could be associated with resistance
mechanisms.
 | Table IAnalysis of intracellular signaling
pathway based on iTRAQ. |
Table I
Analysis of intracellular signaling
pathway based on iTRAQ.
|
Pathway/proteins | H3122 ratio
| H3122-AFR ratio
|
|---|
|
Treated/untreated | P-value |
Treated/untreated | P-value |
|---|
| Focal adhesion | | | | |
| Filamin-A | 1.00 | 0.99 | 1.04a | 0.05a |
| Filamin-A | 1.33 | 0.66 | 2.00a | 0.0001a |
| Paxillin | 1.36a | 0.03a | 1.36a | 0.002a |
| Crk II | 22.28a | 0.03a | 0.34 | 0.70 |
| Crk L | 0.99 | 0.91 | 0.98 | 0.33 |
| MLCP | 1.13 | 0.79 | 1.19 | 0.72 |
| GSK3β | 2.70a | 0.03a | 2.75a | 0.04a |
| GSK3α | 9.82a | 0.04a | 0.85 | 0.12 |
| Actinib | 3.25 | 0.6 | 1.43 | 0.59 |
| Insulin signaling
pathway | | | | |
| FAS | 1.03a | 0.003a | 0.96b | 0.001b |
| PP1 | 9.12a | 0.004a | 0.32b | 0.015b |
| ACC | 3.7a | 0.002a | 0.42b | 0.018b |
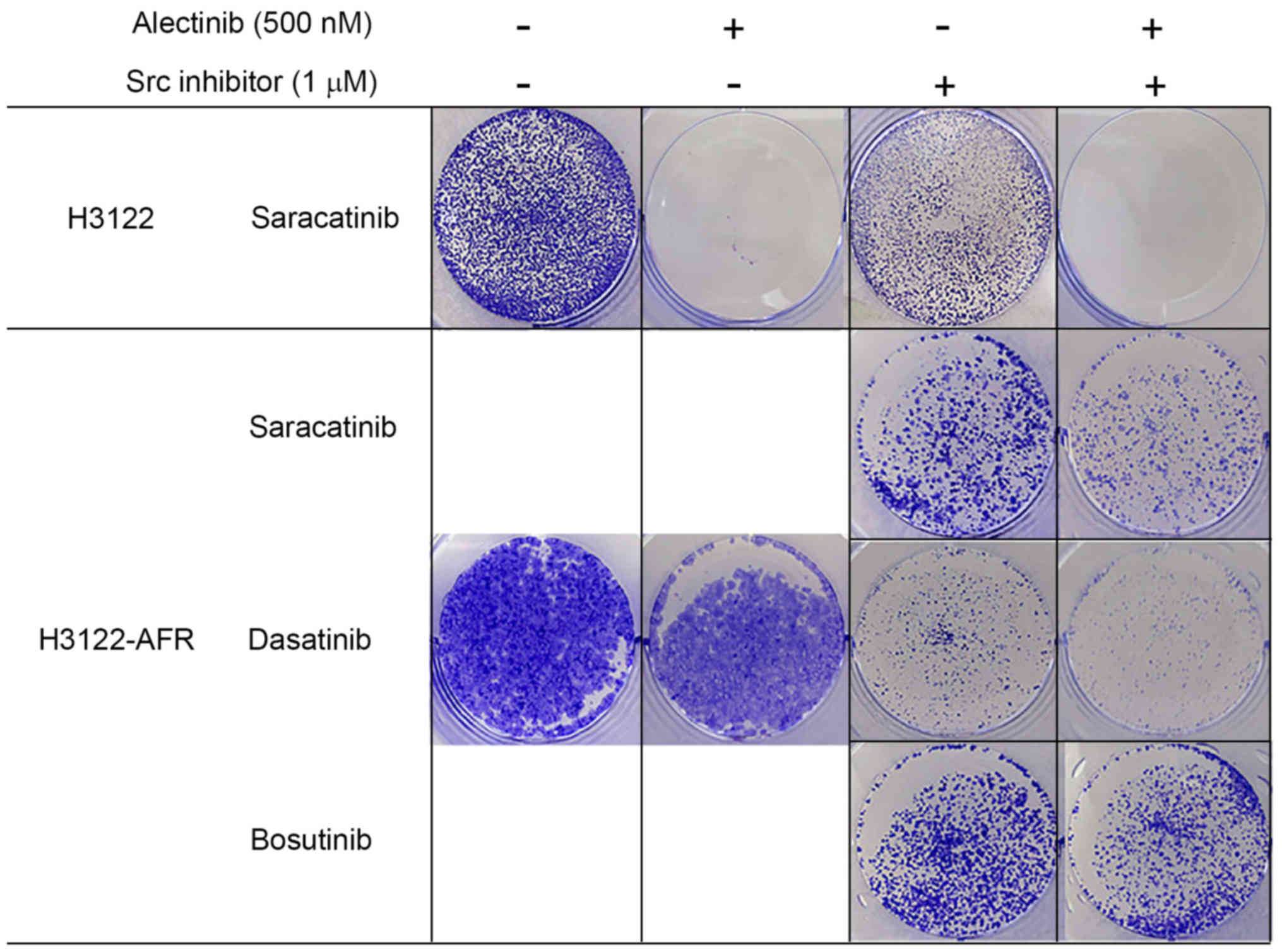
Activation of Src signaling mediates
acquired resistance to alectinib in H3122-AFR cells
Src was reported to confer resistance to crizotinib,
but not alectinib in a cell line-derived model and resistant cancer
cell lines derived from ALK TKI-resistant patients (11).
We hypothesized that the Src protein could be a
therapeutic target in H3122-AFR. To test this hypothesis, Src
phosphorylation was inhibited with the Src inhibitor, saracatinib.
In colony formation assays, the growth of H3122-AFR cells was
inhibited more potently by combined treatment with alectinib and
saracatinib, compared with alectinib or saracatinib alone (Fig. 3). When H3122-AFR cells were treated
with the other Src inhibitors, dasatinib or bosutinib, tumor growth
was also inhibited with each compound alone (Fig. 3). These in vitro data are
consistent with the therapeutic potential of Src inhibitors for
combatting resistance to ALK inhibitors.
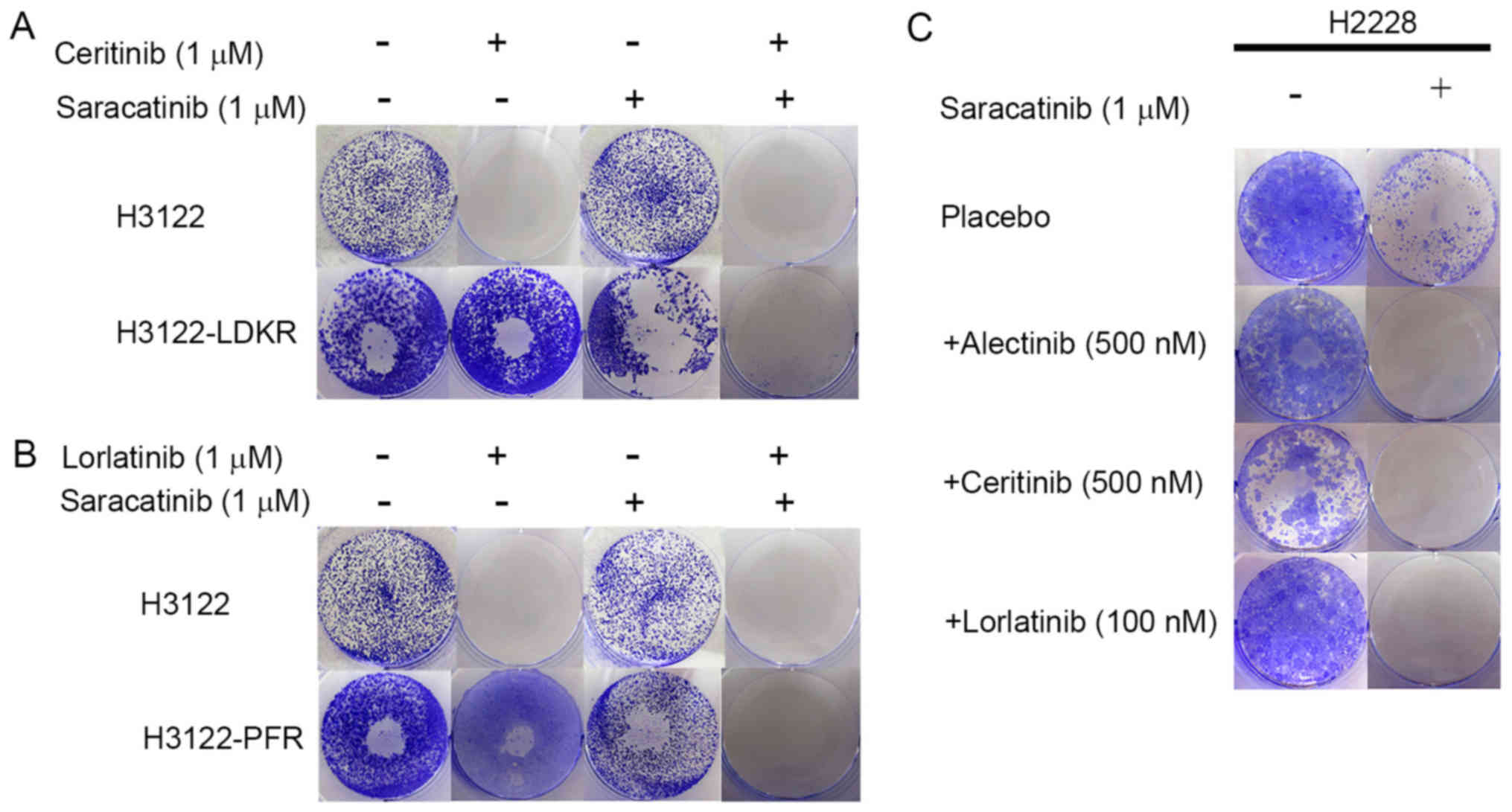
Dual blockade of ALK and phospho-Src in
other ALK resistant cells
To study whether activation of Src signaling is a
mechanism of acquired resistance to ceritinib or lorlatinib, we
employed colony formation assays to confirm the combined effects of
each ALK inhibitor and saracatinib. In the H3122-LDKR and H3122-PFR
cell lines, dual blockade of ALK and phosphor-Src inhibited tumor
growth more effectively compared with each ALK inhibitor alone, or
saracatinib alone (Fig. 4A and
B).
The H2228 cell line is resistant to ALK TKIs
compared with the H3122 cell line due to pre-existing activation of
EGFR signaling (data not shown). Colony formation assays showed
that combined treatment with each ALK inhibitor and saracatinib
effectively inhibited H2228 cell growth (Fig. 4C). This result showed that the
combined use of ALK and Src inhibitors could be effective in
different contexts of ALK inhibitor resistance.
Inhibition of Src overcomes resistance by
inhibition of alternative RTKs in H3122-AFR cells
Previous reports showed that Src is a common node
downstream of multiple resistance pathways (19,20).
We confirmed that H3122-AFR cells had higher phosphorylation levels
of Src, paxillin and crk II, as well as crkL, P130Cas and FAK
compared with H3122 by western blotting (data not shown). We
assessed the roles of these Src pathway related proteins in the
development of ALK inhibitor resistance by studying the combined
effect of alectinib and saracatinib on Src signaling in H3122-AFR
cells by western blotting. When tumors were treated with
saracatinib alone, the phosphorylation levels of Src pathway
proteins decreased (Fig. 5A). We
also found that EGFR phosphorylation was decreased following
treatment with saracatinib alone. On the other hand, ALK
phosphorylation levels decreased in response to alectinib alone.
Combined treatment with alectinib and saracatinib induced a
reduction in AKT phosphorylation (Fig.
5B). In addition, we confirmed that phospho-RTK activity was
higher in untreated H3122-AFR cells compared with H3122 cells, and
RTK activity decreased in H3122-AFR cells in response to
saracatinib alone (Fig. 5C).
Next we analyzed phospho-RTK signaling in H3122 and
H3122-AFR cells using human phospho-RTK arrays. These data showed a
decrease in ALK phosphorylation in H3122-AFR cells, and an increase
in the phosphorylation of multiple RTKs in H3122-AFR cells compared
with H3122 cells (data not shown). In addition, there was a
decrease in the phosphorylation of multiple RTKs, especially the
members of EGFR/HER, insulin receptor (IR) and HGFR families in
H3122-AFR cells after combined treatment with alectinib and
saracatinib (Fig. 5D).
We also carried out two-color immunofluorescence
staining to study the effects of combined ALK and Src inhibition on
H3122-AFR cells. These data addressed the expression of
phospho-tyrosine proteins and total Src in H3122-AFR cells, and
also revealed the co-localization of phospho-tyrosine proteins and
Src. Double immunofluorescence staining showed markedly diminished
phospho-RTK and Src signals in H3122-AFR cells after combined
treatment with alectinib and aaracatinib (Fig. 5E). Taken together, these data
indicate that Src inhibition overcomes alectinib resistance in
H3122-AFR cells in vitro.
Combined effects of alectinib and
saracatinib in H3122-AFR xenograft models
To evaluate the combined effect of alectinib and
saracatinib in vivo, H3122-AFR xenograft tumors were
established in nude mice. The combined treatment with alectinib and
saracatinib resulted in significant tumor regression compared with
treatment with each compound alone (Fig. 6A). During the treatment, body
weight or individual appearance were not changed. At 22 days after
treatment, when the average size of tumors in the control group
exceeded 500 mm3, the percent change in tumor volume was
significantly decreased by combined treatment, while the volume of
some tumors increased in response to monotherapy (Fig. 6B).
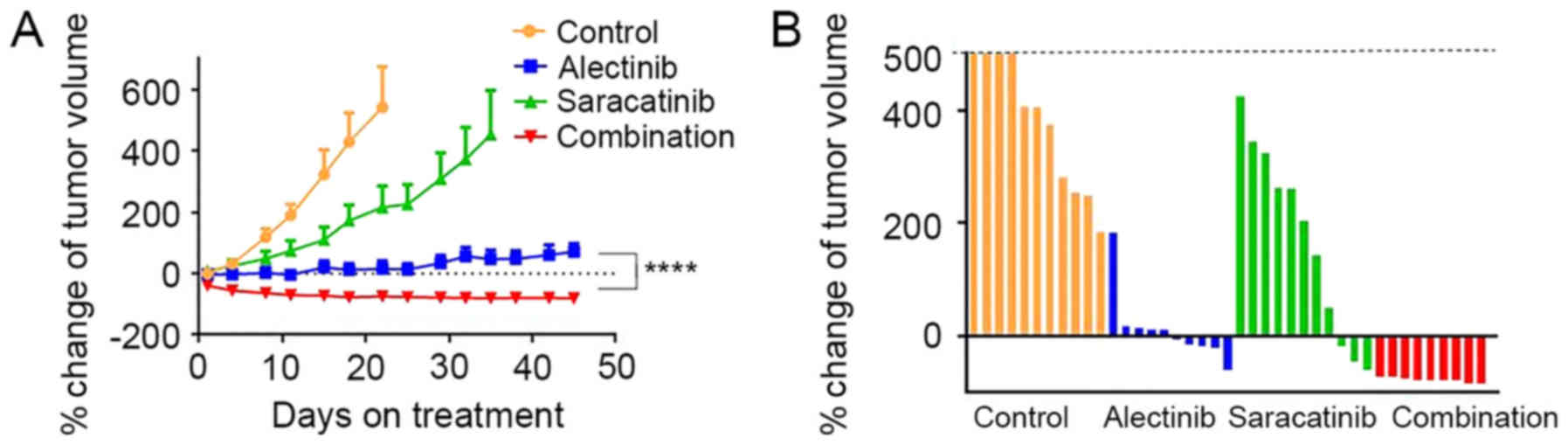 | Figure 6The combined effect of alectinib and
saracatinib treatment in H3122-AFR xenograft models. (A) Nude mice
bearing H3122-AFR xenografts were treated with vehicle only
(Control; yellow, n=11), alectinib (blue, 20 mg/kg, n=10),
saracatinib (green, 50 mg/kg, n=11), combination (red, alectinib 20
mg/kg + saracatinib 50 mg/kg, n=9) for the indicated number of
days. Average percent change in tumor volume relative to initial
tumor volume is shown. The measurements are presented as means ±
SEM. ****P<0.0001 for alectinib vs. combination
treatment group. (B) Percent change in tumor volume at 22 days
after treatment for the individual tumors in each treatment
group. |
Discussion
We identified the activation of Src signaling as a
mechanism of acquired resistance to ALK inhibitors in NSCLC cells.
Our data also showed that RTK activation and downstream PI3K/AKT
signaling can be effectively blocked by inhibiting Src.
The activation of alternative survival pathways is a
common mechanism driving the development of drug resistance in
cancer cells. Indeed, the activation of Src is involved in the
resistance to the crizotinib used for the treatment of NSCLC and
other cancers (11).
iTRAQ-based quantitative proteomic analysis has
contributed to the discovery of novel therapeutic targets for
cancer (21,22) and recently An et al
(12) identified CrkL activation
as an NSCLC resistance mechanism by proteomics analysis. However,
the role of Src activation in acquired resistance to alectinib,
ceritinib and lorlatinib has not been previously reported. We used
iTRAQ proteomics and Hsp90-inhibitors to identify chaperone
peptides/proteins in ALK resistant cell lines. The screening
results revealed that the focal adhesion pathway, involving
paxillin and crkII activation, could be involved in ALK resistance.
Previous reports identified interactions between Src and proteins
in the focal adhesion pathway (23,24)
and in the present study, ALK resistant cells had higher
phosphorylation levels of Src-related proteins suggesting that this
pathway could be a therapeutic target in ALK resistant cell
lines.
Previous studies demonstrated that Src interacts
with multiple RTKs and facilitates activation of downstream
pathways (25–28). In this study, combined treatment
with ALK and Src inhibitors effectively reduced PI3K/AKT mediated
survival and proliferation signaling. We showed that dual blockade
of ALK and Src inhibited alternative RTKs which were involved in
ALK resistance in H3122-AFR cells. Activation of EGFR, IR and HGFR
signaling are known mechanisms of resistance to alectinib (29,30).
Phospho-RTK arrays and western blotting results confirmed that
these bypass signaling pathways were active in H3122-AFR cells
compared to H3122 cells. Activation of these alternative survival
pathways is likely to be primarily responsible for mediating ALK
resistance in H3122-AFR cells because secondary ALK mutations and
ALK amplification were not identified in this cell line.
Phospho-RTK arrays also showed that combined
treatment with ALK and Src inhibitors led to decreased
phosphorylation of multiple RTKs. Although the levels of
phospho-RTK inhibition were not dramatic, the efficacy of combined
treatment was greater than the efficacy of ALK or Src inhibitors
alone in vitro and in vivo. These results suggest
that Src is a common node downstream of multiple resistance
pathways in ALK-NSCLCs, which is consistent with previous
investigations of RET rearranged NSCLCs and breast cancer (19,20).
Although it is important to validate our findings
using clinical specimens, our data suggest that targeting Src
signaling may be an effective approach for the treatment of
ALK-NSCLCs that are resistant to ALK inhibitors.
Acknowledgments
The present study was supported by a Grant-in-Aid
for Young Scientists [(B) 26860595] from the Ministry of Education,
Culture, Sports, Science and Technology.
References
|
1
|
Lee YJ, Kim JH, Kim SK, Ha SJ, Mok TS,
Mitsudomi T and Cho BC: Lung cancer in never smokers: Change of a
mindset in the molecular era. Lung Cancer. 72:9–15. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oxnard GR, Binder A and Jänne PA: New
targetable oncogenes in non-small-cell lung cancer. J Clin Oncol.
31:1097–1104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koivunen JP, Mermel C, Zejnullahu K,
Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D and
Thomas R: EML4-ALK fusion gene and efficacy of an ALK kinase
inhibitor in lung cancer. Clin Cancer Res. 14:4275–4283. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwak EL, Bang YJ, Camidge DR, Shaw AT,
Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seto T, Kiura K, Nishio M, Nakagawa K,
Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, et
al: CH5424802 (RO5424802) for patients with ALK-rearranged advanced
non-small-cell lung cancer (AF-001JP study): A single-arm,
open-label, phase 1–2 study. Lancet Oncol. 14:590–598. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shaw AT and Engelman JA: Ceritinib in
ALK-rearranged non-small-cell lung cancer. N Engl J Med.
370:2537–2539. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katayama R, Shaw AT, Khan TM,
Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT,
Benes C, et al: Mechanisms of acquired crizotinib resistance in
ALK-rearranged lung cancers. Sci Transl Med. 4:120ra172012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaguchi N, Lucena-Araujo AR, Nakayama S,
de Figueiredo-Pontes LL, Gonzalez DA, Yasuda H, Kobayashi S, Costa
DB and Dual ALK: Dual ALK and EGFR inhibition targets a mechanism
of acquired resistance to the tyrosine kinase inhibitor crizotinib
in ALK rearranged lung cancer. Lung Cancer. 83:37–43. 2014.
View Article : Google Scholar :
|
|
10
|
Niederst MJ and Engelman JA: Bypass
mechanisms of resistance to receptor tyrosine kinase inhibition in
lung cancer. Sci Signal. 6:re62013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crystal AS, Shaw AT, Sequist LV, Friboulet
L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A,
Greninger P, et al: Patient-derived models of acquired resistance
can identify effective drug combinations for cancer. Science.
346:1480–1486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
An R, Wang Y, Voeller D, Gower A, Kim IK,
Zhang YW and Giaccone G: CRKL mediates EML4-ALK signaling and is a
potential therapeutic target for ALK-rearranged lung
adenocarcinoma. Oncotarget. 7:29199–29210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sang J, Acquaviva J, Friedland JC, Smith
DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, et
al: Targeted inhibition of the molecular chaperone Hsp90 overcomes
ALK inhibitor resistance in non-small cell lung cancer. Cancer
Discov. 3:430–443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Katayama R, Khan TM, Benes C, Lifshits E,
Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA and Shaw
AT: Therapeutic strategies to overcome crizotinib resistance in
non-small cell lung cancers harboring the fusion oncogene EML4-ALK.
Proc Natl Acad Sci USA. 108:7535–7540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sequist LV, Gettinger S, Senzer NN,
Martins RG, Jänne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R,
Hafeez N, et al: Activity of IPI-504, a novel heat-shock protein 90
inhibitor, in patients with molecularly defined non-small-cell lung
cancer. J Clin Oncol. 28:4953–4960. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tonack S, Jenkinson C, Cox T, Elliott V,
Jenkins RE, Kitteringham NR, Greenhalf W, Shaw V, Michalski CW,
Friess H, et al: iTRAQ reveals candidate pancreatic cancer serum
biomarkers: Influence of obstructive jaundice on their performance.
Br J Cancer. 108:1846–1853. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Z, Sasaki T, Tan X, Carretero J,
Shimamura T, Li D, Xu C, Wang Y, Adelmant GO, Capelletti M, et al:
Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung
adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res.
70:9827–9836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ross PL, Huang YN, Marchese JN, Williamson
B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et
al: Multiplexed protein quantitation in Saccharomyces cerevisiae
using amine-reactive isobaric tagging reagents. Mol Cell
Proteomics. 3:1154–1169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S, Huang WC, Li P, Guo H, Poh SB,
Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, et al: Combating
trastuzumab resistance by targeting SRC, a common node downstream
of multiple resistance pathways. Nat Med. 17:461–469. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang CW, Jang KW, Sohn J, Kim SM, Pyo KH,
Kim H, Yun MR, Kang HN, Kim HR, Lim SM, et al: Antitumor activity
and acquired resistance mechanism of dovitinib (TKI258) in
RET-rearranged lung adenocarcinoma. Mol Cancer Ther. 14:2238–2248.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subbannayya Y, Syed N, Barbhuiya MA, Raja
R, Marimuthu A, Sahasrabuddhe N, Pinto SM, Manda SS, Renuse S,
Manju HC, et al: Calcium calmodulin dependent kinase kinase 2 - a
novel therapeutic target for gastric adenocarcinoma. Cancer Biol
Ther. 16:336–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu J, Li L, Yu G, Ying W, Gao Q, Zhang W,
Li X, Ding C, Jiang Y, Wei D, et al: The neddylation-cullin 2-RBX1
E3 ligase axis targets tumor suppressor RhoB for degradation in
liver cancer. Mol Cell Proteomics. 14:499–509. 2015. View Article : Google Scholar :
|
|
23
|
Schlaepfer DD and Hunter T: Evidence for
in vivo phosphorylation of the Grb2 SH2-domain binding site on
focal adhesion kinase by Src-family protein-tyrosine kinases. Mol
Cell Biol. 16:5623–5633. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schlaepfer DD and Mitra SK: Multiple
connections link FAK to cell motility and invasion. Curr Opin Genet
Dev. 14:92–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bromann PA, Korkaya H and Courtneidge SA:
The interplay between Src family kinases and receptor tyrosine
kinases. Oncogene. 23:7957–7968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parsons JT and Parsons SJ: Src family
protein tyrosine kinases: Cooperating with growth factor and
adhesion signaling pathways. Curr Opin Cell Biol. 9:187–192. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Biscardi JS, Maa MC, Tice DA, Cox ME, Leu
TH and Parsons SJ: c-Src-mediated phosphorylation of the epidermal
growth factor receptor on Tyr845 and Tyr1101 is associated with
modulation of receptor function. J Biol Chem. 274:8335–8343. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Belsches-Jablonski AP, Biscardi JS, Peavy
DR, Tice DA, Romney DA and Parsons SJ: Src family kinases and HER2
interactions in human breast cancer cell growth and survival.
Oncogene. 20:1465–1475. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Isozaki H, Ichihara E, Takigawa N, Ohashi
K, Ochi N, Yasugi M, Ninomiya T, Yamane H, Hotta K, Sakai K, et al:
Non-small cell lung cancer cells acquire resistance to the ALK
inhibitor alectinib by activating alternative receptor tyrosine
kinases. Cancer Res. 76:1506–1516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tani T, Yasuda H, Hamamoto J, Kuroda A,
Arai D, Ishioka K, Ohgino K, Miyawaki M, Kawada I, Naoki K, et al:
Activation of EGFR bypass signaling by TGFα overexpression induces
acquired resistance to alectinib in ALK-translocated lung cancer
cells. Mol Cancer Ther. 15:162–171. 2016. View Article : Google Scholar
|