Introduction
The signal transducer and activator of transcription
(STAT) proteins, including STAT1-6, share common structural domains
of coiled-coil, DNA-binding, linker, SH2, and transactivation (from
N- to C-terminal), and mediate many aspects of cellular apoptosis,
DNA repair, differentiation, and cell cycle progression (1–3). The
primary activation of STAT is mediated by Janus kinases (JAK),
which transmit extracellular signals to the cytosol and nucleus
through transmembrane receptor/ligands via the JAK-STAT pathway
(4,5). In this process, STAT is modified by
phosphorylation, glutathionylation, and acetylation, which
facilitates STAT activation for transcriptional regulation of
target genes (6–8). STAT activation through these
modifications contributes to specific cellular responses to
cytokines, interleukins, peptide hormones, and growth factors
(9–11).
Recently, it was reported that STAT also has a
cellular nongenomic function. STAT interacts with GRIM-19, a
subunit of mitochondria respiratory complex I, which determines
whether STAT3 is imported into mitochondria (12,13).
Furthermore, there is direct evidence that STAT is present in the
mitochondria of cultured cells and primary tissues. STAT
mitochondrial importation selectively stabilizes and increases
mitochondria respiratory complexes, allowing them to orchestrate
responses to stimuli (14,15). As mitochondria respiratory complex
I and III are thought to be the main source for ROS generation
(16,17), it has been suggested that STAT1
facilitates ROS production and apoptosis (14). Conversely, STAT activation in some
instances seems to depend on ROS signaling (18,19).
STAT1 and STAT3 are activated in fibroblasts and A431 carcinoma
cells within 5 min after H2O2 stimulation.
Therefore, ROS is thought to be a second messenger to regulate STAT
activation (20). These reports
strongly indicate that STAT and ROS form a positive STAT-ROS
feedback loop, but its details are largely unknown.
Interferons (IFNs) belong to a large group of signal
cytokines, which are released from host cells to communicate with
other cells to trigger immune system defenses (21), helping to eliminate foreign bodies
and malignant cells (22). IFNs
are divided into three types: type I, represented by IFN-α and
IFN-β; type II, represented by IFN-γ; and type III, which was more
recently discovered. It is already known that IFN-γ induces
cellular apoptosis mediated by STAT1 activation (23). IFN-β inhibits HepG2 cell viability
via phosphorylation of STAT2 (24). Also, an IFN-α/IFN-γ co-formulation
is involved in the IFN-STAT-pathway and apoptosis in U87MG cells
(25). Some of the stimulated
apoptosis may be explained by the upregulation of mitochondria
respiratory complexes and ROS production (26,27).
However, a pilot study of clinical IFN-α administration in melanoma
patients indicated that a high dose of IFN is not necessary for
optimal activation of immune signal transduction (28). This indicates that some latent
events beyond the IFN-STAT pathway may be involved in IFN-induced
mitochondria respiratory complexes, ROS production, and cellular
apoptosis.
In the present study, we directly investigated the
potential STAT-ROS cycle in human cancer cells. We found that the
STAT-ROS cycle extends the effect of IFN-induced cellular
apoptosis. The novel mechanism of the STAT-ROS cycle may be of use
to increase the efficacy of IFN administration clinically.
Materials and methods
Cell culture and reagents
The human breast cancer cell line of MCF7, carcinoma
cell line A431, and cervical cancer cell line HeLa (Sigma,
Shanghai, China) were cultured in RPMI-1640 (Thermo Fisher
Scientific, Shanghai, China) supplemented with 10% FCS
(heat-inactivated), 100 U/ml penicillin, and 100 mg/ml
streptomycin, and were incubated at 37°C in a humidified atmosphere
with 5% CO2.
For some experiments, a combination of recombinant
100 U/ml IFN-α 2a, 100 U/ml IFN-γ (Sigma), and 100 U/ml IFN-β (Sino
Biological, Beijing, China) were used for cell culture according to
a previous report (29).
STAT-overexpression in MCF7 cells was established
using adenovirus transfection. Human STAT-1 adenovirus (Ad-h-STAT1)
and human STAT-3 adenovirus (Ad-h-STAT3) were purchased from Vector
Biolabs (Philadelphia, PA, USA). Briefly, the combined adenoviruses
were diluted in RPMI-1640 (containing 10% FCS) and added to the
cells at 37°C for 24 h. After Ad-STAT1/3 transfection, the media
were replaced with serum-free RPMI-1640 media. After 24 h, cells
were used for experiments.
STAT knockdown in MCF7 cells was established by
transfection with small interfering RNA (siRNA) (Invitrogen,
Carlsbad, CA, USA). STAT1 siRNA (siSTAT1):
5′-GCGGAGACAGCAGAGCGCCUGUAUU-3′; STAT3 siRNA (siSTAT3):
5′-GCCAAUUGUGAUGCUUCCCUGAUUG-3′; and negative control siRNA
(siCont) by using Lipofectamine RNAiMAX reagent (Thermo Fisher
Scientific) according to the manufacturer's instructions.
Twenty-four hours later, cells were used for various experiments.
For H2O2 treatment,
H2O2 (100 µM) was applied to MCF7
cells. N-acetyl-cysteine (NAC) (20 mM) (Sigma) was used for ROS
inhibition.
Mitochondria isolation and
preparation
A large cell culture was prepared for mitochondria
isolation with a 15-cm dish being used per sample. After cells were
collected, cellular mitochondria extract buffer 1 (80 mM sucrose,
10 mM MOPS) was added and cells were homogenized at 1,500 rpm for 2
min. Then cellular mitochondria extract buffer 2 (250 mM sucrose,
20 mM MOPS) was added followed by centrifugation at 2,000 rpm at
4°C for 10 min. The supernatant was collected and centrifuged again
at 15,000 rpm at 4°C for 15 min. The precipitate containing
mitochondria was then dissolved in buffer 2 for protein
quantification. Fifty micrograms of mitochondria were incubated
with 50 µl proteinase K (50 µg/ml) to remove
non-mitochondrial proteins (30).
Then mitochondria were dissolved in cell lysis buffer (CST,
Shanghai, China) for western blotting.
Flow cytometry analysis
We performed FACS analysis to detect apoptosis.
Cells were cultured in 60-mm dishes and exposed to IFN for 48 h.
Cells (2×105 cells/500 µl) were labeled
fluorescently to detect apoptotic and necrotic cells by adding 50
µl binding buffer and 5 µl Annexin V-FITC
(Pharmingen, San Diego, CA, USA) as well as 2 µl of PI
(Cedarlane Laboratories, Hornby, Ontario, Canada) to each sample.
Samples were incubated at room temperature for 15 min after gentle
mixing. A minimum of 10,000 cells within the gated region were
analyzed by flow cytometry (Coulter Epics Altra flow cytometer;
Beckman Coulter, Fullerton, CA, USA).
Extracellular flux analysis
The cellular oxygen consumption rate (OCR) and
extracellular acidification rate (ECAR) were measured by a Seahorse
XF 24 extracellular flux analyzer (Agilent Technologies, Shanghai,
China). At the day of measurement, cells were changed to Flux
condition medium and incubated at 37°C for 1 h. Then cells were
measured by baseline OCR and ECAR 3 times. The average values were
calculated as the cellular OCR and ECAR. For measurement of
cellular OCR response to H2O2,
H2O2 (100 µm) was injected into wells
after the three basic measurements. Measurements were set to
continue until the cellular OCR response returned to the
baseline.
MitoSOX measurement
Cells were cultured in 96-well dishes. At the day of
measurement, cells were washed twice by sterilized PBS, then cells
were changed to 5 µM MitoSOX (M36008, Invitrogen, Shanghai,
China)-containing HBSS medium for another incubation at 37°C for 10
min. After three washes by warmed PBS, cells were measured at
Ex/Em: 510/580 and protected from light. The blank was set by
normal cells without MitoSOX incubation.
TUNEL assay
MCF7 cells were cultured in multi-glass slides.
Before cells were prepared for staining, they were washed by
sterilized PBS (5 min for three times), fixed by acetone (−30°C)
for 20 min, then washed with TBS. After a 1-h blocking by BSA at
room temperature, cells were reacted using a TdT-FragEL DNA
Fragmentation Detection kit (Calbiochem, USA) to quantify
apoptosis. Counterstaining with fluorescence mounting medium
containing DAPI (blue; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) was performed to visualize normal nuclei. Slices were observed
by use of a fluorescence microscope (Olympus FluoView™ FV1200).
Western blotting
Western blotting was performed as previously
described (31). Briefly, cells
were collected in sterilized PBS. Whole cell lysates or isolated
mitochondria were dissolved in cell lysis buffer (CST, #9803).
Total protein was adjusted to 1 mg/ml, and 10 µl was applied
to SDS-PAGE for 90 min, followed by transfer to PVDF membranes for
90 min. Then membranes were blocked in 5% skim milk for >1 h.
After 3 washes by TBST, membranes were incubated in specific
antibodies at 4°C overnight. Membranes were washed by TBST 3 times,
followed by incubation in second antibodies at room temperature for
1 h. After washing by TBST 3 times, a FluorChem E (Cell
Biosciences, Beijing, China) imaging system was used to visualize
the signals. First antibodies: STAT1 (#9172; CST), pSTAT1 (Tyr701)
(#7649, CST), STAT3 (#4904; CST), pSTAT3 (Tyr705) (#4113; CST),
ATP5A (ab110273; Abcam), UQCRC2 (ab103616; Abcam), SDHB (ab14714;
Abcam), NDUFB8 (ab192878; Abcam), GAPDH (ab37168; Abcam), Hsp60
(ab46798; Abcam), caspase 3 (#9662; CST), caspase 9 (#9508; CST).
All first antibodies were diluted 1,000-fold. Second antibodies:
anti-mouse IgG antibody (#7076; CST), anti-rabbit IgG antibody
(#7074; CST). All second antibodies were diluted 2,000-fold.
Statistical analysis
All results are reported as mean values ± standard
error. Comparisons between two groups were performed by unpaired
two-tailed t-tests. Multiple comparisons between more than two
groups were performed by one-way ANOVA with Tukey's multiple
comparisons test between each group. A value of p<0.05 was
considered significant.
Results
IFN induces STAT mitochondria importation
and increases cellular OCR, ROS, and apoptosis
To clarify the role of IFN in cancer cells, we used
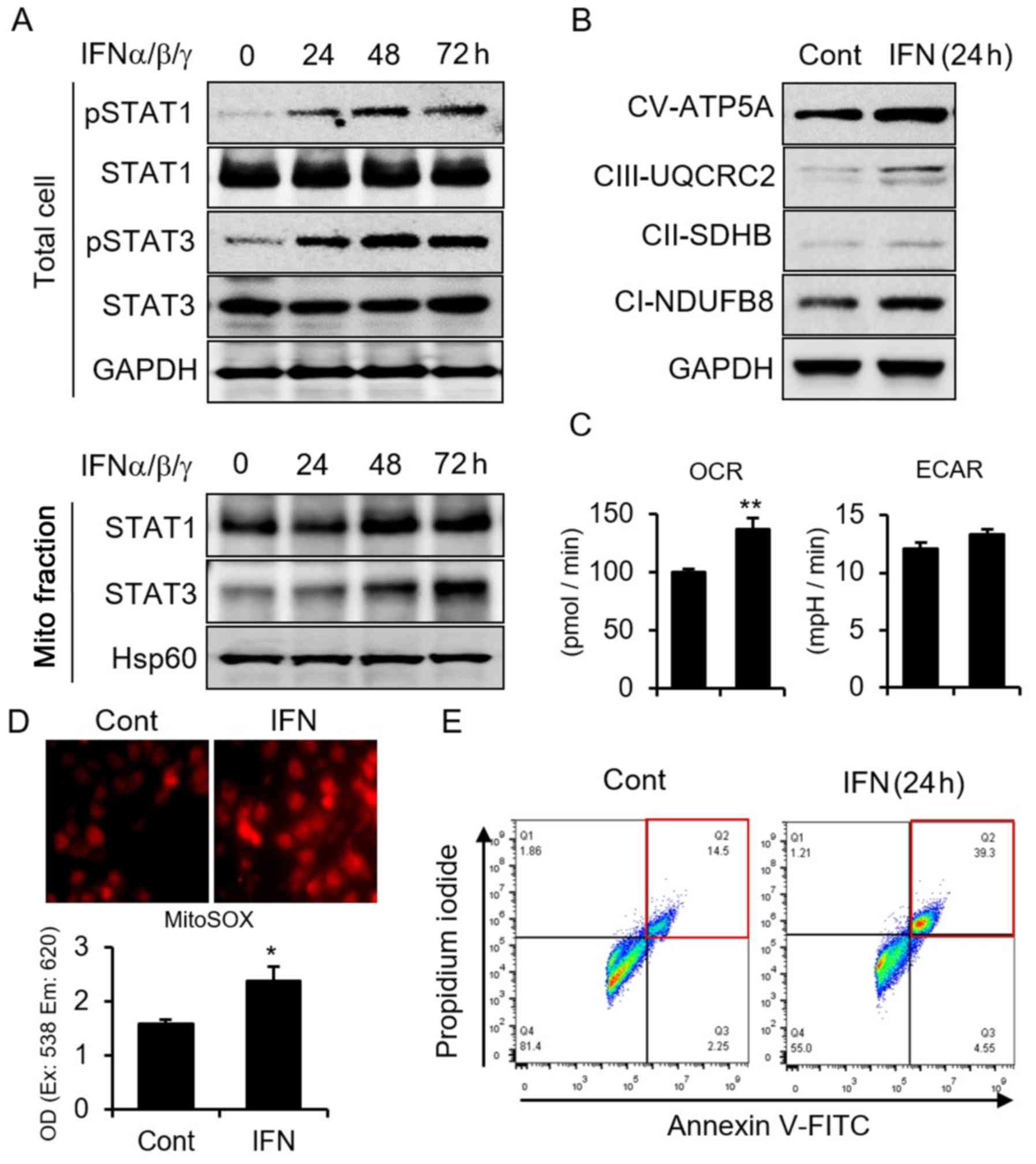
MCF7 breast cancer cells. We first performed a time-course study of
IFN incubation of MCF7 cells and found that IFN induces STAT1 and
STAT3 (STAT1/3) phosphorylation, which reaches a peak after 48 h of
incubation with IFN (Fig. 1A).
Meanwhile, after phosphorylation, we found increased STAT1/3 within
mitochondria, indicating that IFN induces STAT1/3 mitochondria
importation (Fig. 1A).
Additionally, IFN increased mitochondria respiratory complexes with
upregulated OCR (Fig. 1B and C).
Since mitochondria respiratory complexes have been identified as
the main source of ROS (16), we
examined and detected increased ROS generation induced by IFN
(Fig. 1D). The higher ROS thus
induced cancer cells apoptosis (Fig.
1E). These results indicated that IFN incubation induces
STAT1/3 mitochondria importation, OCR, increased ROS, and
subsequent cellular apoptosis.
The feedback between STAT and ROS
indicates the existence of a STAT-ROS cycle
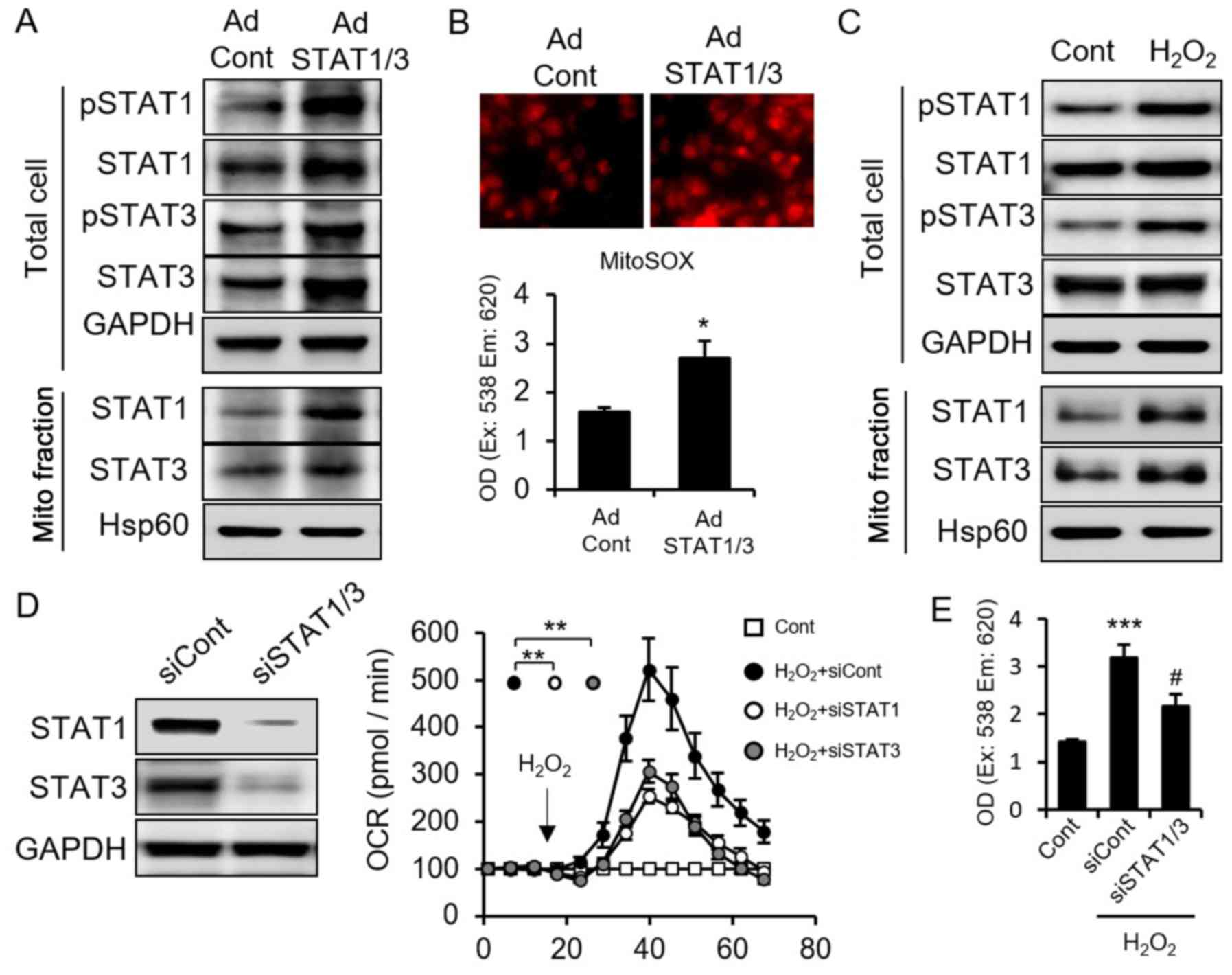
Next, we examined the interaction and relationship
between STAT and ROS. We overexpressed STAT1/3 and confirmed their
increase in both cellular and mitochondria subfractions (Fig. 2A). Cellular ROS level increased
after STAT1/3 overexpression (Fig.
2B), indicating that STAT1/3 induces ROS generation.
Conversely, to clarify whether and how STAT1/3 responds to ROS,
MCF7 cells were incubated with H2O2, a type
of ROS. We found that H2O2 increased STAT1
and STAT3 protein expression and induced mitochondria importation
(Fig. 2C). The mutual feedback
between STAT1/3 and ROS implied the existence of a STAT-ROS
cycle.
Recently, it was reported that production of ROS
4-hydroxynonenal, increases OCR, which is accompanied by more ROS
production (32). Increased ROS
induces more ROS release, known as ROS-induced ROS release (RIRR)
(33). So as STAT1/3 and ROS
induce each other, we next asked whether STAT1/3 mediates RIRR. We
knocked down STAT1/3 using siRNA and measured cellular OCR and ROS
levels. Interestingly, knockdown of STAT1/3 suppressed the OCR
induced by H2O2 treatment (Fig. 2D) as well as suppressed
H2O2-stimulated ROS (Fig. 2E). However, we found knockdown of
STAT1/3 did not block the ROS increase induced by
H2O2, that is because
H2O2 induced mitochondrial ROS include two
parts: one part is the H2O2 itself and the
another part is the generated ROS from mitochondria stimulated by
H2O2. So, it is correct that STAT1/3
inhibited only parts of the mitochondrial ROS, which maybe the
mitochondrial generated ROS by H2O2. This
result also demonstrated STAT1/3 suppression inhibited RIRR. These
results suggested that ROS induces OCR, and that ROS generation is
partially dependent on STAT1/3 activation. Taken together, we
conclude that STAT1/3 and ROS form an intercellular STAT-ROS cycle,
which amplifies ROS generation and enhances the ROS effect.
The STAT-ROS cycle extends IFN-induced
cancer cell apoptosis
As STAT-ROS cycle exists in MCF7 cancer cells
(Fig. 2), and IFN induces both
STAT1/3 activation and ROS production followed by cellular
apoptosis (Fig. 1), we
hypothesized that the STAT-ROS cycle plays a positive role in
IFN-induced cellular apoptosis. Therefore, we checked whether and
how the STAT-ROS cycle affects IFN-induced cellular apoptosis.
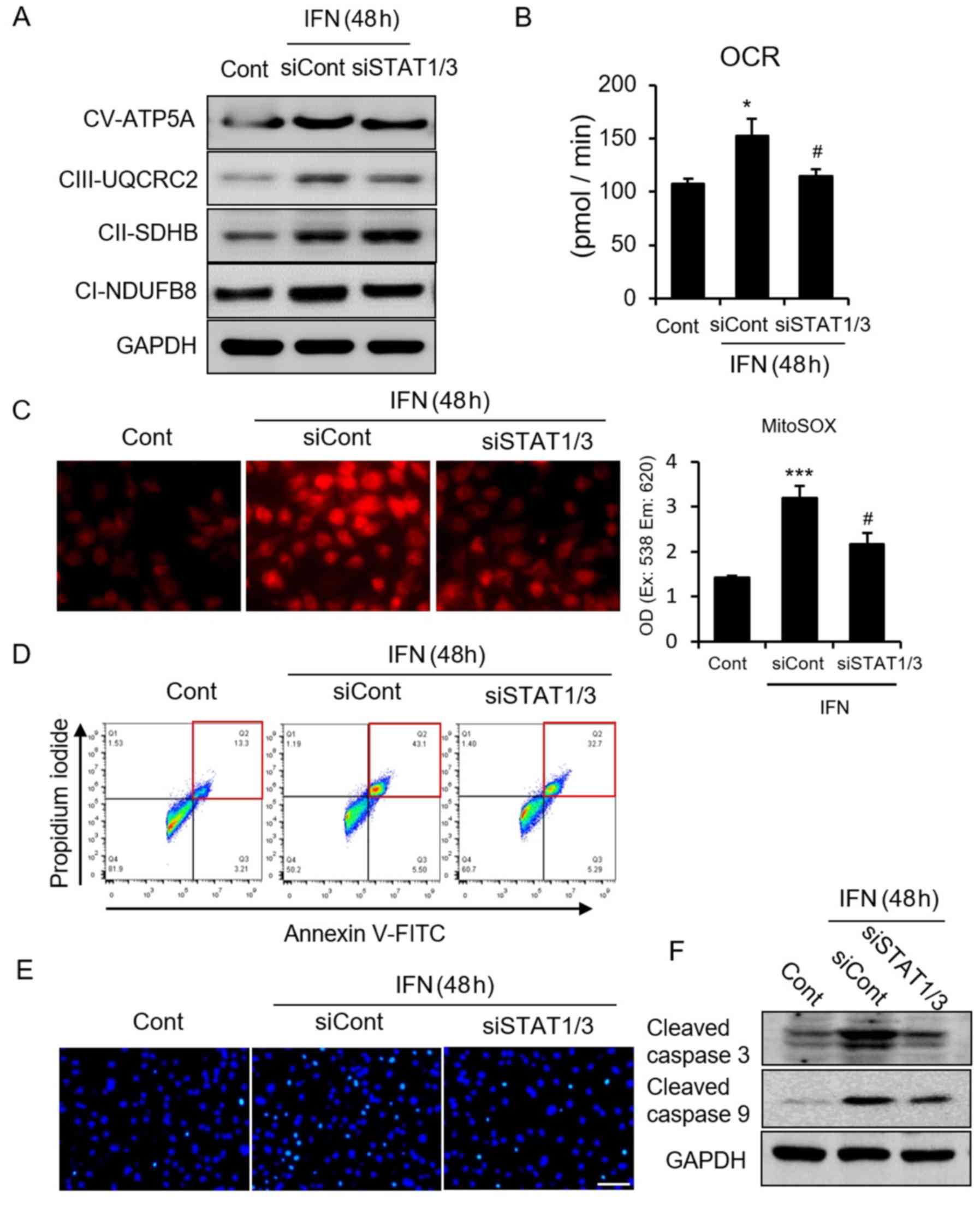
First, we confirmed the central role of STAT in
IFN-induced cellular apoptosis. STAT1/3 deletion inhibited the IFN
incubation-induced increase of mitochondria respiratory complex I
and III (Fig. 3A) and the
consequent increase in OCR and ROS (Fig. 3B and C). Next, to test whether
STAT1/3 deletion could suppress IFN-induced cellular apoptosis, we
performed FACS, TUNEL assays, and looked for caspase activation. We
found that cellular apoptosis was significantly inhibited by
STAT1/3 deletion (Fig. 3D–F),
indicating a crucial role of STAT in IFN-induced ROS-dependent
cellular apoptosis. Next, to clarify the role of ROS in the
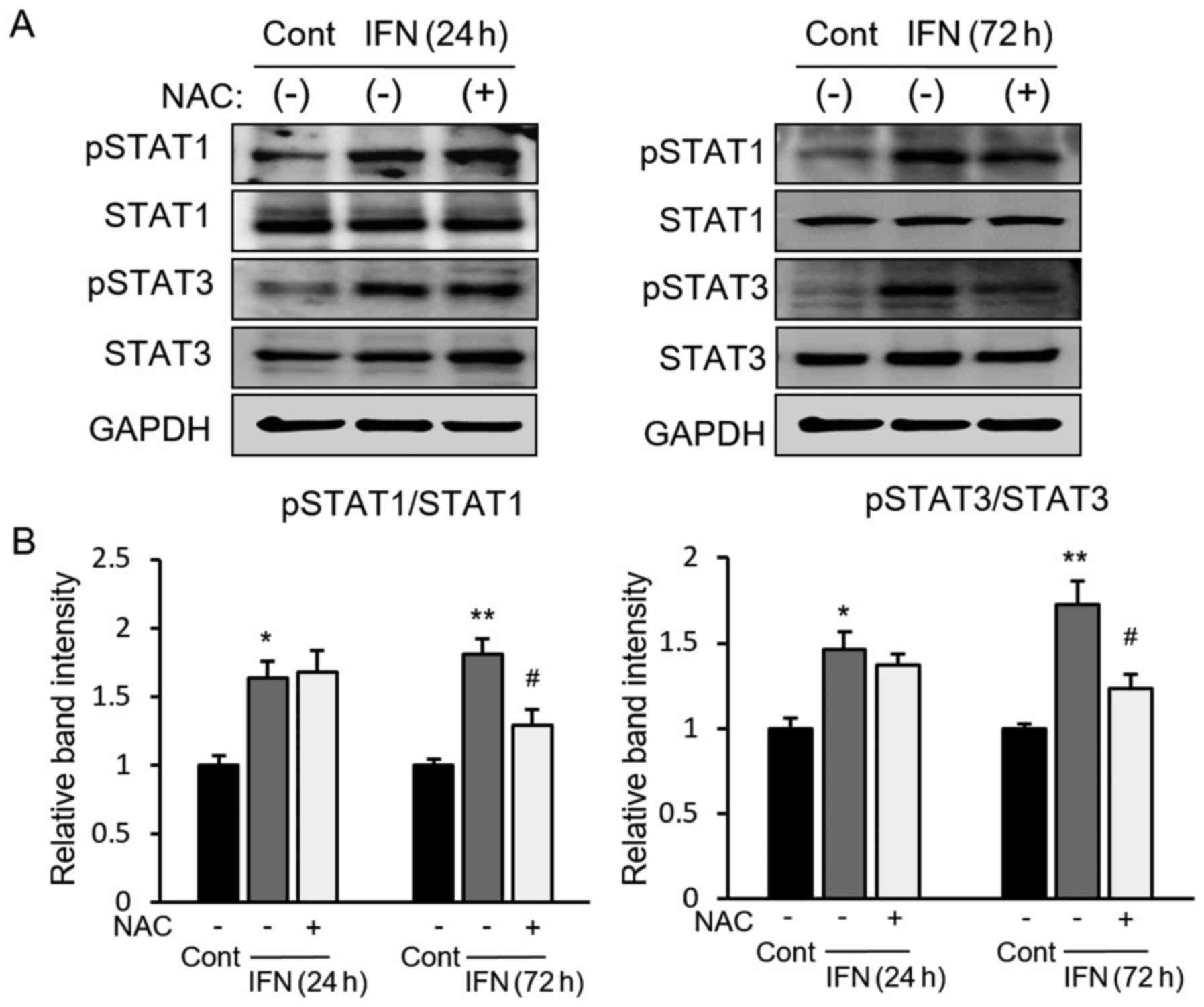
STAT-ROS cycle, we checked STAT1/3 activation after ROS inhibition
under IFN incubation. We found that ROS inhibition by NAC did not
affect the activation of STAT after a relatively short time (24 h)
during IFN incubation; however, significantly suppressed activation
of STAT did occur after a relatively long time (72 h) (Fig. 4A and B). Taken together, these
results suggested that IFN-induced ROS generation is initially
dependent on STAT activation, which induces ROS further and
activated STAT in a later phase of IFN incubation, indicating that
the STAT-ROS cycle extends the effect of IFN on cancer cells.
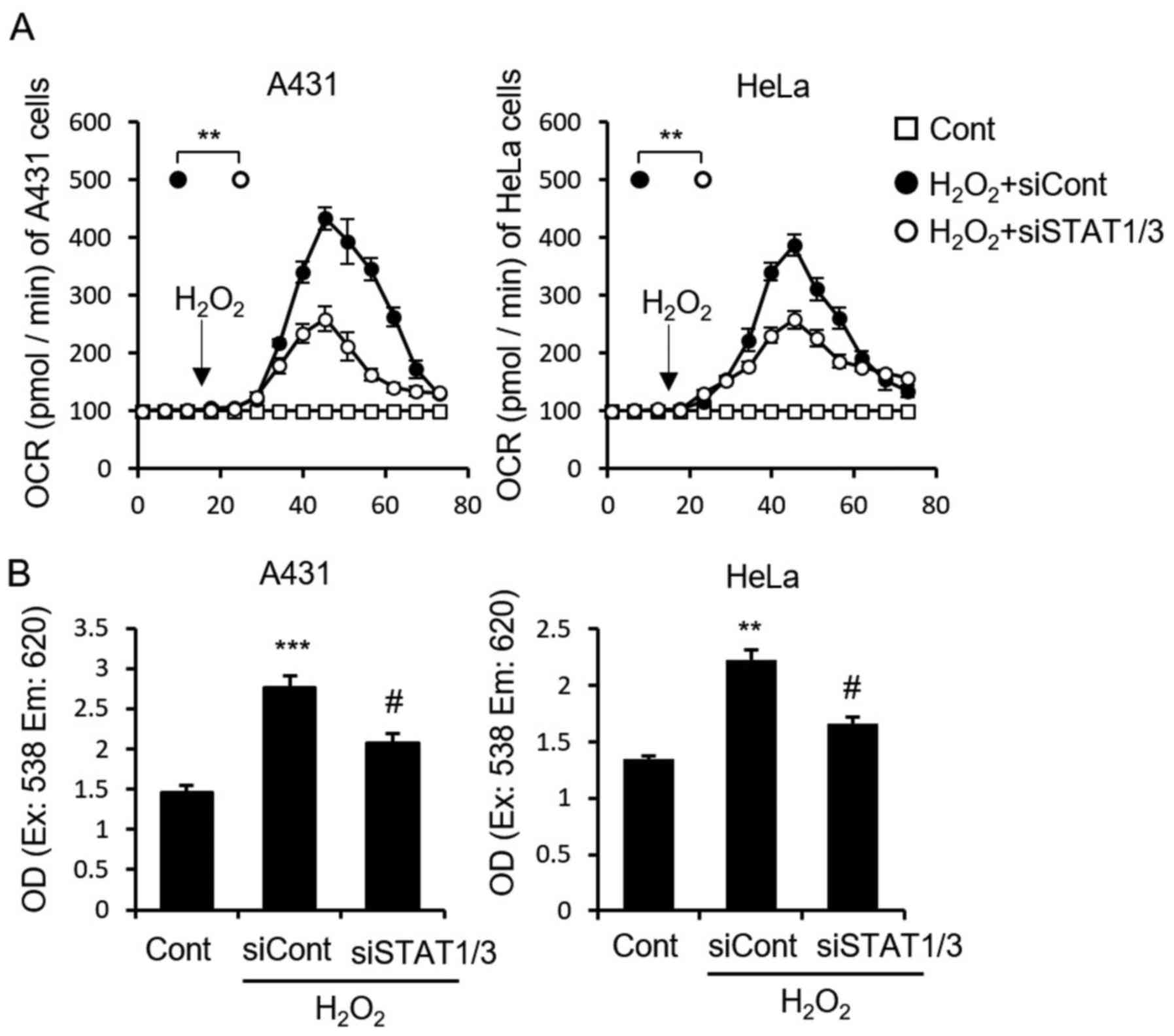
The STAT-ROS cycle exists in A431 and
HeLa cells
We demonstrated that the STAT-ROS cycle extends the
effect of IFN-induced apoptosis in MCF7 cells. It has been reported
that IFN activates STAT1 and induces apoptosis in both A431 and
HeLa cells (34). To determine
whether this cycle is common within cancer cells, we examined the
STAT-ROS cycle in A431 and HeLa cells. As expected, STAT1/3
deletion in both cell lines partially blocked the
H2O2-induced increase in cellular OCR
(Fig. 5A) as well as the
H2O2-induced increase in ROS (Fig. 5B), which were similar to that seen
used in the MCF7 cells (Fig. 2D and
E). These results indicated STAT suppression inhibited
H2O2 induced mitochondrial RIRR, demonstrated
the STAT-ROS cycle exists also in A431 and HeLa cells.
Discussion
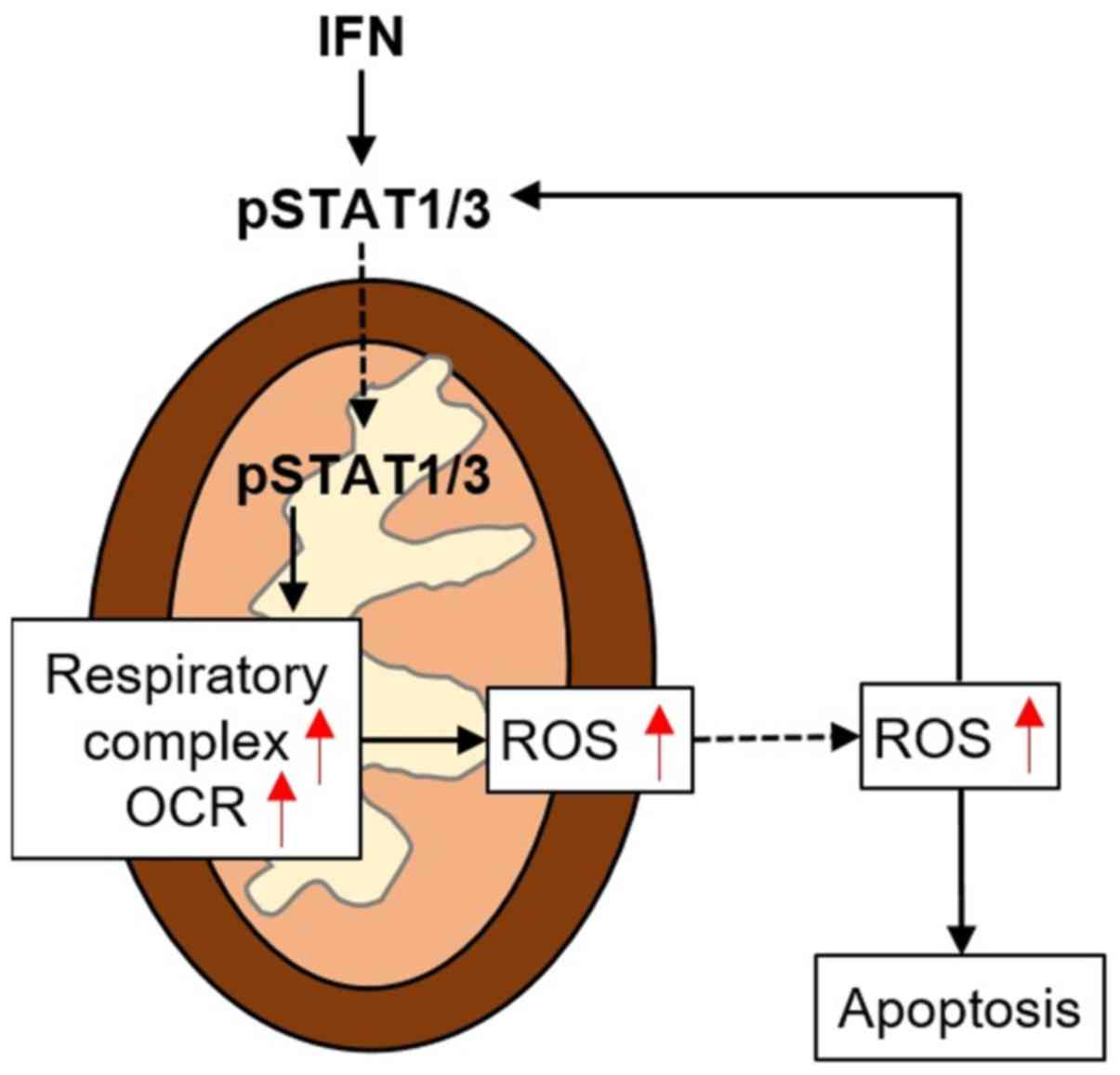
The present study demonstrated a potential cycle
between STAT1/3 and ROS in MCF7 cancer cells. This STAT-ROS cycle
facilitates RIRR to enhance the ROS effect. When STAT1/3 was
exposed to IFN, this cycle extended IFN-induced
mitochondria-dependent cellular apoptosis. Additionally, we also
found that this cycle exists in A431 and HeLa cells, indicating it
is a common mechanism in cancer cells to amplify IFN-induced
cellular apoptosis (Fig. 6).
Chemical cycles are very important for the efficient
usage of reagents and exist universally in vivo. For
example, the tricarboxylic acid and lactic acid cycles. Although
simpler than these classic chemical cycles, the STAT-ROS cycle may
commonly exist in cancer cells, allowing for efficient use of ROS
for further ROS production to damage cancer cells.
ROS-STAT cycle push ROS activity, make ROS more
efficient than without STAT. Although we have domenstrated this
mechanism was involved only in some cancer cells in the present
study, we think this cycle seems not only limited to these cancer
cells but also commonly exists in some normal cell (these
speculation should to be confirmed). So, in our opinion, a bit ROS
can be enlarged by ROS-STAT cycle, which makes ROS generationg and
release more efficient. Clearly, oxidative stress clearance system
was also involved in this cycle. Unquestionably, ROS-STAT cycle
enhances the ROS clearance system, which was suppressed by STAT
knockdown (data not shown). However, we think the activated ROS
clearance system was dependent on ROS level due to ROS-STAT cycle,
but not directly affected by this cycle, because ROS could activate
oxidative stress clearance system directly.
We found that communication between mitochondria and
cytoplasm was necessary for this reactive system. The STAT-ROS
cycle begins with exposure to IFN to induce STAT, but ends with ROS
production. It is thus necessary for ROS to move from mitochondria
to the cytosol, which requires a reversible mitochondria
permeability transition pore (mPTP) opening, an inner membrane
anion channel (IMAC) opening, or mitochondria membrane potential
(Δψm) loss (33).
Interestingly, STAT3 can induce mPTP openings (35,36),
and STAT1 and IRF1 synergistically induce Δψm loss
(14), suggesting that STAT not
only induces ROS production but also creates the necessary
conditions (mPTP or lost Δψm) to facilitate ROS release
from mitochondria. Released ROS induces cellular apoptosis but also
further promotes ROS production through STAT mitochondria
importation. NFκB has been reported to be activated by ROS and
further induces STAT, indicating that NFκB may be involved in
ROS-activated STAT (37). However,
in addition to STAT1 and STAT3, released ROS also activates STAT5
and STAT6 (38,39), though their effect on mitochondria
is largely unknown. Therefore, it is necessary to investigate
whether STAT5 or STAT6 are also involved in mitochondria
importation.
There are two pathways for STAT signaling, the
classical pathway in which phosphorylated STAT is translocated to
the nucleus, and the non-classical pathway in which phosphorylated
STAT is translocated to mitochondria (40). Interestingly, these two distinct
pathways are linked into an integrated system as STAT, in the
classical pathway, it induces the transcription of mitochondria
genes, while in the non-classical pathway, STAT-mitochondria
importation further promotes mitochondrial activities such as ATP
production and ROS generation (40). Although the two distinct activities
of phosphorylated STAT lead to some loss of mitochondria
importation, nucleus-translocated STAT supports mitochondria
biogenesis and activity to promote the STAT-ROS cycle.
STAT activation in cancer cells is common (41–47).
STAT activates the autonomous proliferation of SUM-102PT and MDA468
human breast cancer cells through an autocrine/paracrine
interaction with HB-EGF (41), and
in pancreatic cancer cells activates BxPC-3, AsPC-1, Capan-2,
MiaPaCa-2, Panc-1, and HPDE-6 (42,43).
It is activated in myeloma and lung cancer cells (44) as well as in prostate cancer cells
(45,46). Although in most of these cancer
cells, STAT phosphorylation is involved in tumor growth, and thus
suppression of STAT activation should facilitate cancer cells
apoptosis, the STAT/ROS cycle should extend the effect of ROS and
facilitate tumor apoptosis (14,18,19).
The role of STAT in both tumorigenesis and tumor suppression may be
explained by differences in the classical and non-classical
pathways (48), namely genomic vs.
non-genomic effects. It is therefore very important to understand
how STAT classical or genomic pathway and non-classical or
non-genomic pathway are mediated or balanced in specific cells. We
hypothesize that the non-classical pathway, and not the classical
pathway, is involved in STAT activation to induce cancer cell
apoptosis. Considering this, the STAT/ROS cycle should only exist
in mitochondria or be confined to specific intracellular
locations.
In conclusion, we demonstrated the existence of a
STAT/ROS cycle in some cancer cells (MCF7, A431 and HeLa). The
STAT/ROS cycle involves STAT phosphorylation with mitochondria
importation, mitochondria respiratory complex increase, and ROS
production and release from mitochondria. This cycle extends the
effect of IFN and facilitates IFN-induced cancer cell apoptosis.
This novel concept may provide new methods for improving IFN
therapy.
Acknowledgments
We thank our colleagues for useful suggestions and
comments. This study was supported by a grant from the National
Natural Science Foundation of China (81372785).
References
|
1
|
Arbuthnot P, Capovilla A and Kew M:
Putative role of hepatitis B virus X protein in
hepatocarcinogenesis: Effects on apoptosis, DNA repair,
mitogen-activated protein kinase and JAK/STAT pathways. J
Gastroenterol Hepatol. 15:357–368. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beebe K, Lee WC and Micchelli CA: JAK/STAT
signaling coordinates stem cell proliferation and multilineage
differentiation in the Drosophila intestinal stem cell lineage. Dev
Biol. 338:28–37. 2010. View Article : Google Scholar
|
|
3
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aaronson DS and Horvath CM: A road map for
those who don't know JAK-STAT. Science. 296:1653–1655. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schindler C and Darnell JE Jr:
Transcriptional responses to polypeptide ligands: The JAK-STAT
pathway. Annu Rev Biochem. 64:621–651. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuan ZL, Guan YJ, Chatterjee D and Chin
YE: Stat3 dimerization regulated by reversible acetylation of a
single lysine residue. Science. 307:269–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie Y, Kole S, Precht P, Pazin MJ and
Bernier M: S-glutathionylation impairs signal transducer and
activator of transcription 3 activation and signaling.
Endocrinology. 150:1122–1131. 2009. View Article : Google Scholar :
|
|
8
|
Ray S, Boldogh I and Brasier AR: STAT3
NH2-terminal acetylation is activated by the hepatic acute-phase
response and required for IL-6 induction of angiotensinogen.
Gastroenterology. 129:1616–1632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Horvath CM: The Jak-STAT pathway
stimulated by interleukin 6. Sci STKE. 2004:tr92004.PubMed/NCBI
|
|
10
|
Leonard WJ: Role of Jak kinases and STATs
in cytokine signal transduction. Int J Hematol. 73:271–277. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levy JB, Schindler C, Raz R, Levy DE,
Baron R and Horowitz MC: Activation of the JAK-STAT signal
transduction pathway by oncostatin-M cultured human and mouse
osteoblastic cells. Endocrinology. 137:1159–1165. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lufei C, Ma J, Huang G, Zhang T,
Novotny-Diermayr V, Ong CT and Cao X: GRIM-19, a death-regulatory
gene product, suppresses Stat3 activity via functional interaction.
EMBO J. 22:1325–1335. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tamminen P, Anugula C, Mohammed F,
Anjaneyulu M, Larner AC and Sepuri NB: The import of the
transcription factor STAT3 into mitochondria depends on GRIM-19, a
component of the electron transport chain. J Biol Chem.
288:4723–4732. 2013. View Article : Google Scholar
|
|
14
|
Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY,
Park YS, Kwon Y, Choi KH, Jo I, Park SI, et al: The role of
STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial
transmembrane potential during hepatic cell death induced by
LPS/d-GalN. J Mol Biol. 369:967–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB,
Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et
al: Function of mitochondrial Stat3 in cellular respiration.
Science. 323:793–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL
and Lesnefsky EJ: Production of reactive oxygen species by
mitochondria: Central role of complex III. J Biol Chem.
278:36027–36031. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Batandier C, Guigas B, Detaille D, El-Mir
MY, Fontaine E, Rigoulet M and Leverve XM: The ROS production
induced by a reverse-electron flux at respiratory-chain complex 1
is hampered by metformin. J Bioenerg Biomembr. 38:33–42. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HS, Cho IH, Kim JE, Shin YJ, Jeon JH,
Kim Y, Yang YM, Lee KH, Lee JW, Lee WJ, et al: Ethyl pyruvate has
an anti-inflammatory effect by inhibiting ROS-dependent STAT
signaling in activated microglia. Free Radic Biol Med. 45:950–963.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu T, Castro S, Brasier AR, Jamaluddin M,
Garofalo RP and Casola A: Reactive oxygen species mediate
virus-induced STAT activation: Role of tyrosine phosphatases. J
Biol Chem. 279:2461–2469. 2004. View Article : Google Scholar
|
|
20
|
Simon AR, Rai U, Fanburg BL and Cochran
BH: Activation of the JAK-STAT pathway by reactive oxygen species.
Am J Physiol. 275:C1640–C1652. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parkin J and Cohen B: An overview of the
immune system. Lancet. 357:1777–1789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiladjian JJ, Giraudier S and Cassinat B:
Interferon-alpha for the therapy of myeloproliferative neoplasms:
Targeting the malignant clone. Leukemia. 30:776–781. 2016.
View Article : Google Scholar
|
|
23
|
Cao ZH, Zheng QY, Li GQ, Hu XB, Feng SL,
Xu GL and Zhang KQ: STAT1-mediated down-regulation of Bcl-2
expression is involved in IFN-γ/TNF-α-induced apoptosis in NIT-1
cells. PLoS One. 10:e01209212015. View Article : Google Scholar
|
|
24
|
Ethiraj P, Veerappan K, Samuel S and
Sivapatham S: Inhibitory effects of interferon-β on hepatocellular
carcinoma HepG2 via Akt/STAT phosphorylation. Fundam Clin
Pharmacol. 29:278–285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bello C, Vazquez-Blomquist D, Miranda J,
Garcia Y, Novoa LI, Palenzuela D and Bello I: Regulation by
IFN-α/IFN-γ co-formulation (HerberPAG®) of genes
involved in interferon-STAT-pathways and apoptosis in U87MG. Curr
Top Med Chem. 14:351–358. 2014. View Article : Google Scholar
|
|
26
|
Zídek Z, Jansa P and Kmoníčková E:
Activation of respiratory complex II by interferon-gamma and its
inhibition by pyrimidine derivatives. Neuro Endocrinol Lett.
35(Suppl 2): 141–148. 2014.
|
|
27
|
Huang G, Chen Y, Lu H and Cao X: Coupling
mitochondrial respiratory chain to cell death: An essential role of
mitochondrial complex I in the interferon-beta and retinoic
acid-induced cancer cell death. Cell Death Differ. 14:327–337.
2007. View Article : Google Scholar
|
|
28
|
Kim KB, Eton O, East MJ, Hodges C,
Papadopoulos NE, Grimm EA and Bedikian AY: Pilot study of
high-dose, concurrent biochemotherapy for advanced melanoma.
Cancer. 101:596–603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henry SC, Schmidt EA, Fessler MB and
Taylor GA: Palmitoylation of the immunity related GTPase, Irgm1:
Impact on membrane localization and ability to promote
mitochondrial fission. PLoS One. 9:e950212014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang JY, Deng W, Chen Y, Fan W, Baldwin
KM, Jope RS, Wallace DC and Wang PH: Impaired translocation and
activation of mitochondrial Akt1 mitigated mitochondrial oxidative
phosphorylation Complex V activity in diabetic myocardium. J Mol
Cell Cardiol. 59:167–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian Z, Miyata K, Kadomatsu T, Horiguchi
H, Fukushima H, Tohyama S, Ujihara Y, Okumura T, Yamaguchi S, Zhao
J, et al: ANGPTL2 activity in cardiac pathologies accelerates heart
failure by perturbing cardiac function and energy metabolism. Nat
Commun. 7:130162016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sansbury BE, Riggs DW, Brainard RE,
Salabei JK, Jones SP and Hill BG: Responses of hypertrophied
myocytes to reactive species: Implications for glycolysis and
electrophile metabolism. Biochem J. 435:519–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chin YE, Kitagawa M, Kuida K, Flavell RA
and Fu XY: Activation of the STAT signaling pathway can cause
expression of caspase 1 and apoptosis. Mol Cell Biol. 17:5328–5337.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith CC, Dixon RA, Wynne AM, Theodorou L,
Ong SG, Subrayan S, Davidson SM, Hausenloy DJ and Yellon DM:
Leptin-induced cardioprotection involves JAK/STAT signaling that
may be linked to the mitochondrial permeability transition pore. Am
J Physiol Heart Circ Physiol. 299:H1265–H1270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lemoine S, Zhu L, Legallois D, Massetti M,
Manrique A and Hanouz JL: Atorvastatin-induced cardioprotection of
human myocardium is mediated by the inhibition of mitochondrial
permeability transition pore opening via tumor necrosis factor-α
and Janus kinase/signal transducers and activators of transcription
pathway. Anesthesiology. 118:1373–1384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dvorak K and Dvorak B: Role of
interleukin-6 in Barrett's esophagus pathogenesis. World J
Gastroenterol. 19:2307–2312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Santambrogio P, Erba BG, Campanella A,
Cozzi A, Causarano V, Cremonesi L, Gallì A, Della Porta MG,
Invernizzi R and Levi S: Over-expression of mitochondrial ferritin
affects the JAK2/STAT5 pathway in K562 cells and causes
mitochondrial iron accumulation. Haematologica. 96:1424–1432. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park SJ1, Lee JH, Kim HY, Choi YH, Park
JS, Suh YH, Park SM, Joe EH and Jou I: Astrocytes, but not
microglia, rapidly sense H2O2 via STAT6
phosphorylation, resulting in cyclooxygenase-2 expression and
prostaglandin release. J Immunol. 188:5132–5141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jesel M, Isch-Treussard C, Poenaru S and
Isch F: Electromyography in the study of thyrotoxic myopathies. Rev
Electroencephalogr Neurophysiol Clin. 3:183–192. 1973.In French.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sartor CI, Dziubinski ML, Yu CL, Jove R
and Ethier SP: Role of epidermal growth factor receptor and STAT-3
activation in autonomous proliferation of SUM-102PT human breast
cancer cells. Cancer Res. 57:978–987. 1997.PubMed/NCBI
|
|
42
|
Sahu RP and Srivastava SK: The role of
STAT-3 in the induction of apoptosis in pancreatic cancer cells by
benzyl isothiocyanate. J Natl Cancer Inst. 101:176–193. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thoennissen NH, Iwanski GB, Doan NB,
Okamoto R, Lin P, Abbassi S, Song JH, Yin D, Toh M, Xie WD, et al:
Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–5884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liby K, Voong N, Williams CR, Risingsong
R, Royce DB, Honda T, Gribble GW, Sporn MB and Letterio JJ: The
synthetic triterpenoid CDDO-Imidazolide suppresses STAT
phosphorylation and induces apoptosis in myeloma and lung cancer
cells. Clin Cancer Res. 12:4288–4293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mora LB, Buettner R, Seigne J, Diaz J,
Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, et
al: Constitutive activation of Stat3 in human prostate tumors and
cell lines: Direct inhibition of Stat3 signaling induces apoptosis
of prostate cancer cells. Cancer Res. 62:6659–6666. 2002.PubMed/NCBI
|
|
46
|
Ni Z, Lou W, Leman ES and Gao AC:
Inhibition of constitutively activated Stat3 signaling pathway
suppresses growth of prostate cancer cells. Cancer Res.
60:1225–1228. 2000.PubMed/NCBI
|
|
47
|
Burke WM, Jin X, Lin HJ, Huang M, Liu R,
Reynolds RK and Lin J: Inhibition of constitutively active Stat3
suppresses growth of human ovarian and breast cancer cells.
Oncogene. 20:7925–7934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bromberg J: Stat proteins and oncogenesis.
J Clin Invest. 109:1139–1142. 2002. View Article : Google Scholar : PubMed/NCBI
|