Introduction
Glioblastoma (GBM) is one of the most common and
fatal type of brain tumors. Current clinical treatment for GBM
includes maximal surgical resection followed by post-operative
radiotherapy and adjuvant chemotherapy (1,2).
Despite recent advances in treating other solid tumors, treatment
for GBM still remains palliative, with a very poor prognosis and a
median survival rate of 12–15 months (3). The best mean survival time with
successful tumor resection, radiotherapy and temozolomide (TMZ)
chemotherapy may reach up to 18 months (4). The main cause of treatment failure is
resistance to radiotherapy and chemotherapy (5). Recent research has applied the cancer
stem cell theory of carcinogenesis to tumors, suggesting the
existence of a small subpopulation of glioblastoma stem-like cells
(GSCs) within GBM (6). GSCs are
thought to contribute to tumor progression, treatment resistance
and tumor recurrence, and therefore targeting GSCs has emerged as a
powerful GBM treatment strategy (7,8).
Accumulating evidence has revealed that several molecular markers,
such as Wnt, Hedgehog, Notch, transforming growth factor-β (TGF-β),
epidermal growth factor receptor (EGFR), telomerase and efflux
transporters, are important for self-renewal and differentiation of
GSCs (9,10). Although they might be useful for
targeted therapy in GSCs, finding novel therapeutic targets and
agents to eradicate GSCs can provide a promising treatment strategy
that significantly improves GBM patient survival and quality of
life.
In a previous study, terpestacin from fungal
metabolites was discovered as a new angiogenesis inhibitor with a
unique bicyclo sesterterpene structure (11). The deconvolution of its cognate
target protein using a target identification method has revealed
that terpestacin binds to the ubiquinol-cytochrome c
reductase binding protein (UQCRB) of complex III in mitochondrial
respiratory chain (12).
Terpestacin binding to UQCRB resulted in inhibition of
hypoxia-induced ROS generation and such inhibition blocked
hypoxia-inducible factor (HIF) activation and tumor angiogenesis
in vivo (12). Furthermore,
UQCRB-mediated mitochondrial ROS played a critical role in hypoxic
signaling in tumor cells as well as vascular endothelial growth
factor (VEGF) signaling in endothelial cells (13). Therefore, UQCRB has become a new
therapeutic target for anti-angiogenic and antitumor drug
development. More recently, based on a target-based screen with
structural information on the binding mode of terpestacin and
UQCRB, novel synthetic small molecules targeting UQCRB were
developed (14,15). The compounds specifically bound to
UQCRB and inhibited mitochondrial ROS-mediated hypoxic signaling,
resulting in potent suppressive effects against key angiogenic
processes of endothelial cells activated by VEGF in vitro as
well as antiangiogenic and antitumor activities in vivo,
without inducing cytotoxicity (14,15).
In the present study, to explore a new therapeutic
strategy targeting GSCs by regulation of mitochondrial function,
the effects of mitochondrial UQCRB inhibitors against GSCs were
investigated. Our results showed that the UQCRB inhibitors
effectively suppressed the self-renewal capacity and metastatic
potential of GSCs at subtoxic concentrations. Particularly, the
inhibitory action of the UQCRB inhibitors against GSCs was
associated with blocking of c-Met signaling pathways and subsequent
reduction of the expression levels of GSC markers, through the
regulation of mitochondrial function in GSCs. In addition, UQCRB
depletion in GSCs phenocopied all the effects of the UQCRB
inhibitors, suggesting that UQCRB and its inhibitors could be a new
therapeutic target and lead compounds for eliminating cancer stem
cells in GBM.
Materials and methods
Materials
UQCRB inhibitors, 1c (A1893) and 1f (A1938), were
synthesized and characterized as described previously (15). The compounds were dissolved in
dimethyl sulfoxide (DMSO) at a concentration of 100 mM as a stock
solution and then further diluted with culture media for
appropriate working doses. The negative control groups were treated
with equal volumes of DMSO. Gelatin and laminin were purchased from
Sigma-Aldrich (St. Louis, MO, USA), and Matrigel® was
obtained from BD Biosciences (San Jose, CA, USA). Anti-CD133 and
anti-HIF-1α antibodies were purchased from Miltenyi Biotec GmbH
(Bergisch Gladbach, Germany) and BD Biosciences, respectively.
Anti-Nanog, anti-Sox2, anti-Oct4, anti-phospho-Met, anti-Met,
anti-phospho-Stat3, anti-Stat3, anti-phospho-Akt, anti-Akt,
anti-phospho-Erk1/2, anti-Erk1/2, anti-VEGF and anti-β-actin
antibodies were obtained from Cell Signaling Technology (Danvers,
MA, USA).
Cell culture
Human glioblastoma cell lines, U87MG and U373MG,
were obtained from the Korean Cell Line Bank (KCLB). The cells were
cultured in minimum essential medium (Gibco, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (Gibco) and 1%
penicillin-streptomycin-amphotericin B (Lonza, Walkersville, MD,
USA), and maintained at 37°C in a humidified 5% CO2
incubator. To enrich glioblastoma stem-like cells (GSCs), the cells
grown in serum-based media were suspended in Dulbecco's modified
Eagle's medium/Nutrient Mixture F-12 (Gibco) containing 1X B-27
serum-free supplement (Gibco), 5 µg/ml heparin
(Sigma-Aldrich), 2 mM L-glutamine (Gibco), 20 ng/ml epidermal
growth factor (EGF; Gibco), 20 ng/ml basic fibroblast growth factor
(bFGF; Koma Biotech, Seoul, Korea) and 1% penicillin/streptomycin
(Gibco). The serum-free media with EGF and bFGF were added to the
cells twice a week. Neurospheres were passaged every 7 days by
dissociating with Accutase (Millipore, Temecula, CA, USA).
Cell growth assay
Cell growth was evaluated by WST-1 assay, a
water-soluble tetrazolium salt method. Neurosphere cells were
dissociated with Accutase and seeded into 96-well culture plate at
a density of 3×103 cells/well using the serum-free media
with EGF and bFGF. After 7 days of exposure to UQCRB inhibitors, 10
µl WST-1 reagent solution (Dogen, Korea) was added to each
well, and the cells were incubated for additional 3 h at 37°C. The
absorbance was measured at a wavelength of 450 nm using a
microplate reader (Thermo Scientific, Vantaa, Finland).
Cell viability assay
Cell viability was evaluated by trypan blue
exclusion assay. Dissociated neurosphere cells were seeded at a
density of 1×105 cells/well in 12-well culture plate
using the serum-free media with EGF and bFGF. UQCRB inhibitors were
added to each well and the cells were incubated for up to 7 days.
The cells were stained with trypan blue and counted using a
hemocytometer. Cell viability was calculated as the number of
viable cells divided by the total number of cells.
Neurosphere formation assay
Neurosphere cells were dissociated with Accutase,
and seeded into 96-well culture plate at a density of 50 cells per
well using the serum-free media with EGF and bFGF. The cells were
treated with UQCRB inhibitors and cultured for 1–2 weeks. The
number of neurospheres in each well was counted under an optical
microscope (Olympus, Center Valley, PA, USA). Data were presented
as the percentage of sphere-forming cells relative to DMSO-treated
control.
Migration assay
The ibidi culture inserts (ibidi GmbH, Martinsried,
Germany), which consist of two chambers separated by a
500-µm divider, were used for the migration assay. The
inserts were placed into a laminin-coated 24-well culture plate
using sterile tweezers. Neurosphere cells were dissociated with
Accutase, and single-cell suspensions were prepared at a density of
5×105 cells/ml, of which 70 µl was transferred to
each chamber. After cell attachment for 24 h, the culture inserts
were removed by using sterile tweezers. The cells were washed with
phosphate-buffered saline (PBS) to remove detached cells and
incubated with the GSC culture media in the absence or presence of
UQCRB inhibitors for up to 48 h. The perimeter of the central
cell-free zone was confirmed under an optical microscope
(Olympus).
Invasion assay
Cell invasion was assayed using a
Transwell® chamber system with polycarbonate filter
inserts with a pore size of 8.0 µm (Corning Costar, Acton,
MA, USA). The lower side of the filter was coated with 10 µl
gelatin (1 mg/ml) and the upper side was coated with 10 µl
Matrigel (3 mg/ml). The GSCs (2×105) were placed in the
upper chamber of the filter, and UQCRB inhibitors were added to the
lower chamber filled with the serum-free media containing EGF and
bFGF. The chamber was incubated at 37°C for 48 h, and the cells
were subsequently fixed with methanol and stained with
hematoxylin/eosin. The total number of cells that invaded the lower
chamber of the filter was counted using an optical microscope
(Olympus).
Western blot analysis
Cell lysates were separated by 10% SDS-PAGE
electrophoresis and the separated proteins were transferred to
polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA)
using standard electroblotting procedures. The blots were blocked
and immunolabeled with primary antibodies against CD133, Nanog,
Sox2, Oct4, phospho-Met (Tyr1234/1235), Met, phospho-Stat3
(Tyr705), Stat3, phospho-Akt (Ser473), Akt, phospho-Erk1/2
(Thr202/Tyr204), Erk1/2, HIF-1α, VEGF and β-actin overnight at 4°C.
Immunolabeling was detected with an enhanced chemiluminescence
(ECL) kit (Bio-Rad, Hercules, CA, USA), according to the
manufacturer's instructions.
Measurement of reactive oxygen species
(ROS)
Intracellular ROS and mitochondrial ROS levels were
detected with 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA) and MitoSOX™ Red mitochondrial superoxide
indicator (Molecular Probes, Eugene, OR, USA), respectively. For
the assays, the GSCs seeded at a density of 5×104
cells/well in 96-black well culture plate were treated with UQCRB
inhibitors for 48 h. After incubation with H2DCFDA (10
µM) or MitoSOX Red (5 µM) for 10 min, the
fluorescence intensity was detected using a multimode microplate
reader (Thermo Scientific) at the excitation/emission wavelengths
of 495/529 nm for intracellular ROS and 510/580 nm for
mitochondrial ROS, respectively. The fluorescent images were also
obtained using an Optinity KI-2000F fluorescence microscope (Korea
Lab Tech, Seong Nam, Korea).
Mitochondrial membrane potential
determination
The mitochondrial membrane potential was detected
using the fluorescent, lipophilic dye, JC-1
(5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide, Sigma-Aldrich). At hyperpolarized membrane potentials, this
dye forms a red fluorescent J-aggregate, whereas at depolarized
membrane potentials, this dye remains in its green fluorescent
monomeric form. The GSCs were seeded in 24-black well culture plate
at a density of 5×104 cells/well and treated with UQCRB
inhibitors for 48 h. The cells were incubated with JC-1 (5
µM) for 20 min and the images were obtained using an
Optinity KI-2000F fluorescence microscope.
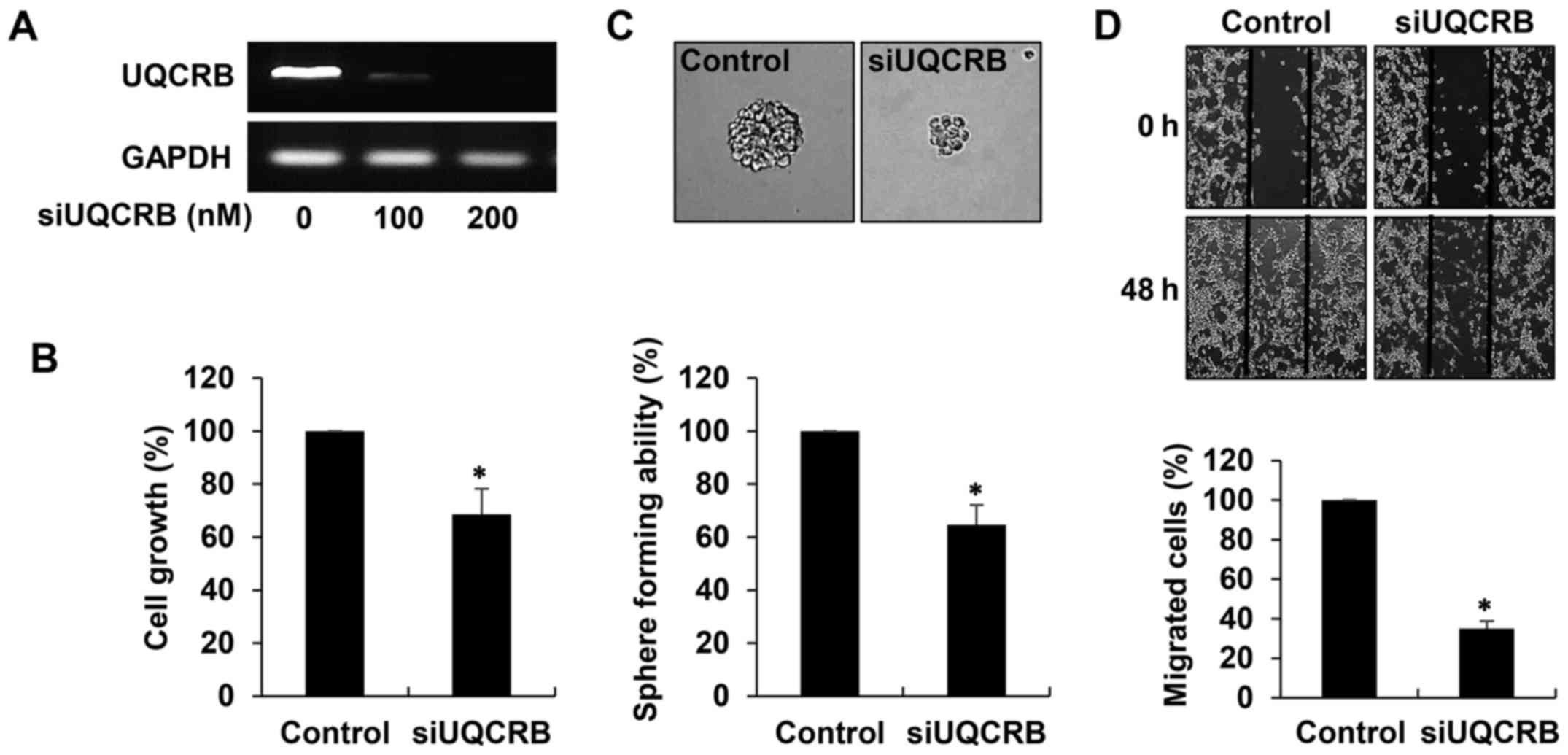
UQCRB silencing
Human UQCRB-specific siRNA (siUQCRB) was constructed
as described previously (13). The
sense and antisense sequences of this siRNA were 5′-GGG UUA AUG CGA
GAU GAU ACA AUA U-3′ and 5′-AUA UUG UAU CAU CUC GCA UUA ACC C-3′,
respectively. For depletion of UQCRB mRNA, U87MG GSCs seeded in
laminin-coated culture plate with the GSC culture media were
transfected with either scrambled negative siRNA or UQCRB siRNA
using Lipofectamine 3000 transfection reagent (Invitrogen, Grand
Island, NY, USA) according to the manufacturer's instructions.
Interference of UQCRB mRNA was validated through reverse
transcriptase-polymerase chain reaction (RT-PCR) analysis using
specific primers for UQCRB (sense, 5′-ATGGCTGGTAAGCAGGCC-3′;
anti-sense, 5′-CTTCTTTGCCCATTCTTC-3′).
Statistical analysis
The results are expressed as the mean ± standard
error (SE). Student's t-test was used to determine statistical
significance between the control and test groups. A P-value of
<0.05 was considered statistically significant.
Results
The effect of UQCRB inhibitors on the
proliferation of GSCs
All established glioma cell lines, like most other
cancer cell lines, are grown in media containing serum, whereas
GSCs are grown as neurospheres in serum-free media, since serum
causes irreversible differentiation of primary tumor cells
(16,17). Accordingly, in this study, GSCs
were propagated under an optimal condition using serum-free media
supplemented with basic FGF and EGF (18). In order to determine the
therapeutic effect of mitochondrial UQCRB inhibitors against GSCs,
two synthetic small molecules that are known to target UQCRB, 1c
(A1893) and 1f (A1938), were analyzed in the present study
(15) (Fig. 1).
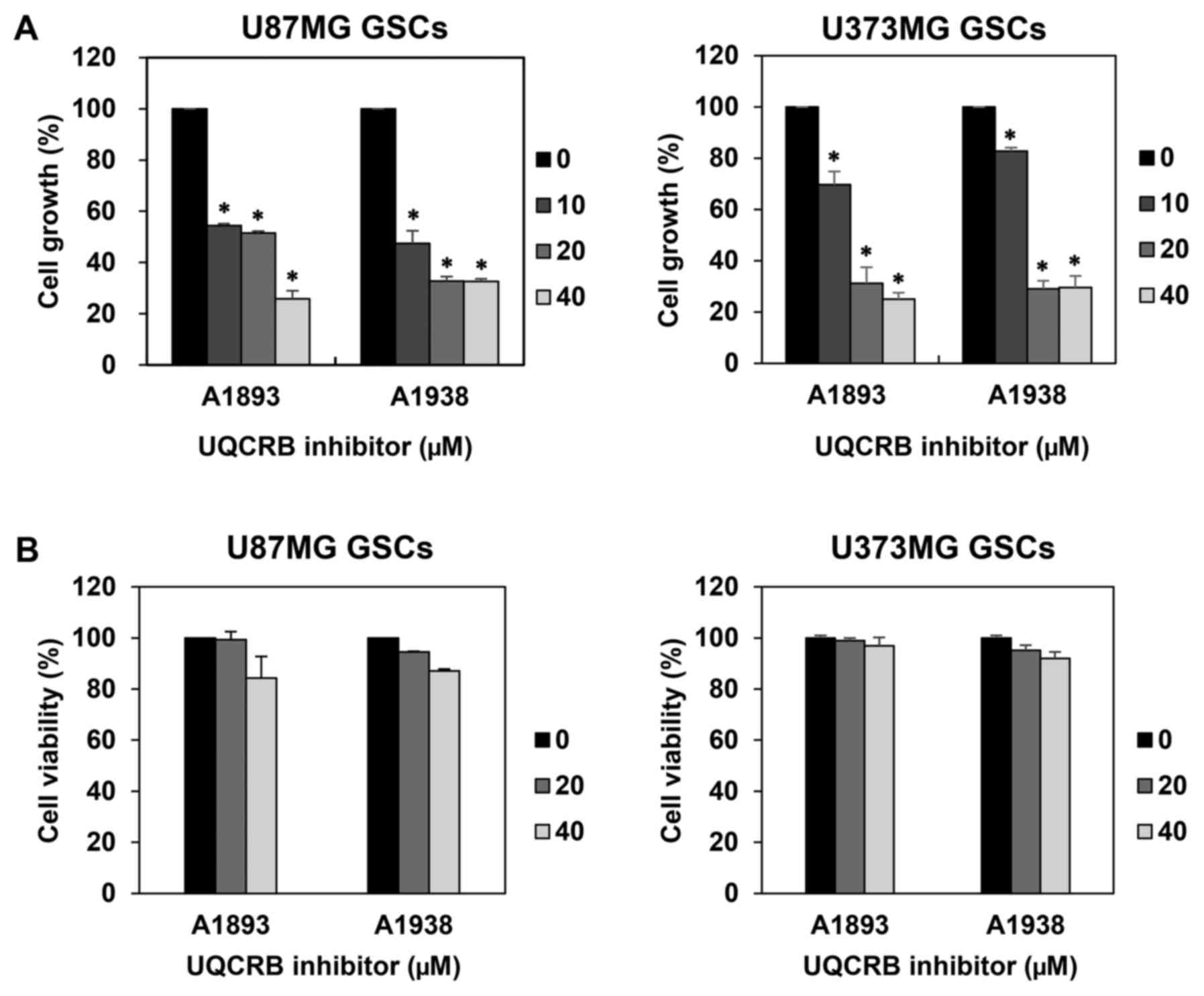
We first examined the effect of UQCRB inhibitors on
the proliferation of GSCs derived from U87MG and U373MG cells using
a water-soluble tetrazolium salt method. The UQCRB inhibitors
suppressed the proliferation of both GSCs in a dose-dependent
manner (Fig. 2A). To further
evaluate whether the GSC growth inhibition by UQCRB inhibitors was
due to cytotoxic effects, a viability assay was performed using the
trypan blue exclusion method. The viability of GSCs exceeded 80%
relative to DMSO-treated control cells, even after treatment with
40 µM of UQCRB inhibitors for 7 days (Fig. 2B). Therefore, the UQCRB inhibitors
can inhibit the proliferation of U87MG and U373MG GSCs at subtoxic
concentrations.
The effect of UQCRB inhibitors on the
stemness of GSCs
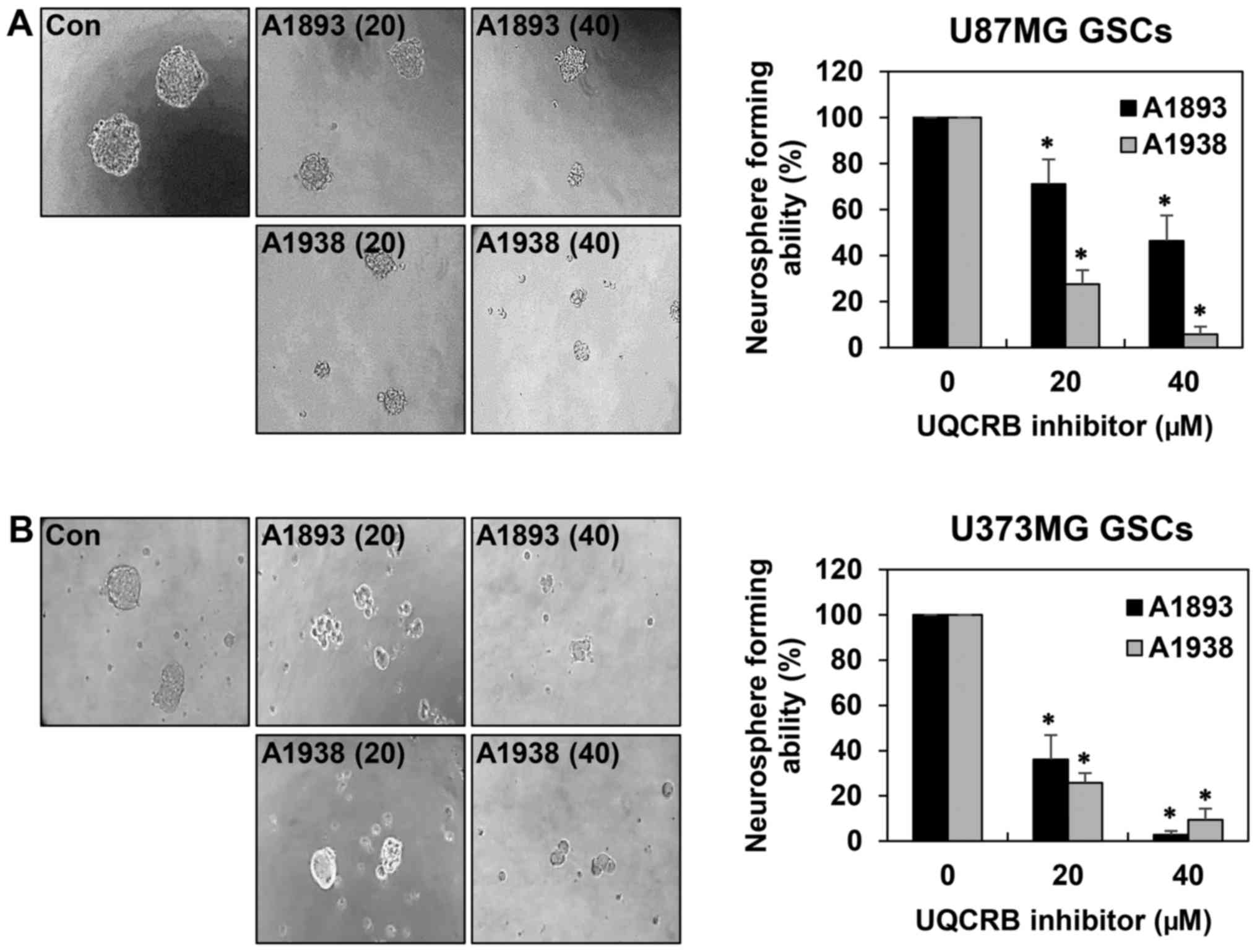
Sphere-forming assays have been widely used to
evaluate the stemness of cells, their capacity for self-renewal and
differentiation, both of which are instrumental in cancer cell
formation (6,19). As shown in Fig. 3, the neurosphere formation ability
of both GSCs was markedly suppressed by treatment with the UQCRB
inhibitors. These results indicate that UQCRB inhibitors can reduce
the self-renewal capacity of GSCs.
The effect of UQCRB inhibitors on the
migration and invasion of GSCs
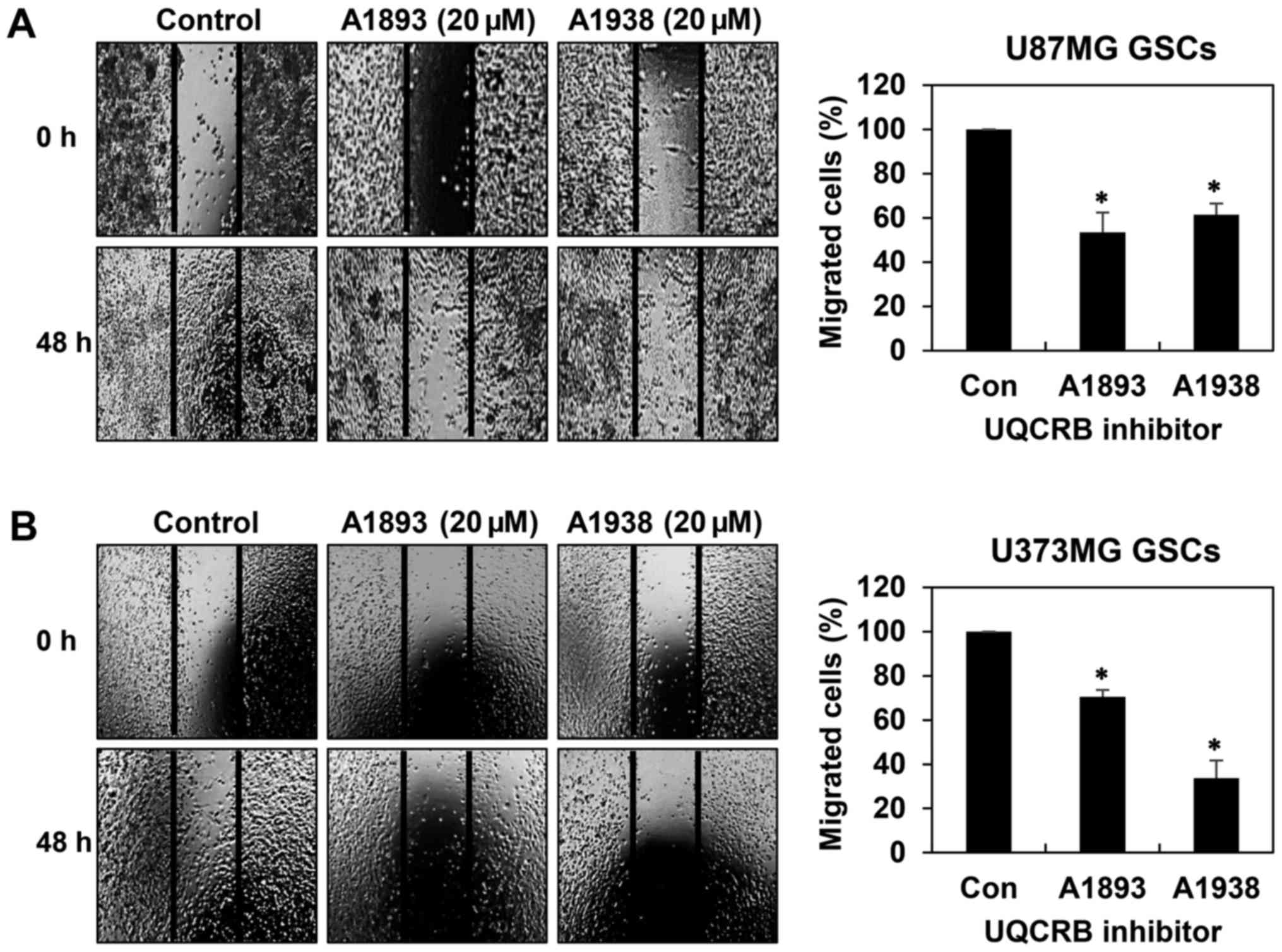
GSCs have been implicated in accelerating tumor
metastasis (20,21). We thus assessed whether UQCRB
inhibitors have an effect on the migration and invasion of GSCs. We
first confirmed their effect on the migration of both GSCs in 48 h
after treatment when compared to control conditions using wound
healing assay. The UQCRB inhibitors led to significant reduction of
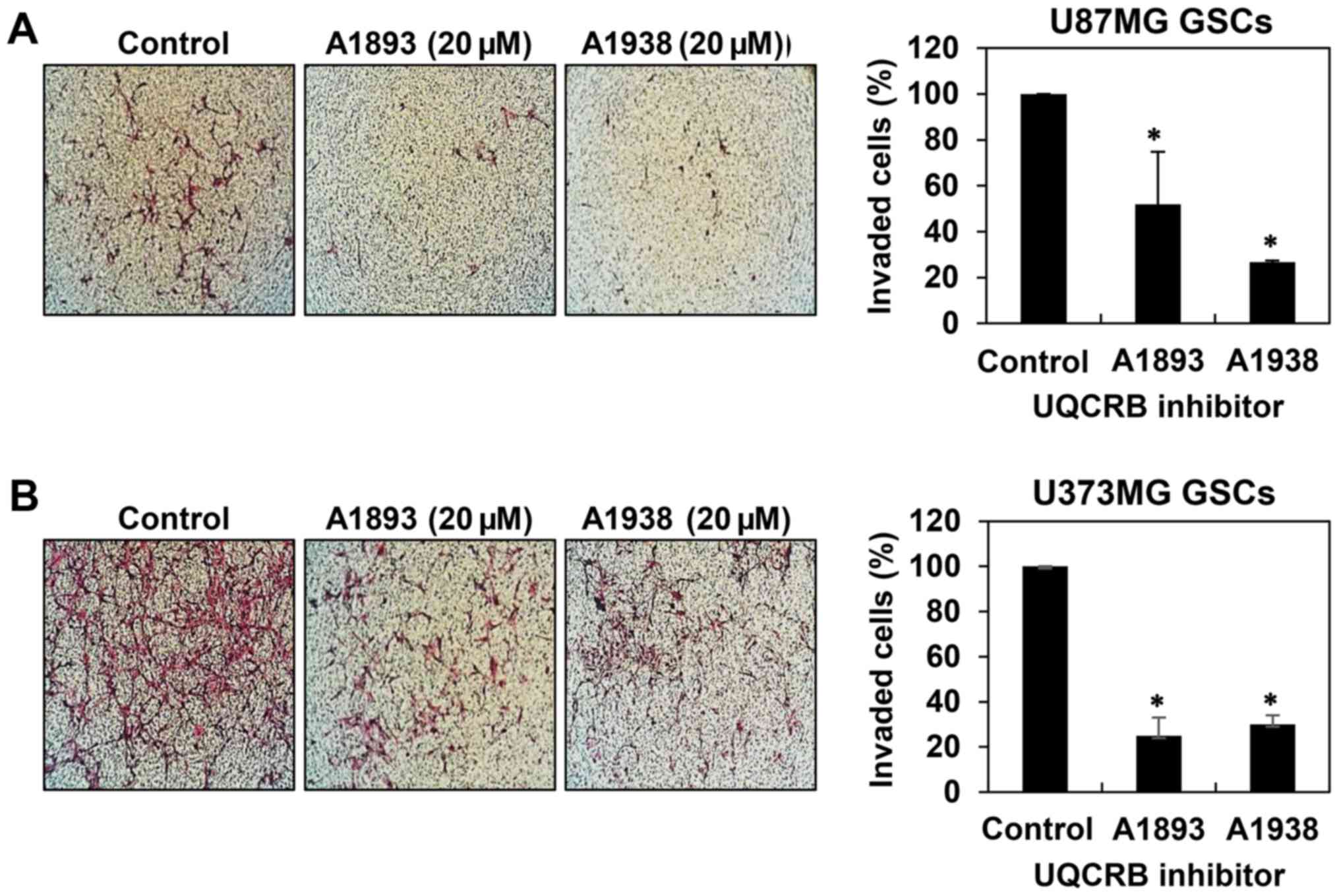
cell migration in U87MG and U373MG GSCs (Fig. 4). Next, invasion assay was
performed by employing the Matrigel-coated transwell chamber
system. The UQCRB inhibitors distinctly decreased the invasion of
U87MG and U373MG GSCs when compared with controls (Fig. 5). These data demonstrate that UQCRB
inhibitors suppress the metastatic capability of GSCs.
The effect of UQCRB inhibitors on the
c-Met signaling pathway in GSCs
Recent studies have shown that c-Met signaling
induces glioma malignancy by increasing GSC population through
upregulation of stemness supporting transcription factors such as
Sox2, Klf4, c-Myc, Oct4 and Nanog (22,23).
We thus investigated the effect of UQCRB inhibitors on the c-Met
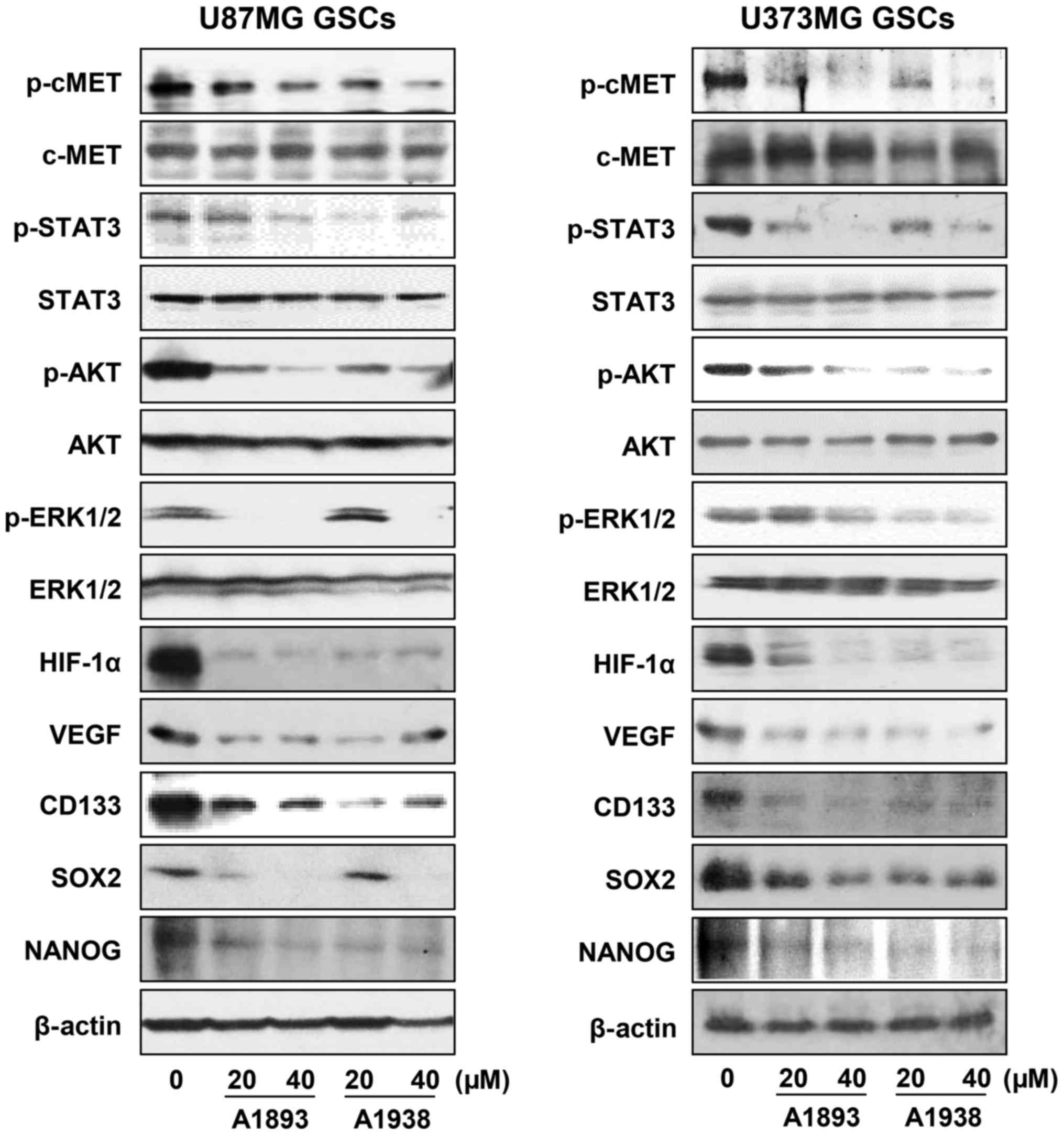
signaling cascade in U87MG and U373MG GSCs. Treatment with the
UQCRB inhibitors effectively inhibited the phosphorylation of c-Met
and its downstream signal transduction effectors including STAT3,
Akt and ERK1/2, without affecting the total protein levels, in
either GSC (Fig. 6). Recent
reports have revealed that hypoxia-inducible factors (HIFs)
stimulate specific signaling pathways and transcription factors
that control cancer stem cell self-renewal and multipotency and are
highly expressed in GSCs (24,25).
The UQCRB inhibitors significantly reduced the protein levels of
HIF-1α as well as its transcriptional target gene, VEGF in GSCs
(Fig. 6). They also decreased the
expression of GBM stemness markers such as CD133, Nanog and Sox2,
which are known to be regulated by c-Met and HIFs (Fig. 6). Taken together, these data
suggest that the suppressive effect of UQCRB inhibitors on the
proliferation, stemness, migration and invasiveness of GSCs might
be partly associated with the downregulation of c-Met signaling and
HIF activity.
The effect of UQCRB inhibitors on the
mitochondrial function in GSCs
Recent studies have indicated that ROS contribute to
cancer stem cell-like properties and play pivotal roles in
tumorigenesis, metastasis and resistance to radiation and
chemotherapy (26–28). Since UQCRB functions as a
mitochondrial ROS mediator (12,13),
we examined whether UQCRB inhibitors affect intracellular ROS
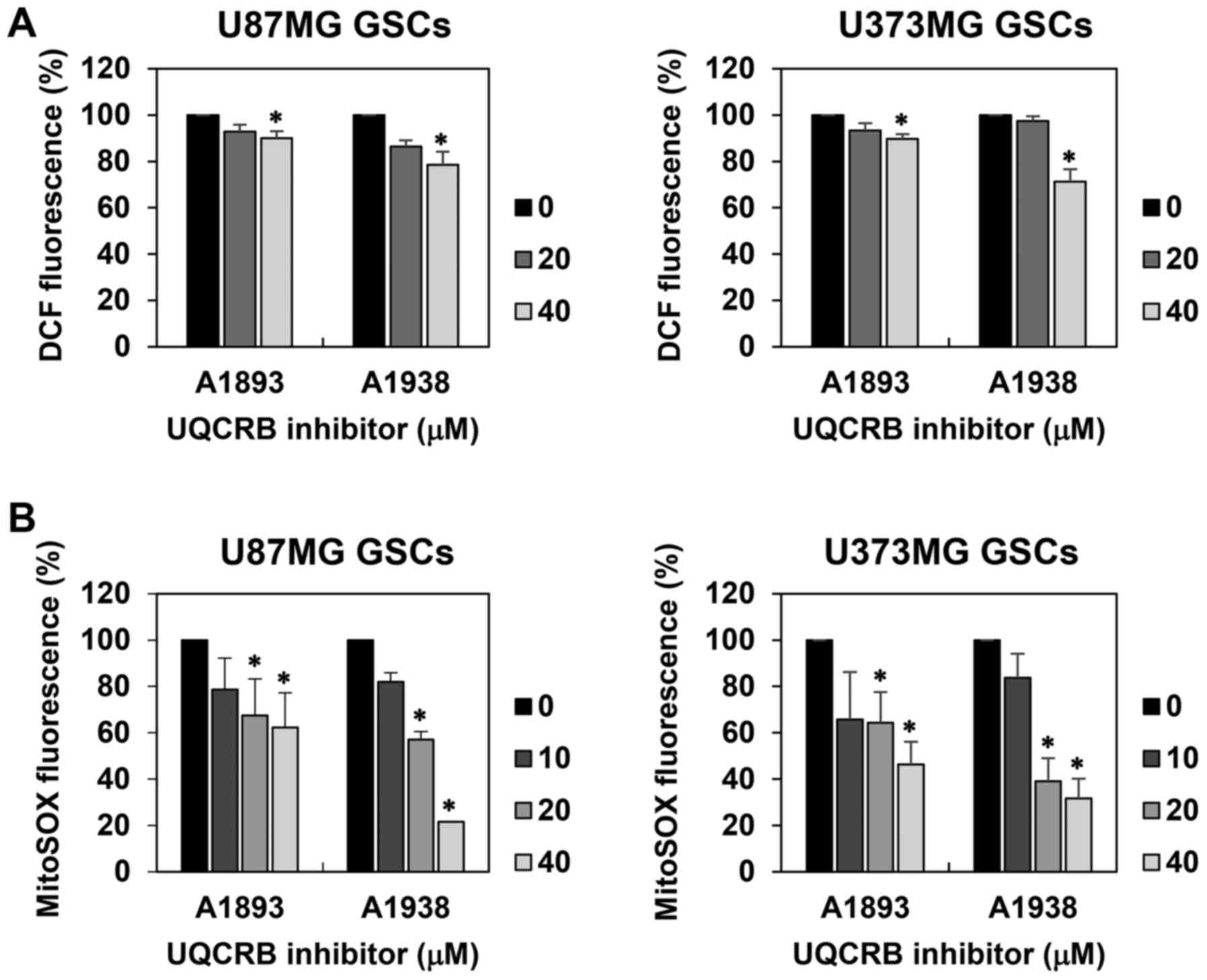
levels in U87MG and U373MG GSCs using a fluorescent probe for ROS
measurement, 2′,7′-dichlorofluorescein. The UQCRB inhibitors
reduced the intracellular ROS levels in both GSCs (Fig. 7A). Moreover, mitochondrial ROS
levels in GSCs were measured using a MitoSOX™ Red mitochondrial
superoxide indicator. The UQCRB inhibitors noticeably inhibited the
mitochondrial ROS generation in both GSCs (Fig. 7B).
Next, to investigate whether UQCRB inhibitors cause
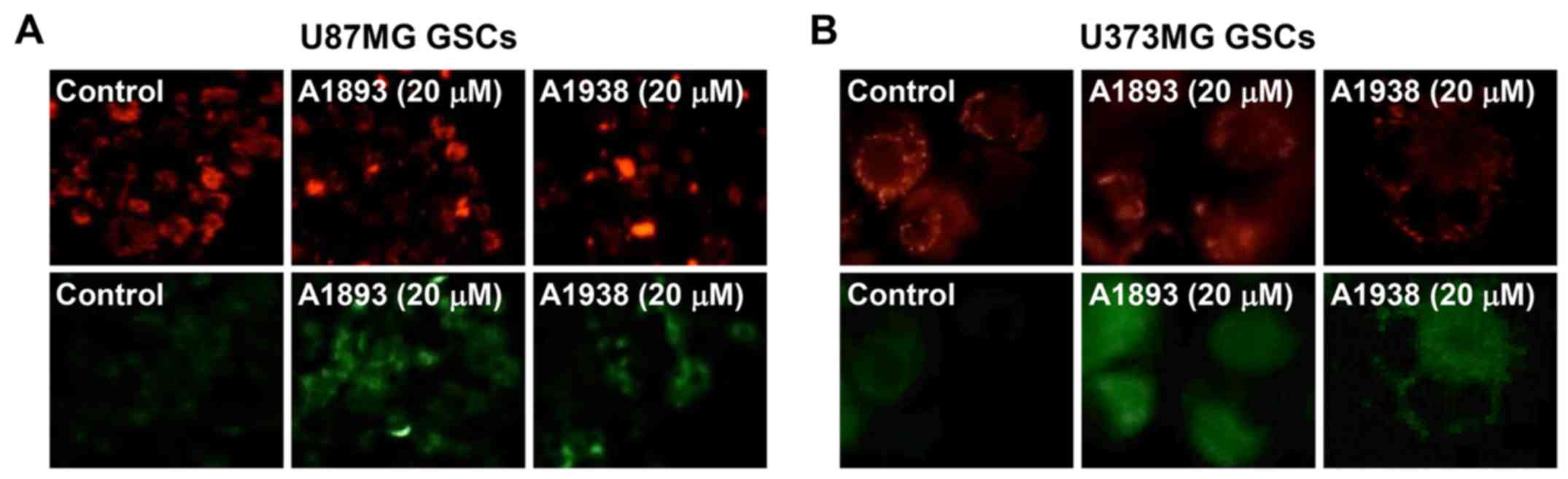
mitochondrial dysfunction in GSCs, the mitochondrial membrane
potential was measured using the JC-1 fluorescent marker. Treatment
with the UQCRB inhibitors led to a decrease in red fluorescence and
an increase in green fluorescence in U87MG and U373MG GSCs,
indicating that they cause a loss of the mitochondrial membrane
potential in the cells (Fig. 8).
These data demonstrate that UQCRB inhibitors may modulate the
mitochondrial function in GSCs, especially at mitochondrial ROS
generation.
The effect of UQCRB knockdown on the
cancer stem cell-like phenotypes of GBM cells
To elucidate the role of UQCRB in the maintenance of
GSCs, we performed UQCRB depletion experiments in U87MG GSCs. The
cells were transfected with either UQCRB siRNA (siUQCRB) or
scrambled negative siRNA, and knockdown of the UQCRB gene was
confirmed through RT-PCR analysis (Fig. 9A). We first investigated the effect
of UQCRB knockdown on the cancer stem cell-like phenotypes of GBM
cells. As shown in Fig. 9B, UQCRB
knockdown significantly inhibited the growth of U87MG GSCs. In
addition, the neurosphere formation and migration abilities of the
GSCs were noteworthily suppressed by UQCRB knockdown (Fig. 9C and D). The results suggest that
mitochondrial UQCRB positively regulates the cancer stem cell-like
properties of GBM cells.
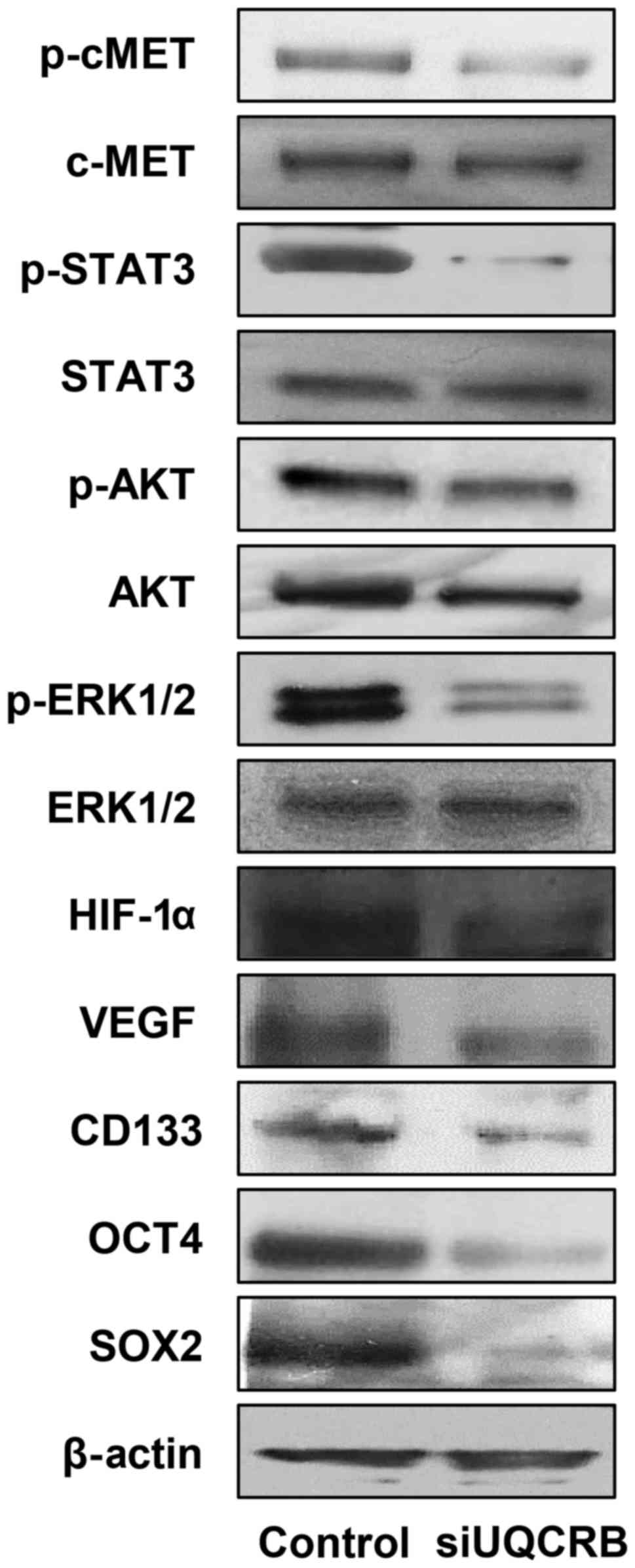
We next investigated the effect of UQCRB knockdown
on the c-Met signaling, HIF and stemness markers in U87MG GSCs.
UQCRB depletion reduced the activation of c-Met and its downstream
signal transduction mediators, STAT3, Akt and ERK1/2 (Fig. 10). UQCRB knockdown also decreased
the expression of HIF-1α and VEGF. The down-regulation of c-Met
signaling and HIF activity consequently led to a reduction in the
expression of GBM stemness markers such as CD133, Oct4 and Sox2
(Fig. 10).
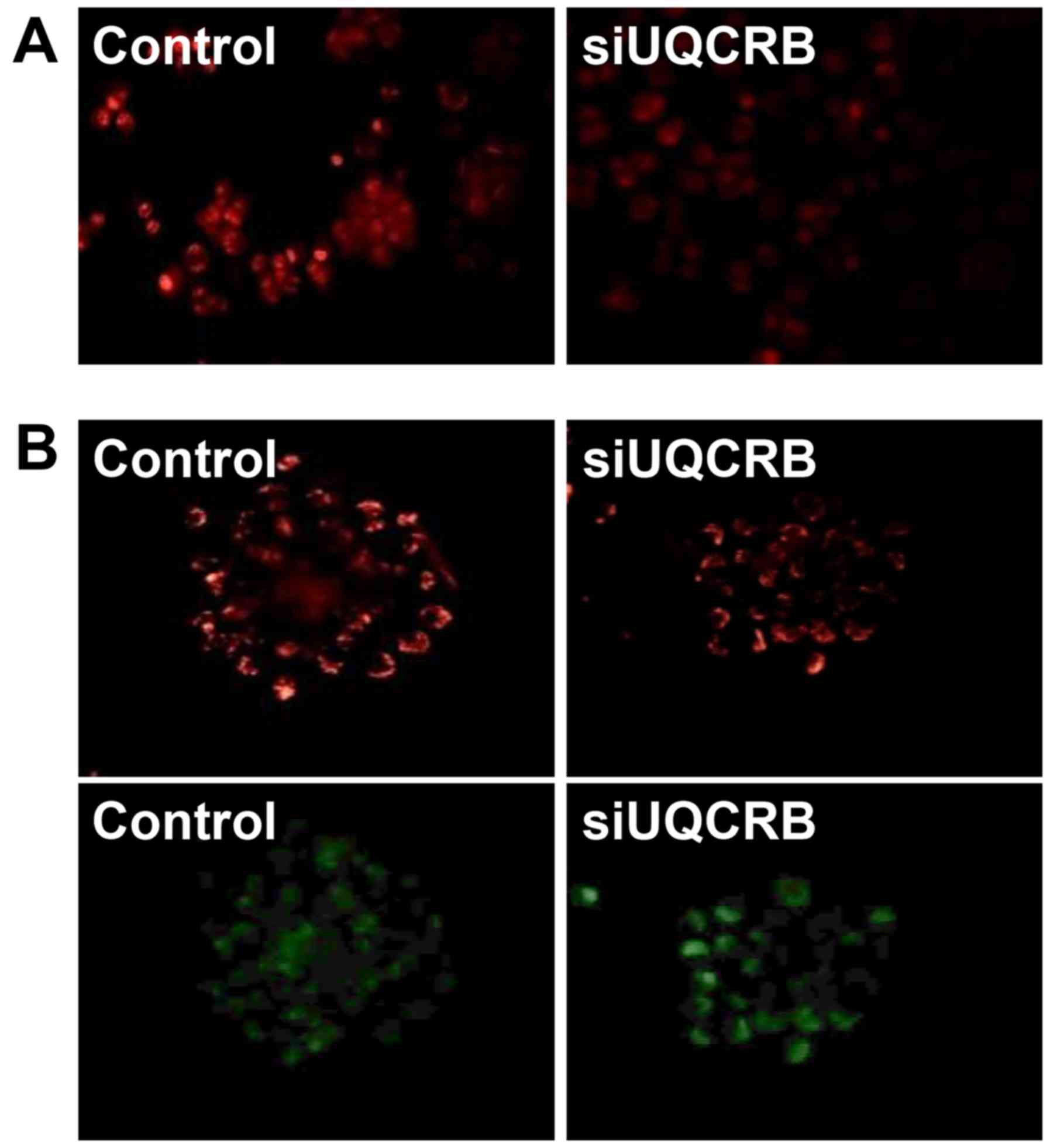
Furthermore, we assessed whether the inhibition of
the GSC characteristics by UQCRB depletion is associated with the
regulation of mitochondrial function. UQCRB knockdown significantly
suppressed the mitochondrial ROS generation and diminished the
mitochondrial membrane potential in U87MG GSCs (Fig. 11). These results strongly
demonstrate that UQCRB might function as a crucial mediator of GSC
maintenance through the modulation of mitochondrial ROS generation
in GSCs.
Discussion
Glioblastoma (GBM) is the most common and aggressive
primary brain tumor in adults with poor prognosis (1–3). GBM
is highly heterogeneous in nature and contains a small but highly
tumorigenic and self-renewing population of cancer stem cells
(6). Glioblastoma stem cells
(GSCs) have been shown to contribute to tumor propagation and
resistance to current therapeutic modalities (7,8).
Recent studies of human GBM have elucidated the genetic alterations
common in this tumor, but much remains unknown about specific
signaling pathways that regulate GSCs (29,30).
The activated c-Met receptor tyrosine kinase is known to stimulate
cell survival, proliferation and invasion by triggering the
activation of its multiple downstream effectors such as
mitogen-activated protein kinases, Akt and STAT3 (31,32).
The overexpression of c-Met has been observed in aggressive human
cancers including GBM and associated with poor prognosis in cancer
patients (33,34). In particular, activation of c-Met
signaling led to an increase in expression of the stemness
transcriptional regulators, Sox2, Klf4, c-Myc, Oct4 and Nanog
(22). Pharmacological inhibition
of c-Met activity in GSCs prevented the activation of Oct4, Nanog
and Klf4 and potently abrogated the clonogenicity, tumorigenicity
and radioresistance of GSCs both in vitro and in vivo
(23,35). Therefore, therapeutic agents that
block c-Met signaling pathway of GSCs would be an attractive
candidate for the combination with current standard therapy against
GBM.
In recent studies, the ubiquinol-cytochrome c
reductase binding protein (UQCRB) of mitochondrial complex III has
been found to be a crucial mediator of mitochondrial ROS
generation, thereby playing a central role in tumor angiogenesis
via upregulation of both hypoxic and VEGF signaling (12,13).
In addition, small molecules targeting UQCRB resulted in the
inhibition of mitochondrial ROS generation, and such inhibition
consequently blocked hypoxia-inducible factor (HIF) activation,
vascular endothelial growth factor receptor 2 (VEGFR2) signal
transduction and tumor angiogenesis in vivo (14,15,36).
Therefore, UQCRB has begun to emerge as a promising therapeutic
target for antiangiogenic and anticancer therapy.
It has been recently reported that HIF-1 induces the
overexpression and constitutive activation of c-Met (37). Furthermore, in melanoma cells,
HIF-1α stabilization by mitochondrial ROS promoted the
c-Met-dependent prometastatic effects, such as enhancement of
spreading on extracellular matrix, motility and invasion, as well
as growth of metastatic colonies and the ability to form
capillary-like structures by vasculogenic mimicry (38). Recent data revealed the involvement
of ROS in the activation of the epithelial-mesenchymal transition
(EMT) which is related to the acquisition and maintenance of stem
cell-like characteristics, in several cancer models (39,40).
Taken together, mitochondrial UQCRB may be an upstream molecular
target to control activation of ROS/HIF/c-Met axis and thus UQCRB
inhibitors could be a new therapeutic agent to eradicate cancer
stem cells.
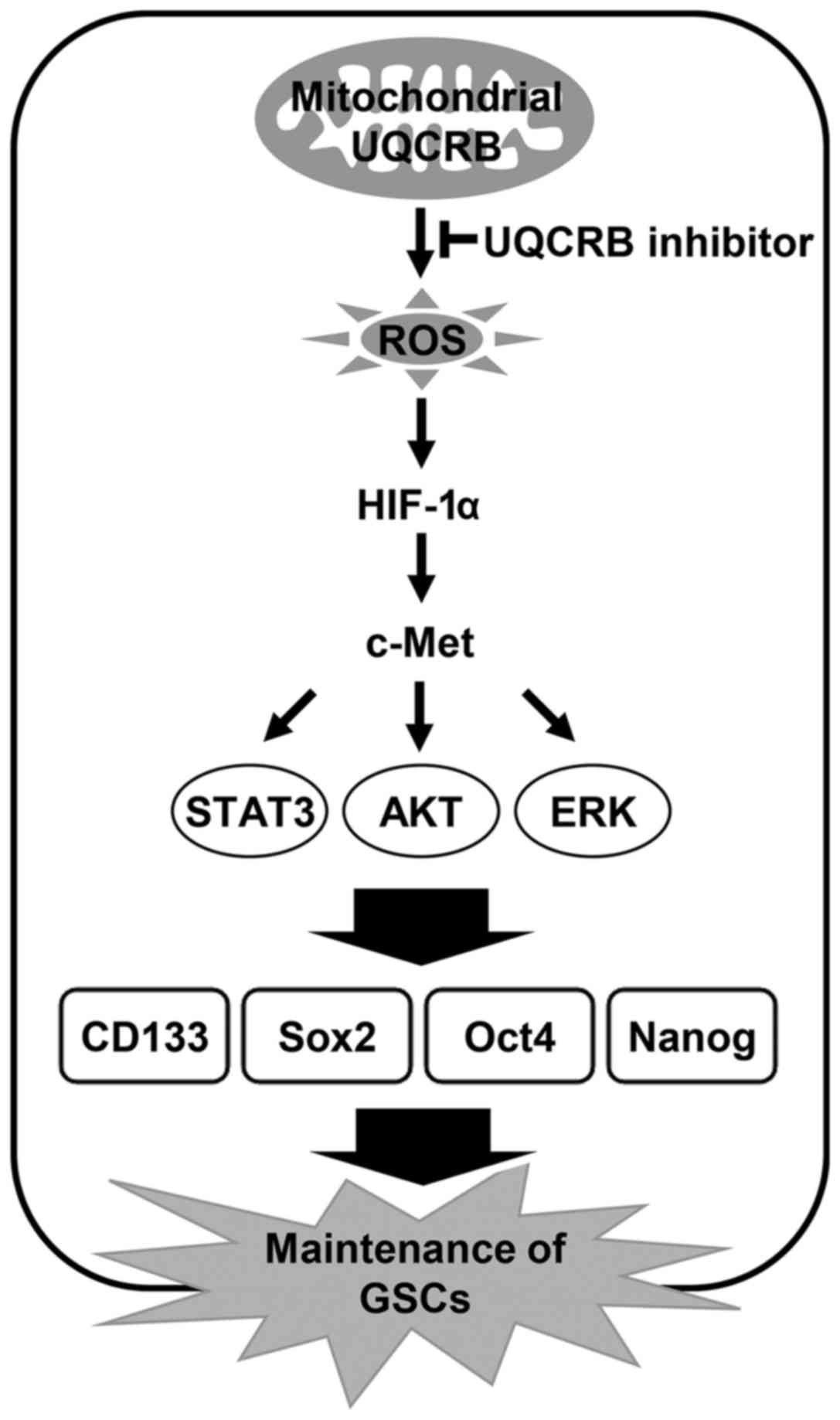
The present study indicates that UQCRB inhibitors
suppress cancer stem cell-like phenotypes in human glioblastoma
cells by downregulating mitochondrial ROS/HIF-1α/c-Met pathway
(Fig. 12). Moreover, knockdown of
UQCRB gene significantly inhibited the proliferation, neurosphere
formation and migration of GSCs, as well as mitochondrial ROS
generation, HIF-1α accumulation, c-Met signaling activation and GSC
stemness marker expression. In conclusion, these findings suggest
that targeting UQCRB function associated with regulation of mROS
generation could be an attractive therapeutic strategy for
interrupting the c-Met signaling that is important for GSC
maintenance. Accordingly, our results provide new insight into the
role of mitochondrial UQCRB in the activation of c-Met signaling of
GSCs and the underlying mechanism of UQCRB inhibitors to eliminate
GSCs.
Acknowledgments
This study was supported by 'Cooperative Research
Program for Agriculture Science and Technology Development (Project
no. PJ01188001)' Rural Development Administration, Republic of
Korea, the Basic Science Research Program through the National
Research Foundation of Korea (NRF) funded by the Ministry of
Education (NRF-2016R1D1A1B03932956), and the Brain Korea 21 Plus
Project, Republic of Korea. This study was also supported in part
by NRF grant 2012M3A9D1054520 and 2015K1A1A2028365.
References
|
1
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
Classification of Tumors of the Central Nervous System: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lutterbach J, Guttenberger R and
Pagenstecher A: Gliosarcoma: A clinical study. Radiother Oncol.
61:57–64. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stavrovskaya AA, Shushanov SS and
Rybalkina EY: Problems of glioblastoma multiforme drug resistance.
Biochemistry (Mosc). 81:91–100. 2016. View Article : Google Scholar
|
|
6
|
Sundar SJ, Hsieh JK, Manjila S, Lathia JD
and Sloan A: The role of cancer stem cells in glioblastoma.
Neurosurg Focus. 37:E62014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu G, Yuan X, Zeng Z, Tunici P, Ng H,
Abdulkadir IR, Lu L, Irvin D, Black KL and Yu JS: Analysis of gene
expression and chemoresistance of CD133+ cancer stem
cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar
|
|
9
|
Cho DY, Lin SZ, Yang WK, Lee HC, Hsu DM,
Lin HL, Chen CC, Liu CL, Lee WY and Ho LH: Targeting cancer stem
cells for treatment of glioblastoma multiforme. Cell Transplant.
22:731–739. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kalkan R: Glioblastoma stem cells as a new
therapeutic target for glioblastoma. Clin Med Insights Oncol.
9:95–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jung HJ, Lee HB, Kim CJ, Rho JR, Shin J
and Kwon HJ: Anti-angiogenic activity of terpestacin, a bicyclo
sesterterpene from Embellisia chlamydospora. J Antibiot (Tokyo).
56:492–496. 2003. View Article : Google Scholar
|
|
12
|
Jung HJ, Shim JS, Lee J, Song YM, Park KC,
Choi SH, Kim ND, Yoon JH, Mungai PT, Schumacker PT, et al:
Terpestacin inhibits tumor angiogenesis by targeting UQCRB of
mitochondrial complex III and suppressing hypoxia-induced reactive
oxygen species production and cellular oxygen sensing. J Biol Chem.
285:11584–11595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jung HJ, Kim Y, Chang J, Kang SW, Kim JH
and Kwon HJ: Mitochondrial UQCRB regulates VEGFR2 signaling in
endothelial cells. J Mol Med (Berl). 91:1117–1128. 2013. View Article : Google Scholar
|
|
14
|
Jung HJ, Kim KH, Kim ND, Han G and Kwon
HJ: Identification of a novel small molecule targeting UQCRB of
mitochondrial complex III and its anti-angiogenic activity. Bioorg
Med Chem Lett. 21:1052–1056. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung HJ, Cho M, Kim Y, Han G and Kwon HJ:
Development of a novel class of mitochondrial ubiquinol-cytochrome
c reductase binding protein (UQCRB) modulators as promising
antiangiogenic leads. J Med Chem. 57:7990–7998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Galli R, Binda E, Orfanelli U, Cipelletti
B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F and Vescovi
A: Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Laks DR, Masterman-Smith M, Visnyei K,
Angenieux B, Orozco NM, Foran I, Yong WH, Vinters HV, Liau LM,
Lazareff JA, et al: Neurosphere formation is an independent
predictor of clinical outcome in malignant glioma. Stem Cells.
27:980–987. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kotliarova S, Kotliarov Y, Li A, Su
Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheng L, Wu Q, Guryanova OA, Huang Z,
Huang Q, Rich JN and Bao S: Elevated invasive potential of
glioblastoma stem cells. Biochem Biophys Res Commun. 406:643–648.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Zhang W, Mao XG, Zhen HN, Cao WD
and Hu SJ: Targeting role of glioma stem cells for glioblastoma
multiforme. Curr Med Chem. 20:1974–1984. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Li A, Glas M, Lal B, Ying M, Sang Y,
Xia S, Trageser D, Guerrero-Cázares H, Eberhart CG, et al: c-Met
signaling induces a reprogramming network and supports the
glioblastoma stem-like phenotype. Proc Natl Acad Sci USA.
108:9951–9956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu
Kang B, Kang YJ, Lathia JD, Cheong KH, Song PH, et al: MET
signaling regulates glioblastoma stem cells. Cancer Res.
72:3828–3838. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Keith B and Simon MC: Hypoxia-inducible
factors, stem cells, and cancer. Cell. 129:465–472. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang
YJ, Zhao L, Chen FH, Wang XT, You QD, et al: HIF-1α is critical for
hypoxia-mediated maintenance of glioblastoma stem cells by
activating Notch signaling pathway. Cell Death Differ. 19:284–294.
2012. View Article : Google Scholar
|
|
26
|
Shi X, Zhang Y, Zheng J and Pan J:
Reactive oxygen species in cancer stem cells. Antioxid Redox
Signal. 16:1215–1228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ding S, Li C, Cheng N, Cui X, Xu X and
Zhou G: Redox regulation in cancer stem cells. Oxid Med Cell
Longev. 2015:7507982015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Im CN, Yun HH, Yoo HJ, Park MJ and Lee JH:
Enhancement of SOX-2 expression and ROS accumulation by culture of
A172 glioblastoma cells under non-adherent culture conditions.
Oncol Rep. 34:920–928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haque A, Banik NL and Ray SK: Molecular
alterations in glioblastoma: Potential targets for immunotherapy.
Prog Mol Biol Transl Sci. 98:187–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: Principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kong DS, Song SY, Kim DH, Joo KM, Yoo JS,
Koh JS, Dong SM, Suh YL, Lee JI, Park K, et al: Prognostic
significance of c-Met expression in glioblastomas. Cancer.
115:140–148. 2009. View Article : Google Scholar
|
|
34
|
Nabeshima K, Shimao Y, Sato S, Kataoka H,
Moriyama T, Kawano H, Wakisaka S and Koono M: Expression of c-Met
correlates with grade of malignancy in human astrocytic tumours: An
immunohistochemical study. Histopathology. 31:436–443. 1997.
View Article : Google Scholar
|
|
35
|
Kim B, Jung N, Lee S, Sohng JK and Jung
HJ: Apigenin inhibits cancer stem cell-like phenotypes in human
glioblastoma cells via suppression of c-Met signaling. Phytother
Res. 30:1833–1840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jung HJ and Kwon HJ: Exploring the role of
mitochondrial UQCRB in angiogenesis using small molecules. Mol
Biosyst. 9:930–939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Comito G, Calvani M, Giannoni E, Bianchini
F, Calorini L, Torre E, Migliore C, Giordano S and Chiarugi P:
HIF-1α stabilization by mitochondrial ROS promotes Met-dependent
invasive growth and vasculogenic mimicry in melanoma cells. Free
Radic Biol Med. 51:893–904. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Radisky DC, Levy DD, Littlepage LE, Liu H,
Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et
al: Rac1b and reactive oxygen species mediate MMP-3-induced EMT and
genomic instability. Nature. 436:123–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cannito S, Novo E, Compagnone A, Valfrè di
Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A,
Bozzo F, et al: Redox mechanisms switch on hypoxia-dependent
epithelial-mesenchymal transition in cancer cells. Carcinogenesis.
29:2267–2278. 2008. View Article : Google Scholar : PubMed/NCBI
|