Introduction
Focal adhesion kinase 1 (FAK1) is a non-receptor
tyrosine kinase encoded by the protein tyrosine kinase 2
(PTK2) gene and a multifunctional regulator of cell
signaling for diverse cellular processes, including cell migration,
growth factor signaling, cell cycle progression and cell survival
(1,2). FAK1 and proline-rich tyrosine kinase
2 (PYK2, a.k.a. FAK2) are of the FAK family, and intensive studies
of FAK1 have demonstrated its role in tumor malignancy. FAK1 was
initially identified as a protein tyrosine kinase that is
phosphorylated in response to local cell attachment, and it was
recently found to be functionally phosphorylated at Tyr residues
397, 576 and 577 via interactions with integrins or other growth
factor receptors (1–3). In particular, the phosphorylation of
Tyr397 is known to lead to the binding and activation of SH2 domain
proteins, such as Src family proteins, phosphoinositide 3-kinase,
phospholipase C-γ, Grb7 and SH3 domain proteins, to transduce the
signals for cell adhesion/migration, actin polymerization, and cell
survival (4,5).
In the tumor microenvironment, FAK1 is known to
promote the tumor progression and metastasis by controlling the
cell movement, invasion, survival and gene expression for the
epithelial-to-mesenchymal transition (6). In fact, data from the Cancer Genome
Atlas database show that the expression of PTK2 mRNA is
elevated in approximately 37% of serious ovarian tumors and 26% of
invasive breast cancers, and these increased levels are correlated
with poor overall survival (7–9). In
addition, FAK1 is reported to be overexpressed in many malignant
tumors, including non-small cell lung cancers, exerting an
oncogenic effect (10,11). These reports imply that FAK1 is
highly associated with tumor cell development and malignancy and
that the inhibition of FAK1 activity may be an effective
therapeutic approach for inhibiting the growth and metastasis of
tumor cells.
Currently, small molecule FAK1 inhibitors such as
TAE226, PF-562, 271 and GSK-2256098 are being developed as
promising chemotherapeutic agents to prevent tumor growth,
metastasis, vascular permeability and angiogenesis in mouse models
(12–21). Among these inhibitors, TAE226
appears to inhibit FAK1 (IC50, 5.5 nM) as well as other
protein tyrosine kinases such as PYK2, insulin receptor,
insulin-like growth factor-I receptor and c-Met (21). TAE226 suppresses cell proliferation
and invasion and promotes cell death in glioma and ovarian tumor
models (17,22,23).
In combination with docetaxel, a microtubule stabilizer, TAE226
significantly decreases angiogenesis and invasion in ovarian cancer
(17,22,23).
In our previous study, we synthesized a new small molecule
inhibitor of FAK1, 2-[2-(2-methoxy-4-morpholin-4-yl-phenylamino)-5-trifluoromethyl-pyrimidin-4-ylamino]-N-methyl-benzamide
(hereafter abbreviated as MPAP), which blocks the phosphorylation
of Tyr397 by modifying a functional group on the bis-anilino
pyrimidine moiety of TAE226, increasing hydrophobic interactions
with the side-chain of the gatekeeper residue Met499. This
inhibitor exhibits potent kinase inhibition of FAK1, with an
IC50 value of 4.8 nM, which is lower than that of TAE226
(24). Therefore, in this study,
we further investigated the antitumor activity of the novel FAK1
inhibitor, MPAP, on lung cancers as well as determined its effect
when combined with radiation, which is the standard treatment for
patients with lung cancer.
Materials and methods
Cell lines and cell culture
Human lung carcinoma cell lines (A549 and H1299) and
human normal lung fibroblast cell lines (IMR90 and WI38) were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). All cell lines were cultured in RPMI-1640 (Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS; Life Technologies) and maintained at 37°C in a
humidified 5% CO2 atmosphere.
Irradiation (IR)
Cells were uniformly irradiated at room temperature
with various doses of a 137Cs γ-source (Gammacell 3000
ELan; MDS Nordion, Ottawa, ON, Canada) at a dose rate of 3.25
Gy/min. Cells of the control group were simultaneously exposed to
sham IR. Tumor xenograft mice were irradiated with a
60Co γ-source using the Theratron 780 (MDS Nordion)
radiotherapy unit when the tumor volume reached ~100–200
mm3. During IR, mice were covered with a 0.5-cm bolus
and exposed to a single dose of 5 Gy at a dose rate of 0.5296
Gy/min.
MTT assay
Cells (1×106) were seeded in 6-well
plates and incubated for 24 h after various treatments. MPAP was
purchased from 4Chem Laboratory Co., Ltd. (Suwon, Korea). Then, 5
mg/ml of MTT was added to the cell culture medium, and cells were
further incubated for 2–4 h. After the supernatant was removed,
dimethyl sulfoxide (DMSO) was added to the cells and mixed by
shaking for 15 min. Cell viability was determined by measuring the
absorbance at 570 nm using a microplate reader.
Colony formation assay
Cells (500/well) were seeded in a 6-well plate and
cultured under various conditions for 12 days. Cells were fixed and
stained with 1% methylene blue in absolute methanol solution for 10
min. Colony density was quantified by densitometry.
Cell cycle analysis
Cells were harvested and then fixed in 75% ethanol
for 1 h. Cells were then washed with phosphate-buffered saline
(PBS) and resuspended in PBS buffer containing 100 mg/ml RNase A
(Sigma-Aldrich, Darmstadt, Germany) and 50 mg/ml propidium iodide
(PI; Life Technologies), followed by incubation at 37°C for 30 min.
The percentage of cells in each cell cycle stage (G1/G0, S or G2/M)
was analyzed with a FACSCalibur cell analyzer (BD Biosciences, San
Jose, CA, USA) and FlowJo software version 10.1r5.
Western blot analysis
Cells were harvested and washed with ice-cold PBS.
Whole cell lysates were prepared using 2X SDS/Sample buffer.
Samples were then separated by 8–12% SDS-PAGE, transferred to a
nitrocellulose membrane, and probed with the indicated antibodies,
followed by ECL detection. The antibodies used in the present study
were FAK1 (D2R2E; Cell Signaling Technology, Danvers, MA, USA),
pFAK1 (T397) (D20B1; Cell Signaling Technology), pFAK1 (T576/577)
(Cell Signaling Technology), pFAK1 (T925) (Cell Signaling
Technology), pSrc (T416) (Cell Signaling Technology), RB (4H1; Cell
Signaling Technology), phospho-RB (S807/811) (D20B12; Cell
Signaling Technology), p53 (DO-1, Santa Cruz Biotechnology, Dallas,
TX, USA), p21 (12D1; Cell Signaling Technology), cyclin D1 (92G2;
Cell Signaling Technology), CDK4 (K9G3E; Cell Signaling Technology)
and β-actin (Sigma-Aldrich).
Xenograft tumor model
Mice were maintained in an animal facility, and all
animal experiments were performed according to the Korean National
Guidelines of Laboratory Animal Experiments; protocols were
approved by the Institutional Animal Care and Use Committee of the
Korea Institute of Radiological and Medical Sciences. BALB/c nude
mice (CAnN.Cg-Foxn1nu/CrljOri, 6 weeks old,
female) were purchased from Orient Bio Inc. (Gyunggi-do, Korea).
A549 cells (1×106) were injected subcutaneously into
BALB/c nude mice. When the tumor volume reached an average of
100–200 mm3, MPAP was intraperitoneally introduced once
at 10 mg/kg with or without IR at a dose of 5 Gy. Tumor size was
measured periodically 2–3 times per week. Tumor volume was
calculated according to following formula: Tumor volume
(mm3) = (width)2 × length/2.
Statistical analysis
Statistical significance was determined by the
analysis of variance (ANOVA). Non-parametric analyses using the
Tukey's multiple comparison test were used when appropriate. A
P<0.05 was considered significant. Data are presented as means ±
SD (standard deviation) of three independent experiments.
Results
A new FAK1 inhibitor, MPAP, decreases the
viability of lung cancer cells
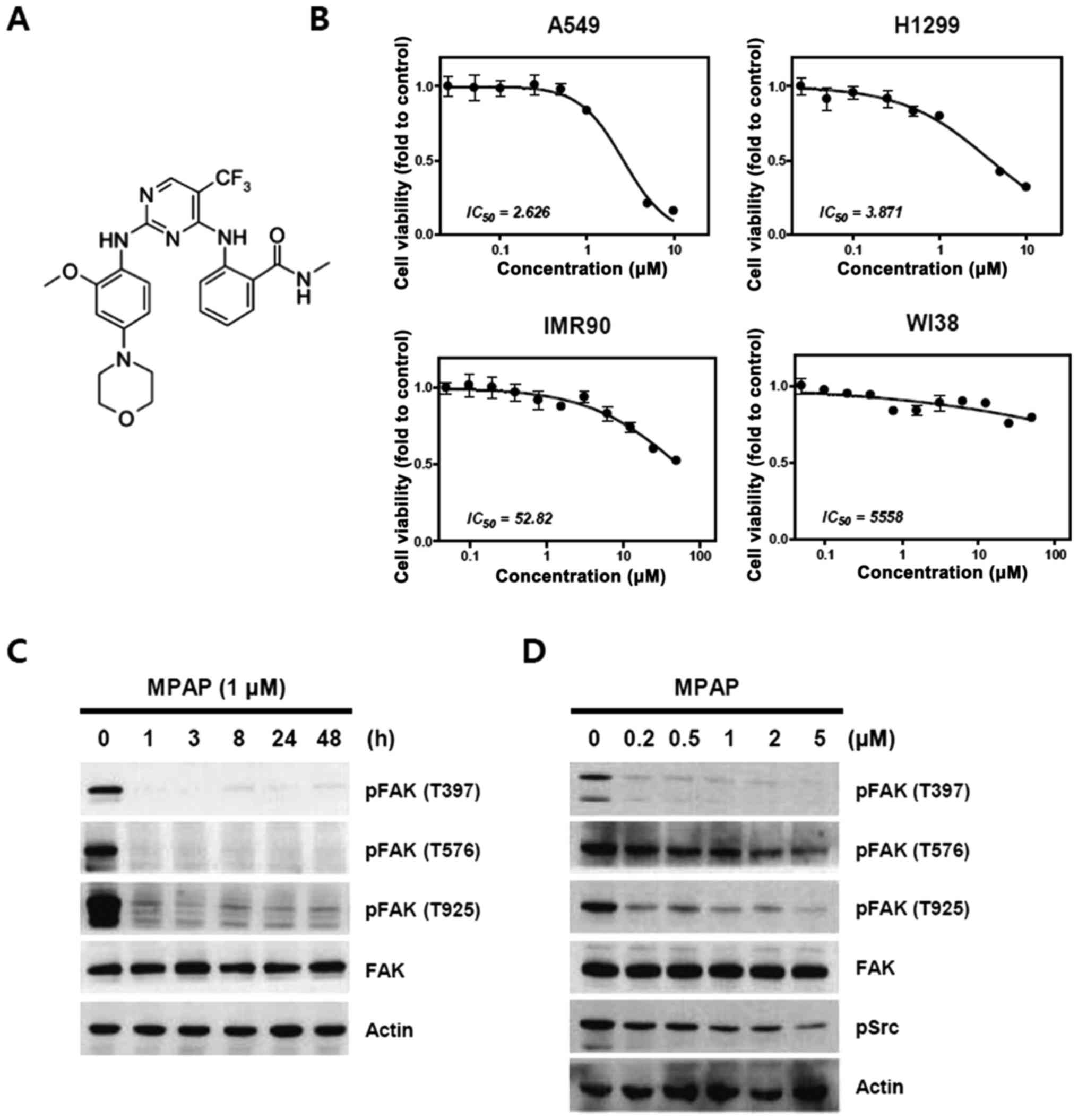
MPAP was synthesized by modifying the functional
group of TAE226 on the bis-anilino pyrimidine moiety with CF3, as
described in a previous report (24) (Fig.
1A). First, we investigated whether MPAP reduces the activation
state of the FAK1 protein and suppresses the growth of lung cancer
cells. Treatment with MPAP sharply decreased the viability of A549
and H1299 lung cancer cells, with above 10- to 1000-fold higher
sensitive than the viability of normal lung fibroblast cells
(Fig. 1B). The IC50
value of MPAP on A549 and H1299 cancer cell viability is 2.626 and
3.871 μM, respectively. We then investigated the treatment
time and dose of MPAP required to inhibit FAK efficiently for
further experiments. As shown in Fig.
1C, 1 μM MPAP markedly
inhibited the phosphorylation of FAK1 at tyrosine residues 397,
576/577 and 925, and this lasted for 48 h; in contrast, expression
levels of FAK1 were not altered. In addition, FAK1 phosphorylation
was also clearly inhibited by MPAP even at low concentrations (0.2
and 0.5 μM) that did not inhibit cell viability (Fig. 1D). Therefore, we applied the dose
0.2 μM in the subsequent experiment to examine the
combinatorial effect of MPAP with IR.
MPAP enhances radiation-induced
clonogenic inhibition in lung cancer cells
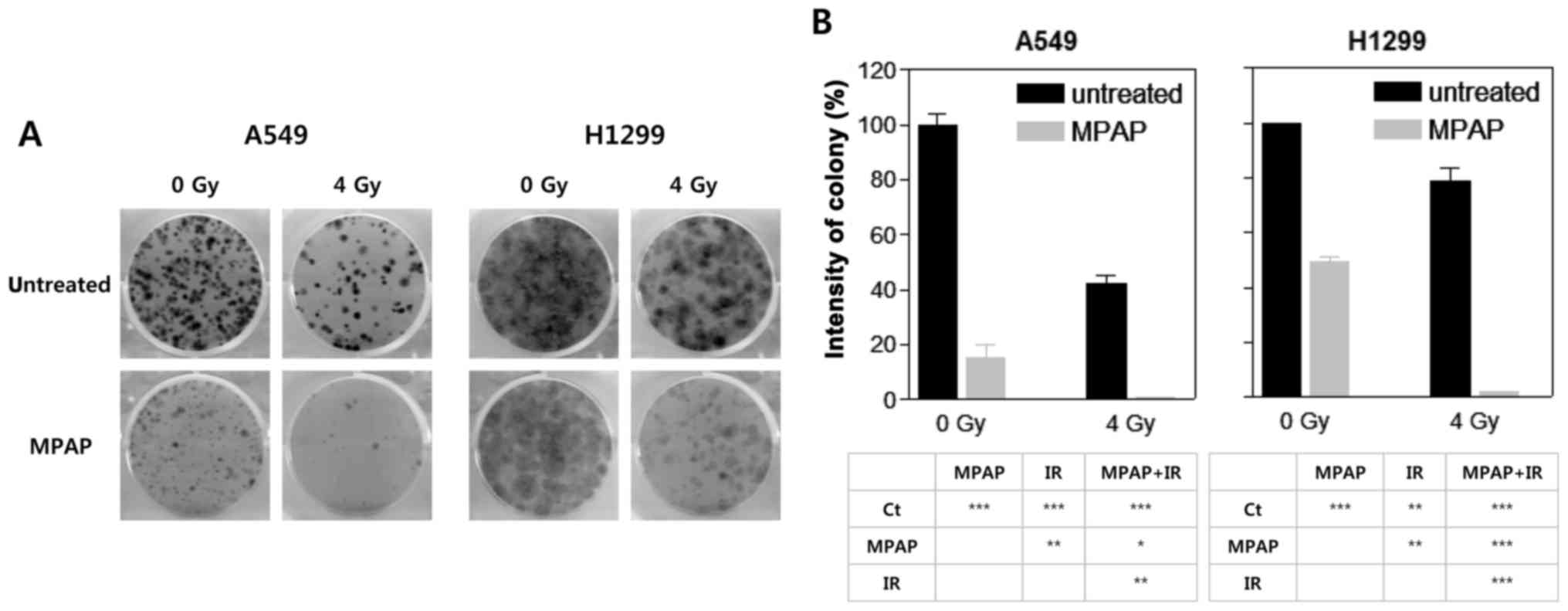
To determine the radiosensitizing potential of MPAP
in lung cancer cells, a colony formation assay was performed with
0.2 μM MPAP in the presence or absence of a 4-Gy dose of IR.
In this experiment, we used two different human lung cancer cell
lines, A549 (wild-type p53) and H1299 (p53-null). In A549 cells,
MPAP or IR alone markedly decreased colony formation, while
complete inhibition was observed with combined MPAP and IR
treatment, indicating that MPAP has a radiosensitizing effect
(Fig. 2A and B, left panel).
Similarly, MPAP or IR alone significantly decreased colony
formation in H1299 cells, but the extent of inhibition was lower
than that in A549 cells. Notably, MPAP combined with IR also
completely inhibited colony formation in H1299 cells (Fig. 2A and B, right panel), suggesting
that the combined effect of IR and MPAP was higher than that noted
in the case of MPAP alone. These data suggest that MPAP has a
radiosensitizing effect that is exhibited in a p53-independent
manner.
Combined MPAP and IR treatment induces G1
arrest
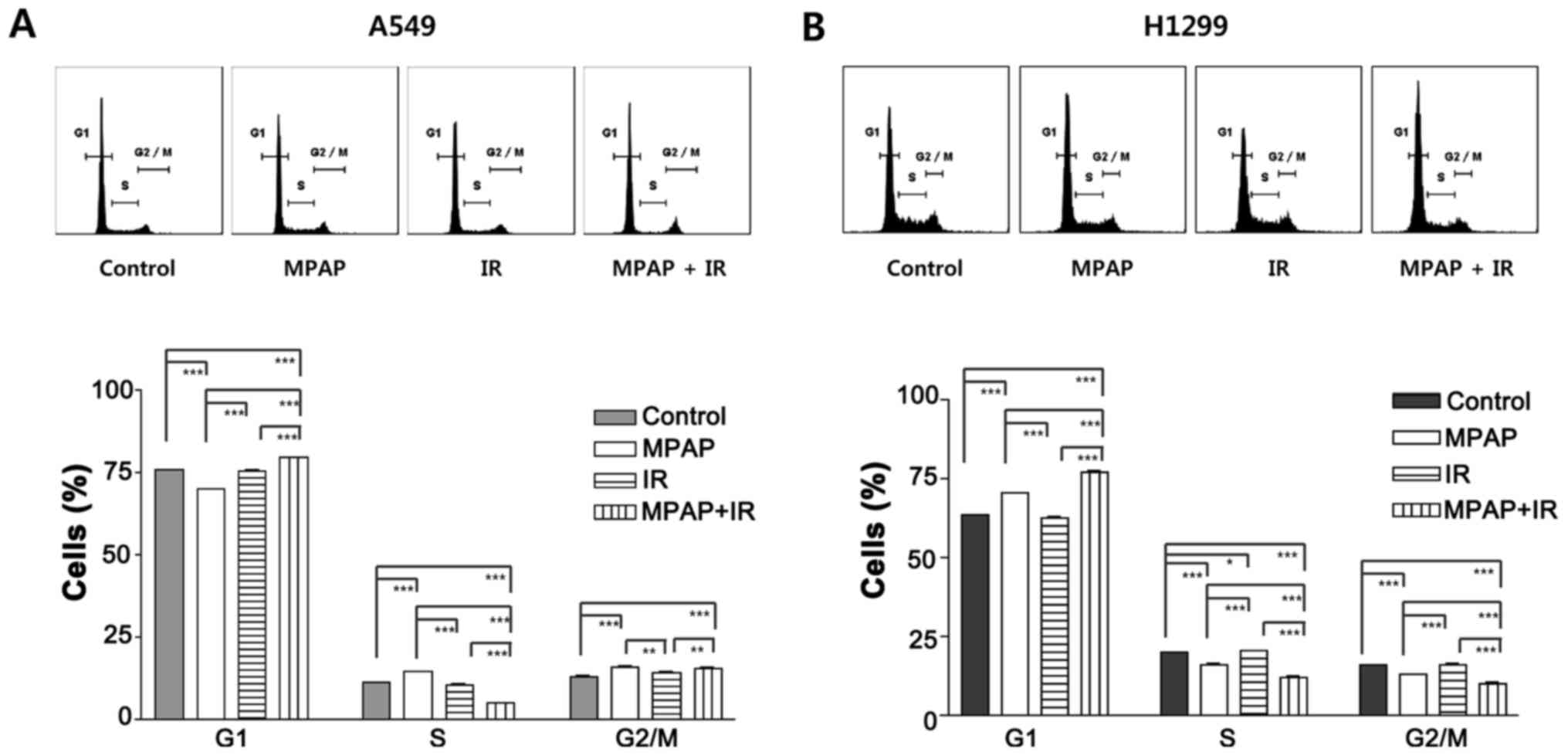
To further investigate how MPAP and IR suppress
tumor cell growth, we performed cell cycle analysis to assess cell
cycle arrest and apoptosis. Apoptosis was not observed in any of
the three treatments (MPAP, IR or combination) in either A549 or
H1299 cells; this is because we used low doses of MPAP and IR to
determine the combinatorial effect, avoiding direct toxicity. In
A549 cells, MPAP treatment slightly increased the percentages of
cells in the S and G2 phases, but IR alone did not affect the cell
cycle distribution (Fig. 3A). The
combination treatment of MPAP and IR significantly decreased the
percentage of S cells and arrested cells in the G1 and G2/M phases
(Fig. 3A). Unlike in A549 cells,
MPAP treatment clearly increased the G1 cell population in H1299
cells, while the combination treatment of MPAP and IR resulted in
an even stronger increase in the G1 cell population (Fig. 3B). These data suggest that combined
treatment with MPAP and IR significantly induces G1 cell cycle
arrest via a p53-independent pathway.
Decreased cyclin D1/CDK4/pRB induces
p53-independent G1 arrest by MPAP and IR
To determine which cell cycle regulators are
involved in MPAP/IR-mediated cell cycle perturbation, we performed
western blot analysis using antibodies for G1 cell cycle-related
proteins. Consistent with the results of Fig. 1C and D, MPAP successfully inhibited
the phosphorylation of FAK1 and decreased the levels of CDK4 and
cyclin D1, key regulators of the G1-to-S phase progression of the
cell cycle. The expression levels of cyclin D1 and CDK4 were
markedly increased by IR treatment of A549 cells with wild-type p53
but were diminished by combined treatment with MPAP and IR. In
addition, the expression of a major G1 checkpoint protein, RB, was
also decreased by IR and decreased to an even greater extent by
combined treatment with MPAP and IR in both cell lines. Levels of
phosphorylation of RB at serine residues 807 and 811 exhibited the
same pattern as RB expression (Fig.
4).
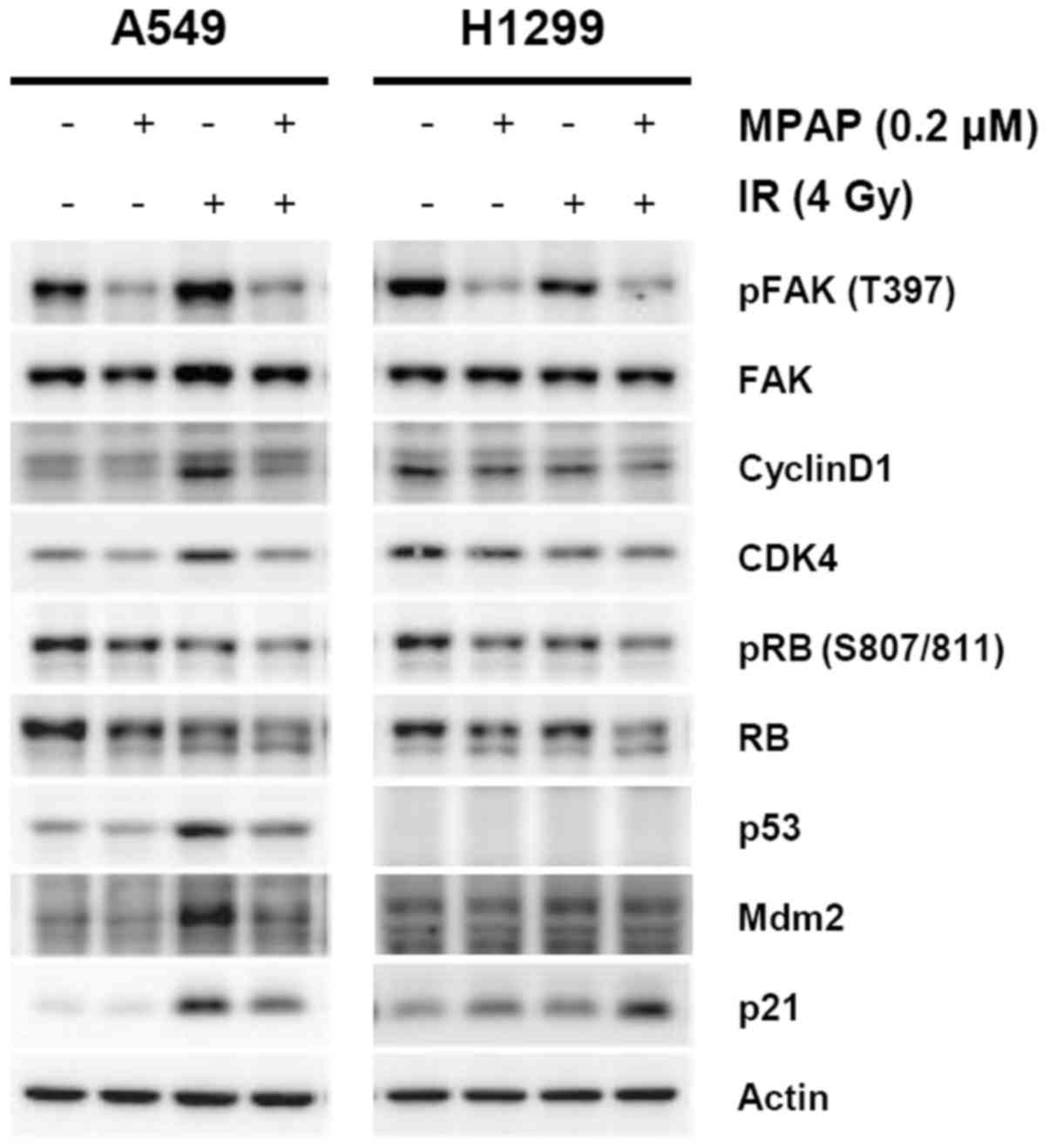 | Figure 4The regulation of cell cycle
regulators for G1/S progression by combined treatment with MPAP and
IR. A549 or H1299 cells were treated with 0.2 μM of MPAP for
3 h and exposed to 4 Gy of γ-radiation, followed by incubation for
24 h. Whole cell lysates were prepared, and levels of phospho-FAK1
(T397), FAK1, phospho-RB (S807/811), RB, p53, Mdm2, p21, cyclin D1,
CDK4 and actin proteins were determined by western blot
analysis. |
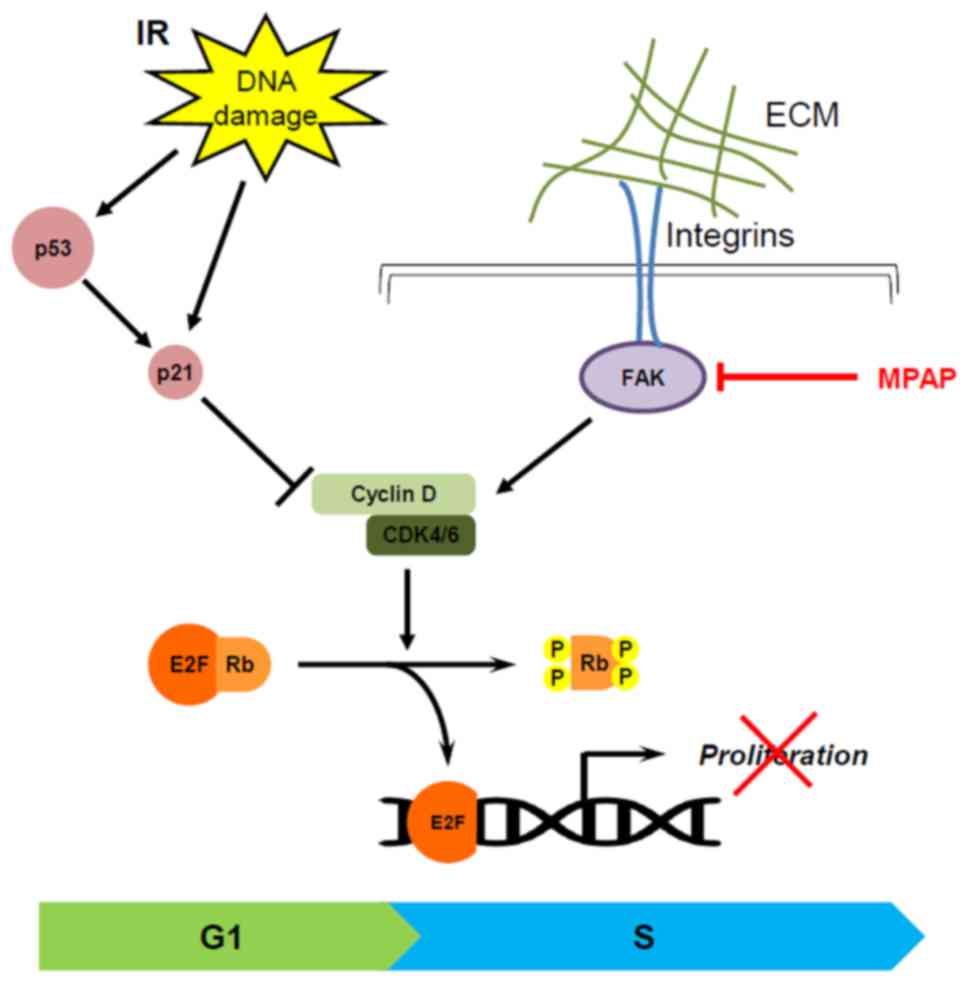
Considering that the p53/p21 pathway is a strong
regulator of G1 cell cycle arrest and an upstream regulator in the
cyclin D1/CDK4/RB signaling axis, levels of p53 and p21 proteins
were determined. The expression levels of p53 and p21 were
increased by IR in A549 cells. Notably, p21 expression was markedly
increased by the combination of MPAP and IR in H1299 p53-null
cells. These data suggest that MPAP/IR-mediated G1 cell cycle
arrest may be attributed to downregulation of the cyclin
D1/CDK4/pRB pathway rather than the p53/p21 pathway.
MPAP confers radiosensitizing effect on
xenograft tumor growth
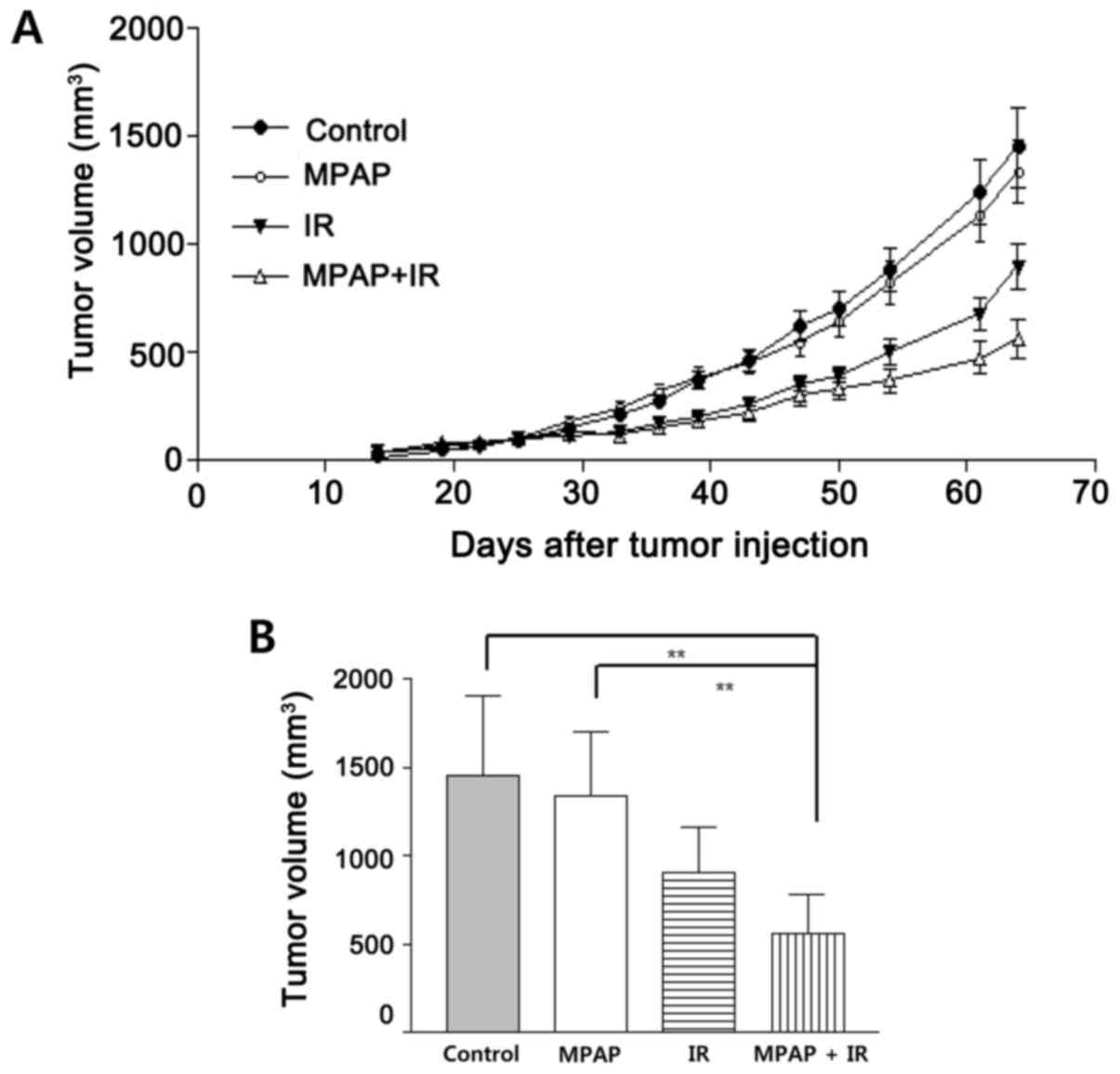
To assess the antitumor effect of MPAP with or
without radiation in vivo, we established xenografts of A549
cells in nude mice. As shown in Fig.
5, treatment with MPAP slightly reduced the tumor size, but
this was not statistically significant. However, 5 Gy of IR
resulted in significant tumor reduction. Combined treatment
suppressed tumor growth better than the IR alone. These data
suggest that MPAP efficiently contributes to radiation-mediated
tumor growth reduction.
Discussion
Generally, radiation therapy is regarded as the most
effective therapeutic approach for cancer treatment and it exhibits
some benefits when compared with chemotherapy (25). However, radioresistance and
recurrence are major limitations in the long-term survival of
patients undergoing radiation therapy (26). In addition, radiotherapy can cause
various side-effects, such as fatigue, nausea, vomiting, appetite
loss, emotional changes, hair loss, and in particular, tissue
fibrosis. Thus, a novel therapeutic approach that reduces these
side-effects would improve radiotherapy. As a possible approach, we
propose a combination treatment of a reduced dose of ionizing
radiation along with a radiosensitizer, such as MPAP. As we
mentioned earlier, several compounds that inhibit FAK activation
have been developed by pharmaceutical companies. The mother
compound of MPAP, TAE226 has been previously reported to possess
cytotoxic activity against cancer cells. However, TAE226 exhibited
radiosensitizing effects on only two out of seven glioblastoma cell
lines at 10 μM, and exhibited no such effects on lung,
pancreatic, or colorectal cancer cells (27,28).
Therefore, the advantages of MPAP are its lower IC50
value against FAK than that of TAE226, as well as its
radiosensitizing effect on lung cancer cells, which is not
exhibited by TAE226.
In this study, we showed that MPAP could inhibit
cancer cell proliferation and the phosphorylation of FAK1 at
Tyr397, Tyr576/577 and Tyr925 (Fig.
1). Moreover, the combination treatment of MPAP and IR
suppressed cancer cell proliferation in wild-type p53 cells, and
this effect was even higher in a p53-null background (Fig. 2). Several studies have demonstrated
that FAK1 inhibits the transcriptional activity of p53 through
protein-protein interactions, and that p53 also binds to the FAK1
promoter, inhibiting its activity. This feedback mechanism is
well-correlated with the overexpression or upregulation of FAK1 in
p53-mutated cancers (29).
Furthermore, mutation or deletion of the p53 gene is the most
frequent genetic change in human cancer. Therefore, combination
therapy with both a FAK1 inhibitor and radiation could represent a
significant advance in therapeutic strategy.
Radiation can induce apoptotic cell death or cell
cycle arrest to suppress tumor cell survival. Our data showed that
4 Gy of radiation decreased long-term clonogenic survival but did
not alter cell cycle progression according to short-term
analysis.
While the induction of cell cycle arrest by IR was
not specifically observed in this study, this could have simply
been undetected, owing to the fact that we did not perform a
time-kinetic analysis of cell cycle perturbations or investigate
the synchronization of cells. The function of MPAP per se on cell
cycle distributions differed in the two cell lines, leading to S
and G2/M arrest in A549 cells and G1 arrest in H1299 cells.
Although FAK inhibition has been reported to reduce the amount of
cyclin D1 and arrest cells in the G1/S phase (30), cell cycle accumulation in the G1, S
or G2 phases is dependent on cell type, radiation dose, harvest
time after IR, and the method of FAK inhibition, such as gene
deletion, ATP-binding site blocker or multi-kinase inhibitor. This
is supported by the finding that hepatocellular carcinoma cells
transduced with FAK shRNA exhibit G2/M arrest, whereas an inhibitor
of Tyr397 phosphorylation of FAK (PND-1186) induces G1 arrest under
the same conditions, suggesting that the signaling pathways
triggered by FAK kinase activity could be distinct from the
scaffolding function of FAK (31).
In addition, TAE226 induces G2 arrest in glioma cells and increases
apoptosis (23). However, MPAP
significantly enhanced G1 cell cycle arrest when used in
combination with IR in A549 cells, and did so more significantly in
H1299 cells (Fig. 3). G1/S cell
cycle progression is controlled by the phosphorylation of RB, and
the cyclin D1/CDK4 complex is responsible for this regulation
(32). Several studies have
demonstrated that integrin-mediated cell adhesion controls cell
cycle progression by regulating the expression levels and
activities of cyclins, CDKs and CDK inhibitors (33,34).
The activation of FAK1 by integrins accelerates the G1 to S-phase
cell cycle progression primarily by increasing cyclin D1 expression
directly at the transcriptional level in a p21-independent manner
(30). Consistent with previous
reports, the most distinctive changes were observed in RB and pRB
expression by MPAP/IR treatment in both cell lines. In addition,
the fact that the decrease in cyclin D1 and CDK4 expression
mediated by MPAP/IR was clearly shown in p53 null-type H1299 cells,
compared with the mild inhibition in p53 wild-type A549 cells
(Fig. 4). These results suggest
that MPAP/IR-mediated G1 arrest is independent of p53 status and
the alteration of the cyclin D1/CDK4/pRB axis could be involved in
MPAP-mediated radiosensitization. Under genotoxic conditions such
as IR, diverse DNA damage lesions trigger the activation of
multiple intracellular signaling pathways, resulting in cell cycle
arrest, DNA repair, or apoptosis. Numerous studies have
demonstrated that p53/p21 are involved in G1 as well as G2/M arrest
after irradiation, thereby sensitizing tumor cells to radiation
(35–37). As expected, IR markedly increased
levels of the p53 and p21 proteins in p53 wild-type cells, but the
induction of cyclin D1/cdk4 and reduction of pRB were concurrently
observed. These data suggest that diverse signaling pathways are
involved in the regulation of cell cycle progression. In p53-null
H1299 cells, p21 expression was markedly increased by the
combination treatment of MPAP and IR, and these increased levels of
p21 might participate in the G1 cell cycle arrest. The function of
p21 can confer sensitivity to IR, as permanent, replicative death
can result from prolonged G1 arrest (38). Even though p53 is not the only
transcription factor to regulate p21 expression, it remains unknown
how p21 protein levels are elevated under p53-null conditions, and
this is worth investigating in the future.
Finally, the radiosensitizing effect of MPAP was
determined in tumor-transplanted mice. Owing to the powerful
antitumor effect of MPAP itself at doses above 0.5 mg/kg, similar
to that of IR, we did not observe a synergistic effect of the
combination treatment of MPAP and IR (data not shown). When a low
dose of MPAP (10 μg/kg) was administered as a single dose
with IR, the tumor burden was more efficiently suppressed than by
either MPAP or IR alone (Fig. 5).
Overall, these results demonstrate that the combination treatment
of MPAP and IR can efficiently suppress lung tumor growth by the
induction of G1 arrest via the regulation of RB phosphorylation in
a p53-independent manner (Fig. 6).
Considering that the detailed mechanism of the radiosensitizing
effects of MPAP remains to be clarified, our results provide
insights for the development of an effective multi-targeted
approach to the treatment of malignant tumors that exhibit a
p53-null background.
Acknowledgments
The present study was supported by a grant from the
Korea Institute of Radiological and Medical Sciences (KIRAMS),
funded by Ministry of Science, ICT and Future Planning (grant nos.
1711045557, 1711045538, 1711045554 and 50531-2017), and in part by
grant no. 20131610101840 from the Ministry of Trade, Industry &
Energy, Republic of Korea.
References
|
1
|
Parsons JT: Focal adhesion kinase: The
first ten years. J Cell Sci. 116:1409–1416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitra SK, Hanson DA and Schlaepfer DD:
Focal adhesion kinase: In command and control of cell motility. Nat
Rev Mol Cell Biol. 6:56–68. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanks SK, Calalb MB, Harper MC and Patel
SK: Focal adhesion protein-tyrosine kinase phosphorylated in
response to cell attachment to fibronectin. Proc Natl Acad Sci USA.
89:8487–8491. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calalb MB, Polte TR and Hanks SK: Tyrosine
phosphorylation of focal adhesion kinase at sites in the catalytic
domain regulates kinase activity: A role for Src family kinases.
Mol Cell Biol. 15:954–963. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Owen JD, Ruest PJ, Fry DW and Hanks SK:
Induced focal adhesion kinase (FAK) expression in FAK-null cells
enhances cell spreading and migration requiring both auto- and
activation loop phosphorylation sites and inhibits
adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol.
19:4806–4818. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al Cancer
Genome Atlas Research Network: Integrated genomic analyses of
ovarian carcinoma. Nature. 474:609–615. 2011. View Article : Google Scholar
|
|
9
|
Sood AK, Armaiz-Pena GN, Halder J, Nick
AM, Stone RL, Hu W, Carroll AR, Spannuth WA, Deavers MT, Allen JK,
et al: Adrenergic modulation of focal adhesion kinase protects
human ovarian cancer cells from anoikis. J Clin Invest.
120:1515–1523. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Nimwegen MJ and van de Water B: Focal
adhesion kinase: A potential target in cancer therapy. Biochem
Pharmacol. 73:597–609. 2007. View Article : Google Scholar
|
|
11
|
Siesser PMF and Hanks SK: The signaling
and biological implications of FAK overexpression in cancer. Clin
Cancer Res. 12:3233–3237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ward KK, Tancioni I, Lawson C, Miller NL,
Jean C, Chen XL, Uryu S, Kim J, Tarin D, Stupack DG, et al:
Inhibition of focal adhesion kinase (FAK) activity prevents
anchorage-independent ovarian carcinoma cell growth and tumor
progression. Clin Exp Metastasis. 30:579–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen XL, Nam JO, Jean C, Lawson C, Walsh
CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, et al:
VEGF-induced vascular permeability is mediated by FAK. Dev Cell.
22:146–157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walsh C, Tanjoni I, Uryu S, Tomar A, Nam
JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, et al: Oral
delivery of PND-1186 FAK inhibitor decreases tumor growth and
spontaneous breast to lung metastasis in pre-clinical models.
Cancer Biol Ther. 9:778–790. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jean C, Chen XL, Nam JO, Tancioni I, Uryu
S, Lawson C, Ward KK, Walsh CT, Miller NL, Ghassemian M, et al:
Inhibition of endothelial FAK activity prevents tumor metastasis by
enhancing barrier function. J Cell Biol. 204:247–263. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cabrita MA, Jones LM, Quizi JL, Sabourin
LA, McKay BC and Addison CL: Focal adhesion kinase inhibitors are
potent antiangiogenic agents. Mol Oncol. 5:517–526. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Halder J, Lin YG, Merritt WM, Spannuth WA,
Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, et al:
Therapeutic efficacy of a novel focal adhesion kinase inhibitor
TAE226 in ovarian carcinoma. Cancer Res. 67:10976–10983. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stokes JB, Adair SJ, Slack-Davis JK,
Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton
AH, Hwang RF, et al: Inhibition of focal adhesion kinase by
PF-562,271 inhibits the growth and metastasis of pancreatic cancer
concomitant with altering the tumor microenvironment. Mol Cancer
Ther. 10:2135–2145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wendt MK and Schiemann WP: Therapeutic
targeting of the focal adhesion complex prevents oncogenic TGF-beta
signaling and metastasis. Breast Cancer Res. 11:R682009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Slack-Davis JK, Hershey ED, Theodorescu D,
Frierson HF and Parsons JT: Differential requirement for focal
adhesion kinase signaling in cancer progression in the transgenic
adenocarcinoma of mouse prostate model. Mol Cancer Ther.
8:2470–2477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoon H, Dehart JP, Murphy JM and Lim ST:
Understanding the roles of FAK in cancer: Inhibitors, genetic
models, and new insights. J Histochem Cytochem. 63:114–128. 2015.
View Article : Google Scholar :
|
|
22
|
Liu TJ, LaFortune T, Honda T, Ohmori O,
Hatakeyama S, Meyer T, Jackson D, de Groot J and Yung WK:
Inhibition of both focal adhesion kinase and insulin-like growth
factor-I receptor kinase suppresses glioma proliferation in vitro
and in vivo. Mol Cancer Ther. 6:1357–1367. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi Q, Hjelmeland AB, Keir ST, Song L,
Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS and Rich JN:
A novel low-molecular weight inhibitor of focal adhesion kinase,
TAE226, inhibits glioma growth. Mol Carcinog. 46:488–496. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nam KY, Jin DH, No KT and Ahn SK:
Discovery of FAK inhibitors using structure based drug design. Bull
Korean Chem Soc. 35:3156–3157. 2014. View Article : Google Scholar
|
|
25
|
Amin NP, Sher DJ and Konski AA: Systematic
review of the cost effectiveness of radiation therapy for prostate
cancer from 2003 to 2013. Appl Health Econ Health Policy.
12:391–408. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Begg AC, Stewart FA and Vens C: Strategies
to improve radiotherapy with targeted drugs. Nat Rev Cancer.
11:239–253. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hehlgans S, Lange I, Eke I and Cordes N:
3D cell cultures of human head and neck squamous cell carcinoma
cells are radiosensitized by the focal adhesion kinase inhibitor
TAE226. Radiother Oncol. 92:371–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Storch K, Sagerer A and Cordes N:
Cytotoxic and radiosensitizing effects of FAK targeting in human
glioblastoma cells in vitro. Oncol Rep. 33:2009–2016. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Golubovskaya VM and Cance WG: FAK and p53
protein interactions. Anticancer Agents Med Chem. 11:617–619. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao J, Pestell R and Guan JL:
Transcriptional activation of cyclin D1 promoter by FAK contributes
to cell cycle progression. Mol Biol Cell. 12:4066–4077. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gnani D, Romito I, Artuso S, Chierici M,
De Stefanis C, Panera N, Crudele A, Ceccarelli S, Carcarino E,
D'Oria V, et al: Focal adhesion kinase depletion reduces human
hepatocellular carcinoma growth by repressing enhancer of zeste
homolog 2. Cell Death Differ. 24:889–902. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Assoian RK: Anchorage-dependent cell cycle
progression. J Cell Biol. 136:1–4. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Assoian RK and Schwartz MA: Coordinate
signaling by integrins and receptor tyrosine kinases in the
regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev.
11:48–53. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iliakis G, Wang Y, Guan J and Wang H: DNA
damage checkpoint control in cells exposed to ionizing radiation.
Oncogene. 22:5834–5847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stewart N, Hicks GG, Paraskevas F and
Mowat M: Evidence for a second cell cycle block at G2/M by p53.
Oncogene. 10:109–115. 1995.PubMed/NCBI
|
|
37
|
Siles E, Villalobos M, Valenzuela MT,
Núñez MI, Gordon A, McMillan TJ, Pedraza V and Ruiz de Almodóvar
JM: Relationship between p53 status and radiosensitivity in human
tumour cell lines. Br J Cancer. 73:581–588. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Waldman T, Zhang Y, Dillehay L, Yu J,
Kinzler K, Vogelstein B and Williams J: Cell-cycle arrest versus
cell death in cancer therapy. Nat Med. 3:1034–1036. 1997.
View Article : Google Scholar : PubMed/NCBI
|