Introduction
Lipid nanoparticles have been widely used as
pharmaceutical carriers to increase the efficacy of
chemotherapeutics (1). These drug
carriers are expected to passively accumulate in tumors with leaky
vasculature, via the enhanced permeability and retention (EPR)
effect (2). One successful example
is the reformulation of doxorubicin encapsulated into PEGylated
liposomes. Doxorubicin is an anthracycline widely used in the
treatment of various cancers, including metastatic breast cancer,
ovarian cancer, and AIDS-related Kaposi's sarcoma; however, its
efficacy is limited by its toxicity. Liposomal formulation enhances
the therapeutic index of anticancer drugs, either by improving the
pharmacokinetic and pharmacodynamic profiles, or by decreasing the
exposure of normal host tissues.
In order to improve the specificity of nanocarriers,
targeting ligands may be attached to their surface via a PEG spacer
arm. The targeting ligands, which are attached to the distal end of
the spacer arm on the surface of nanocarriers, facilitate access of
the carrier to the targeted site of interaction (3). Delivery of nanoparticle involving the
use of peripherally-conjugated targeting moieties is known as
active targeting. Active targeting is a promising tool for the
treatment of cancer due to its ability to increase therapeutic
effectiveness and reduce potential side effects. Active tumor
targeting has been achieved using various targeting ligands,
including antibodies (4) and their
molecular fragments (5), nucleic
acids aptamers (6), and small
molecules: vitamins (7,8), peptides (9–12),
and carbohydrates (13). The
targeted form of PEGylated liposomal doxorubicin is expected to
become a major part of the next generation of this therapeutic
modality.
The SP94 peptide (SFSIIHTPILPL), a targeting ligand
isolated from phage-displayed selections (10,14),
was reported to possess high and specific affinity for various
human hepatocellular carcinoma cell lines, and exhibit minimal
interaction with healthy hepatocytes and other tissues (10,14).
At the present time, the antigen recognized by SP94 has not been
identified, but it has been immunohistochemically characterized.
SP94 can bind to tumor cells in surgical specimens of
hepatocellular carcinoma, but not to their normal counterparts. The
unknown target molecule recognized by SP94 was reported to be
expressed in ~60% of patients with hepatocellular carcinoma
(10).
In recent years, additional tumor-targeted delivery
platforms modified with SP94 peptide are also being developed,
including mesoporous silica nanoparticle-supported lipid bilayers
(protocells) (14,15), bacteriophage MS2 virus-like
particles (VLPs) (16) and HCC
targeting probe (99mTc/188Re-HYNIC-SP94) for
imaging and therapy (17).
Multivalent binding of SP94 peptide results in a 10,000-fold
greater avidity for human hepatocellular carcinoma than for
hepatocytes, endothelial cells, peripheral blood mononuclear cells,
B-lymphocytes, or T-lymphocytes (14). Exquisite targeting specificity of
SP94 peptide combined with enhanced tumor delivery of
multicomponent cargos permits sensitive discrimination between
target and normal tissue. Thus, SP94 peptide is an ideal model with
which to investigate the mechanism of active tumor targeting.
The potential of SP94 in drug delivery was
subsequently evaluated using SP94-conjugated,
doxorubicin-encapsulated liposomes. It was previously shown that
SP94-LD is more effective than non-targeted LD in treating mice
with xenografts of human hepatocellular carcinoma (10). SP94-targeted PEGylated liposomal
doxorubicin (SP94-LD) is believed to accumulate around cancerous
tissue (via the EPR effect) and bind to the cancer cell surface,
then being internalized by ligand-mediated endocytosis (via active
targeting effect) (18). However,
the role of active targeting in nanoparticle delivery is
controversial, and it is difficult to predict how a targeted
nanoparticle drug will behave in vivo. Herein, we confirm
the mechanisms underlying the enhanced cellular uptake of
SP94-modified nanoparticles, and moreover, investigate the
contribution of SP94 peptide during the cellular internalization of
SP94-targeted nanoparticles.
To date, the only chemotherapeutic agent to exert a
survival benefit in patients with hepatocellular carcinoma is
sorafenib (19,20). In order to evaluate the feasibility
of introducing SP94-targeted nanomedicine into clinical trials
against liver cancer, we examined the pharmacokinetic profile,
biodistribution, and in vivo antitumor activity of
SP94-targted nanomedicine against hepatocellular carcinoma, and
compared these properties to those of free drugs and non-targeted
liposomal drugs. Furthermore, we found synergistic/additive growth
inhibition by combination of doxorubicin and vinorelbine in HCC
cell lines in vitro and in vivo. For in vivo
evaluation, we developed an orthotopic hepatocellular carcinoma
model to recapitulate the tumor growth pattern observed in liver
cancer patients, and used this model to study the influence of the
liver microenvironment on response to the combination therapy.
Materials and methods
Cell lines and cell culture
The Mahlavu and SK-HEP-1 human hepatocellular
carcinoma lines were used in this study. The cell lines were
maintained in DMEM and 10% fetal bovine serum at 37°C in a
humidified atmosphere of 5 % CO2 in air.
Peptide synthesis and labeling
Targeting SP94 (SFSIIHTPILPL) peptides were
synthesized and purified by reverse-phase high-performance liquid
chromatography to >95% purity by the Peptide Synthesis Core
Facility, Institute of Cellular and Organismic Biology, Academia
Sinica. The predicted mass was confirmed by mass spectrometry.
Synthesis of peptide-PEG-DSPE
conjugates
A total of 8.5 mg of NHS-PEG-DSPE
[N-hydroxysuccinimido-carboxyl-polyethyleneglycol (MW,
3400)-derived distearoylphosphatidyl ethanolamine] (NOF Corp.)
dissolved in 0.25 ml of dichloromethane (Sigma-Aldrich) was added
to 0.25 ml of DMSO (Sigma-Aldrich) containing 3.1 mg of peptide.
This was then mixed with 0.011 ml of triethylamine (Sigma-Aldrich)
to catalyze the reaction. The stoichiometric molar ratio of peptide
and NHS-PEG-DSPE was 1.1:1. The reaction was carried out for 72 h
at room temperature. The peptide-PEG-DSPE conjugates were purified
by dialysis with a 2-kDa cut-off membrane (Spectrum), and were then
dried through lyophilization.
Preparation of peptide-liposomal
drugs
A lipid film hydration method was used to prepare
PEGylated liposomes composed of distearoylphosphatidylcholine,
cholesterol, and mPEG2000-DSPE, which were then used to encapsulate
doxorubicin (3:2:0.3 molar ratio) or vinorelbine (3:2:0.15 molar
ratio). The lipid films were hydrated at 60°C in 250 mM ammonium
sulfate or 300 mM ammonium salts of 5-sulfosali-cylic acid
solution, and were extruded through polycarbonate membrane filters
with a pore size of 0.1 µm using high-pressure extrusion
equipment (Lipex Biomembranes, Vancouver, BC, Canada) at 55°C.
Doxorubicin or vinorelbine were encapsulated by a remote loading
method, at concentrations of 1 mg or 3.5 mg per 10 µmol of
phospholipid, respectively. The final concentration of liposome was
estimated by phosphate assay. The peptide-PEG-DSPE was subsequently
incorporated into pre-formed liposomes by co-incubation at 60°C,
the transition temperature of the lipid bilayer, for 0.5 h with
gentle shaking. Sepharose 4B (GE Healthcare) gel filtration
chromatography was used to remove released free drug, unconjugated
peptides, and unincorporated conjugates. Doxorubicin concentrations
in the fractions of eluent were determined by measuring
fluorescence at λEx/Em = 485/590 nm using a spectrofluorometer
(Spectra Max M5, Molecular Devices). Vinorelbine concentrations
were determined using the HPLC method.
Bacteriophage preparation and
labeling
M13 bacteriophages were amplified in Escherichia
coli, and phage titers were determined according to published
procedures. Following titer determination, the bacteriophages were
simultaneously labeled with succinimidyl esters of HiLyte
Fluor™ 750 using a modified procedure (21,22).
Briefly, HiLyte Fluor 750 succinimidyl ester was added to
M13 bacteriophage in 100 µM bicarbonate buffer, pH 8.3. The
resulting solution was incubated for 1 h at room temperature in the
dark. Following incubation, the labeled bacteriophage was
precipitated by addition of a PEG-8000/2.5 M NaCl solution, and
then left to stand on ice for 30 min. The bacteriophage was
pelleted by centrifugation at 10,000 × g for 15 min. After removal
of the supernatant, the pellet was resuspended in DPBS buffer.
Typical dye labeling using this procedure resulted in 400–500
copies of each dye per bacteriophage particle.
In vivo near-infrared fluorescence
imaging
A total of 4×1011 pfu of HiLyte Fluor
750-labeled PC94 phage was diluted in 100 µl saline
solution. The saline solution was injected i.v. into mice bearing
subcutaneous Mahlavu tumors. A control solution of
4×1011 pfu of the HiLyte Fluor 750-labeled
control phage was injected into
NOD.CB17-Prkdcscid/J mice bearing subcutaneous
Mahlavu tumors. In vivo imaging was performed using a
Xenogen IVIS® Imaging System 200. The animal was imaged
at 0.1, 0.5, 6, 24, and 48 h post-injection using a Indocyanine
Green (ICG) Filter set (excitation 710–760 nm, emission 810–875
nm). Organs were dissected and imaged 48 h after injection of
conjugate.
Animal model for in vivo targeting
assay
The dorsolateral flanks of severe combined
immunodeficient mice, NOD. CB17-Prkdcscid/J (4–6
weeks old), were injected s.c. with 5×106 Mahlavu or
SK-HEP-1 cells. Tumors were measured with calipers, and mice were
weighed twice weekly. The tumor volumes were calculated using the
following formula: length × (width)2 × 0.52. All animals
were cared for in a specific pathogen-free room and treated in
accordance with the animal care protocol approved by the Academia
Sinica Animal Committee (approval no. 11-06-190).
Quantitative analysis
Plasma, brain, heart, lung, liver, kidney, or tumor
samples were processed using the extraction procedure, and then
analyzed using the method described by Laginha et al
(23). Result concentrations were
determined relative to the respective calibration curves.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL)
Frozen tumor tissue sections were incubated with
TUNEL reaction mixture (Roche Diagnostics) at 37°C for 1 h. The
slides were counterstained with Hoechst 33258 (Molecular Probes)
and mounted with mounting medium (Vector Laboratories). The slides
were then visualized under a fluorescent microscope. The sections
were analyzed using automated cell acquisition (TissueGnostics),
and TUNEL-positive areas were quantified using MetaMorph software
(Molecular Devices).
CD31 staining
The frozen tumor tissue sections were fixed with
methanol/acetone (1:1), washed with PBS, and immersed in blocking
buffer (1 % bovine serum albumin in PBS), followed by incubation
with rat anti-mouse CD31 (BD Pharmingen). The sections were washed
with PBST0.1 (0.1 % Tween-20 in PBS), and then incubated with
rabbit anti-rat antibody (Stressgen) and immersed in
rhodamine-labeled goat anti-rabbit antibody solution (Jackson
ImmunoResearch). The slides were counter-stained with Hoechst
33258, mounted with mounting medium, and visualized under a
fluorescent microscope.
Complete blood count
Blood was collected by venipuncture of the right
submandibular vein of unanesthetized mice with a lancet. Samples
were drawn into plastic K2 EDTA blood-drawing tubes. Complete blood
counts were performed using the Abbott CELL-DYN 3700 Hematology
Analyzer (Abbott Diagnostics, Abbott Park, IL, USA). The parameters
assessed were as follows: white blood cell (WBC) count, neutrophil
absolute count (NEU), lymphocyte absolute count (LYM), red blood
cell/erythrocyte count (RBC), hemoglobin concentration (HGB),
hematocrit (HCT), red cell distribution width (RDW),
platelet/thrombocyte (PLT), mean platelet volume count (MPV),
plateletcrit (PCT), neutrophil percentage (NEU %), lymphocyte
percentage (LYM %), monocyte percentage (MONO %), monocyte absolute
count (MONO), mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC),
and platelet distribution width (PDW).
Drug combination study
Mahlavu cells were seeded in 96-well plates and
allowed to attach overnight. Cells were treated in 96-well format
in triplicate for each drug concentration combination, and
viability was assessed after 3 days of treatment using the
MTT-based assay. The cytotoxicity of each drug and of their
combinations was assessed by drug response matrix using the
Combenefit software (24). Loewe
synergy score and HSA synergy score were calculated and plotting by
SynergyFinder software (25).
Orthotopic implantation of human
hepatocellular carcinoma in mice
NOD.CB17-Prkdcscid/J mice were
used for HCC implantation. SK-HEP-1 cells were infected with
Lenti-luc virus (lentivirus containing the luciferase gene). The
mice were anesthetized via i.p. injection of Avertin,
2,2,2-Tribromoethanol (Sigma Chemical Co.) at a dose of 250 mg/kg.
Prior to orthotopic implantation, a 1-cm laparotomy was performed,
and orthotopic human hepatocellular carcinoma (HCC) was established
by intrahepatic injection of 105 SK-HEP-1-Luc cells
(luciferase-expressing cells) suspended in 30 µl DMEM into
the left liver lobe. Post-injection bleeding and tumor cell escape
were avoided by short-term local compression. The abdomen was
closed using an absorbable 5-0 vicryl suture, and the skin was
closed with a 5-0 proline suture. For orthotopic therapeutic
studies, implanted mice were treated with different formulations of
anticancer drugs. Tumor progression was monitored by
bioluminescence quantification. Mouse body weight and survival rate
were measured. Animal care was carried out in accordance with the
guidelines of Academia Sinica, Taiwan. The experimental protocols
were approved by the Academia Sinica Institutional Animal Care and
Utilization Committee (approval no. 11-06-190).
Data analysis
All data were expressed as the mean ± standard error
(SEM) of at least 3 independent experiments. Statistical
significance was assessed using a two-tailed Student's t-test with
P<0.05 considered to be statistically significant.
Non-compartmental pharmacokinetic analysis of plasma concentrations
versus time was performed with pharmacokinetic parameters using
WinNonlin software version 5.2 (Pharsight, Mountain View, CA,
USA).
Results
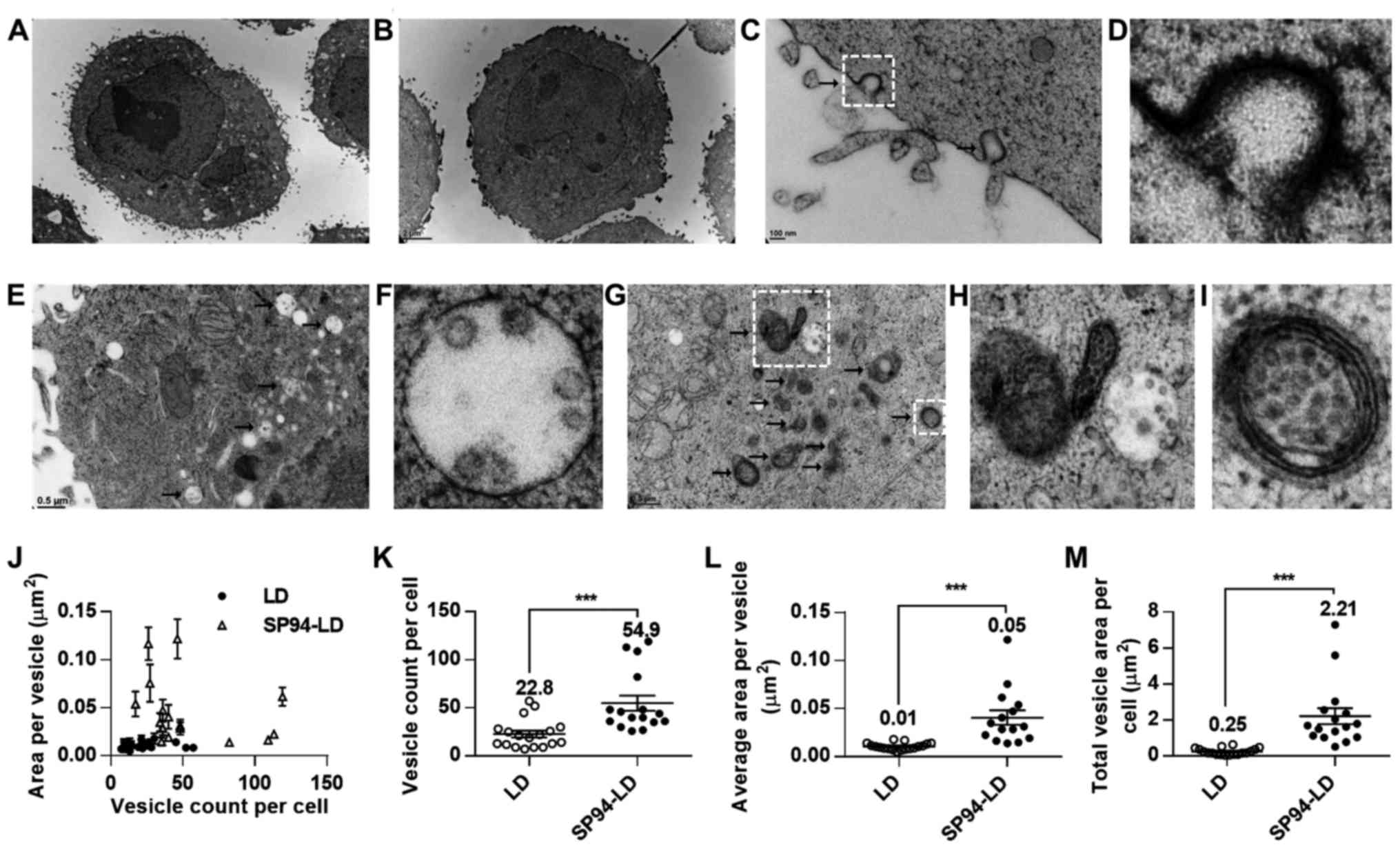
Ultrastructural analysis of in vitro
trafficking of lipid nanoparticles
Intracellular trafficking is a determining factor in
the therapeutic efficiency of nanoparticle-based drug delivery. In
the present study, transmission electron microscopy was used to
investigate the mechanism of cellular uptake of SP94-modified
liposomes. TEM images of SK-HEP-1 cells incubated with SP94-LD or
LD revealed that liposomes were internalized by vesicular
transport, and partially escaped to the cytosol at the perinuclear
region at 37°C (Fig. 1A and B).
After SP94-LD exposure, SK-HEP-1 cells exhibited numerous coated
pit structures (Fig. 1C and D) and
endocytotic vesicles in the cell membrane and cytoplasm (Fig. 1E–I). In contrast, few endocytotic
vesicles were observed at the same magnification in cells incubated
with LD (Fig. 1B). Analysis of the
TEM images revealed that cells incubated with SP94-LD showed a high
amount of internalized vesicles (Fig.
1J–M). The total vesicle area per SK-HEP-1 cell for SP94-LD was
~8.8-fold greater than that for LD (Fig. 1M).
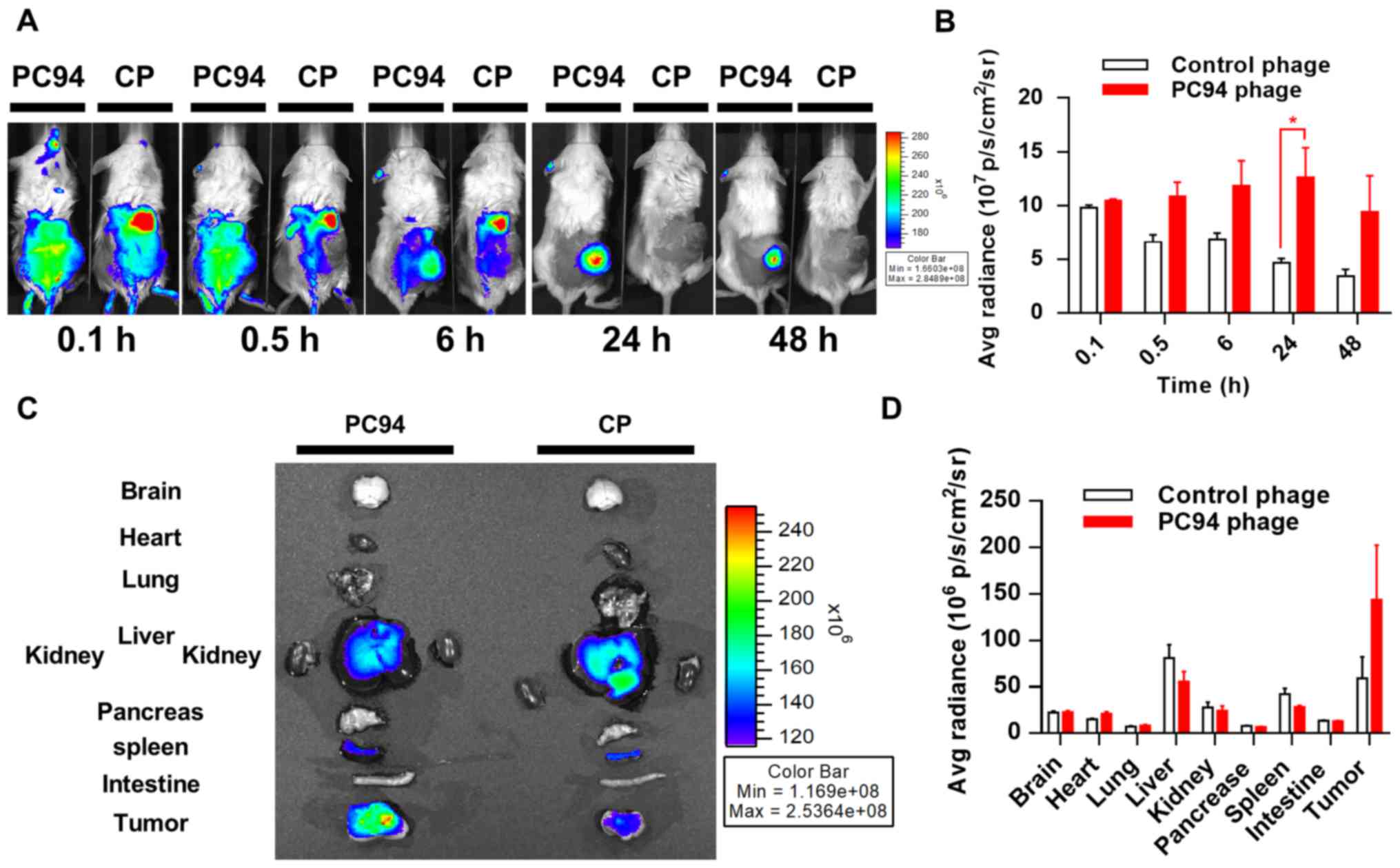
Tumor accumulation and retention of
near-infrared, fluorochrome-labeled, HCC-targeted phage
Molecular imaging plays a critical role in
oncological drug development, as stated in the FDA's Critical Path
Initiative documents (26). To
date, no molecular imaging method has been shown to accurately
detect, characterize, or monitor the response of HCC to treatment.
Labeling of phages with a near-infrared fluorescence tag enables
the distribution of the phages to be efficiently tracked in
vivo. We evaluated the in vivo distribution of PC94
phage using a previously described optical imaging method (22). Mice bearing subcutaneous Mahlavu
tumors were intravenously injected with HiLyte Fluor
750-labeled PC94 phage or control phage, and phages were
monitored at 0.1 and 0.5 h at the initial stages, and again at 6,
24, and 48 h post-injection. Fig.
2A shows representative near-infrared images taken at 0.1, 0.5,
6, 24 and 48 h post-injection from two subjects. Mice injected with
HiLyte Fluor 750-labeled PC94 phage exhibited higher
fluorescent signals in tumors, as compared to tumors in animals
injected with control phage (Fig. 2A
and B).
We proceeded to further evaluate the distribution
profiles of PC94 phage in mice. The accumulation of phages in
Mahlavu tumor peaked at ~24 h and then decreased gradually, but
>80% of the peak level was retained in the tumor by 48 h
(Fig. 2B). The organ accumulation
of phages in Mahlavu tumors was evaluated by near-infrared
fluorescence imaging after sacrifice at 48 h post-injection,
revealing that the SP94 peptide enhanced accumulation in tumor
sites, but not healthy organs including brain, heart, lung, liver,
kidney, pancreas, spleen and intestine (Fig. 2C and D).
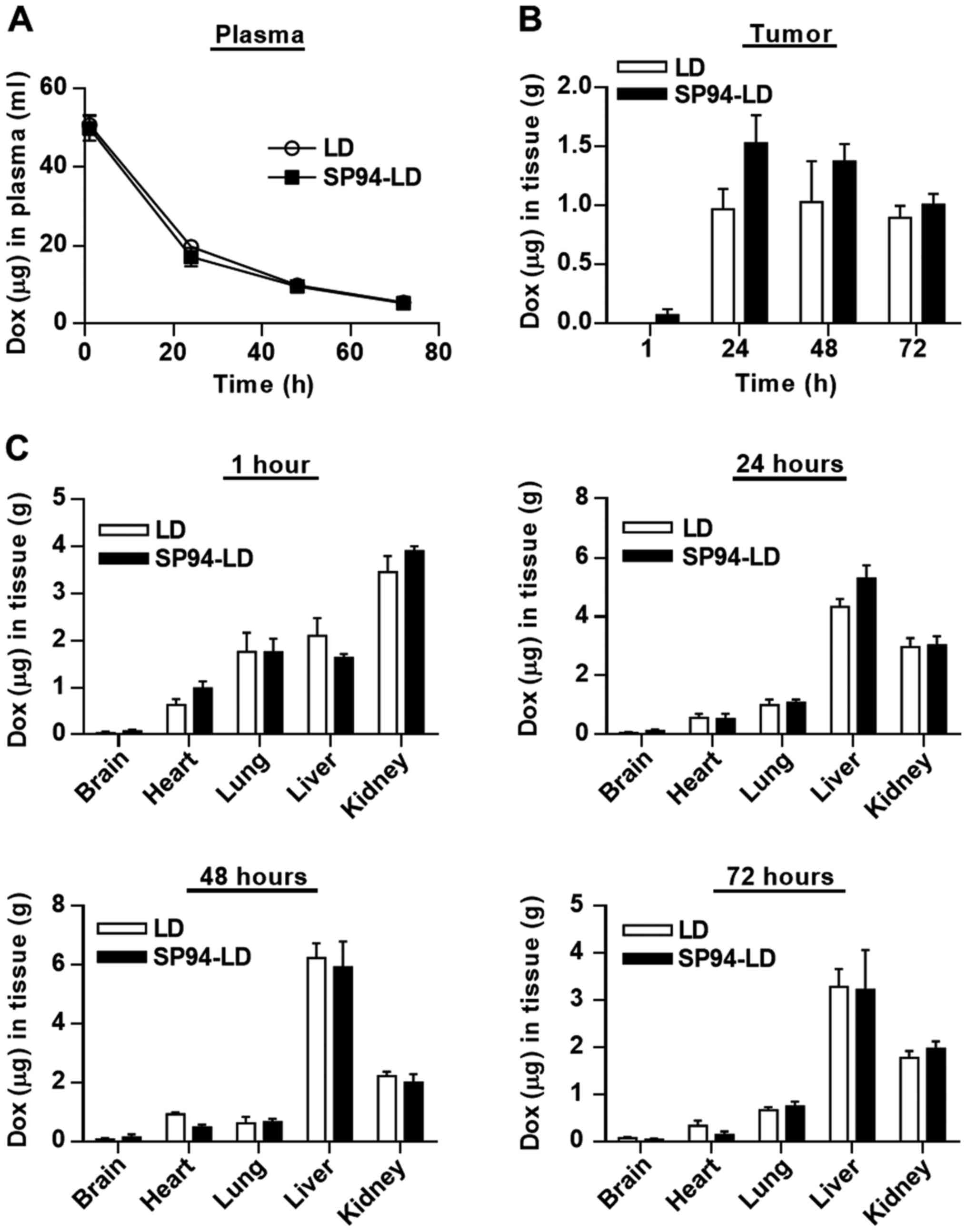
Comparison of the pharmacokinetic and
biodistribution profiles of SP94-LD and LD
For pharmacokinetic analysis, SP94-LD and LD were
administered to NOD.CB17-Prkdcscid/J mice at
matched 2 mg doxorubicin/kg by tail vein injection. Blood samples
were withdrawn at selected time-points, and were examined for
doxorubicin content using a validated fluorescent quantitative
method. The blood profiles of both SP94-LD and LD were similar
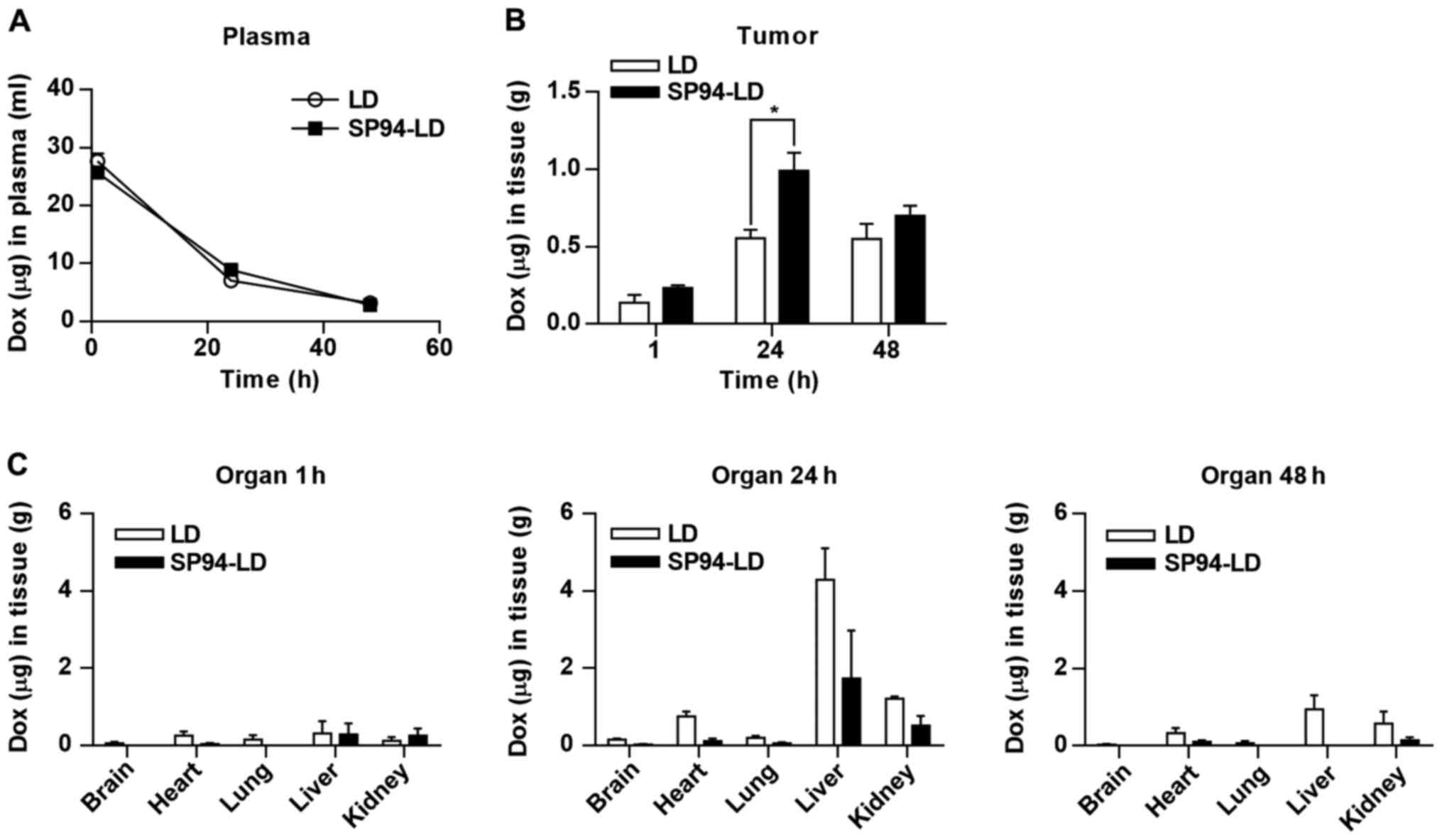
throughout the study, declining progressively over time (Fig. 3A). The time course of SP94-LD and
LD accumulation in tumor and various organs are shown in Fig. 3B and C. The maximum total
doxorubicin concentration in tumor was 1.53±0.47 µg
doxorubicin/g tumor, which occurred at 24 h after SP94-LD
administration; 1.01±0.18 µg/g remained at 72 h (Fig. 3B). Maximum tumor accumulation of LD
at 2 mg/kg (1.03±0.69 µg/g) occurred at 48 h post-injection,
and gradually decreased with time to 0.89±0.69 µg/g at 72 h
post-treatment. At 24 h, tumor accumulation of SP94-LD was 1.6-fold
higher than that of LD. In contrast, distribution of SP94-LD in all
non-malignant tissues was similar to that of LD at all time-points
(Fig. 3C). The tumor doxorubicin
AUC0–72 for SP94-LD was 81.75 µg h/g and the LD
AUC0–72 was 58.23 µg h/g, representing a 1.4-fold
increase in doxorubicin AUC0–72 for mice treated with
the targeted drug.
Intracellular distribution and
accumulation of doxorubicin
To determine the amount of bioavailable drug in
tumor cells, we performed whole body perfusion through the left
ventricle of the heart with DPBS before analyzing biodistribution.
This operation can eliminate blood and liposomes remaining in
vessels and the interstitial space of tumors. After whole body
perfusion, the tumor mass, brain, heart, lung, liver, and kidneys
were harvested, and doxorubicin was quantified. We used
intracellular accumulation of doxorubicin as an indicator of
bioavailability of the liposomal drug. SP94-LD and LD were
administered to tumor (SK-HEP-1)-bearing mice at matched 1 mg
doxorubicin/kg by i.v. injection. The doxorubicin levels were
measured in the blood at different time-points using a fluorescent
quantitative method. The blood profiles of both SP94-LD and LD were
similar (Fig. 4A). Tumor uptake of
SP94-LD at 1 mg/kg gradually increased, before peaking at 0.99±0.24
µg/g at 24 h post-injection, while 0.70±0.13 µg/g
remained at 48 h (Fig. 4B).
Maximum tumor uptake of LD at 1 mg/kg (0.55±0.11 µg/g)
occurred 24 h post-injection, and experienced almost no change over
time, remaining at 0.55±0.19 µg/g at 48 h post-injection. At
24 h, a 1.79-fold higher uptake of SP94-LD was observed as compared
to LD. The tumor doxorubicin AUC0–48 for SP94-LD was
34.45 µg h/g and the LD AUC0–48 was 21.27
µg h/g, representing a 1.62-fold increase in doxorubicin
AUC0–48 when conjugated to SP94. The uptake of both
drugs in most non-malignant tissues was low at 1 h post-injection;
uptake of both drugs by liver and kidney gradually increased by 24
h, and then slowly declined thereafter (Fig. 4C). Interestingly, the retention of
SP94-LD in liver and kidney were lower than that of LD at 24 h
post-injection, although these differences were not statistically
significant (P=0.14 and P=0.06 respectively).
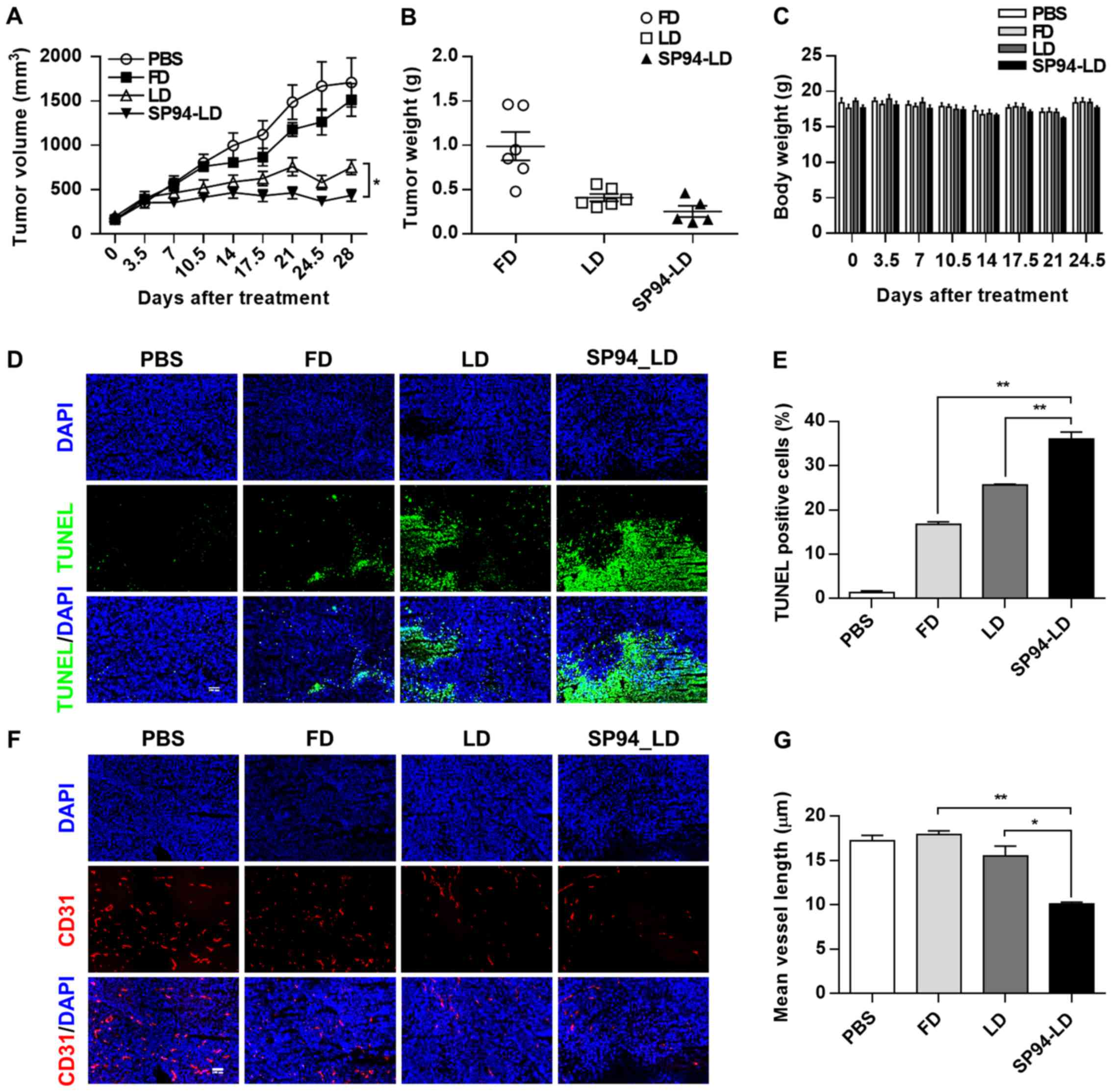
Efficacy of SP94-targeted liposomal
doxorubicin in hepatocellular carcinoma xenograft models
To evaluate the antitumor efficacy of
systemically-administered SP94-LD as compared to LD,
NOD.CB17-Prkdcscid/J were inoculated s.c. with
SK-HEP-1 tumors. Mice bearing hepatocellular carcinoma xenografts
(~100 mm3) were assigned into four groups for different
treatments: A, SP94-LD; B, free doxorubicin (FD); C, LD; and D,
PBS. Treatments were administered through tail vein injection, 1
mg/kg every 3.5 days, for eight doses with a total cumulative dose
of 8 mg/kg. By day 28, administration of SP94-LD had significantly
inhibited tumor growth by 74.6% (P<0.01), whereas treatment with
LD and FD inhibited tumor growth by 56.1 (P<0.01) and 11.6%
(P>0.05), respectively (Fig. 5A and
B), as compared to untreated controls. SP94-LD-mediated
inhibition of growth was more significant than that mediated by LD
(P=0.013). The SP94-LD and LD groups did not exhibit significant
changes in body weight (Fig. 5C)
or complete blood counts (Table I)
during the treatment period.
 | Table IHematological parameters in NOS/SCID
mice treated with different doxorubicin formulations. |
Table I
Hematological parameters in NOS/SCID
mice treated with different doxorubicin formulations.
| Parameter | SP94_LD | LD | FD | PBS |
|---|
| WBC count | 3.10±0.43 | 3.48±0.42 | 7.16±0.75 | 7.72±0.91 |
| NEU count | 2.36±0.41 | 2.69±0.33 | 5.95±0.63 | 6.35±0.91 |
| LYM count | 0.17±0.01 | 0.16±0.02 | 0.54±0.05 | 0.60±0.07 |
| RBC count | 6.29±0.06 | 6.21±0.09 | 7.72±0.22 | 7.52±0.57 |
| HGB value | 10.55±0.22 | 10.53±0.12 | 13.20±0.43 | 13.02±0.88 |
| HCT value | 31.32±1.08 | 30.63±0.31 | 38.82±1.29 | 38.87±2.38 |
| RDW value | 19.40±0.43 | 20.62±0.43 | 23.05±0.51 | 25.63±3.28 |
| PLT count | 1,172.83±85.13 | 1,154.00±71.80 | 940.17±57.09 | 1,047.33±61.99 |
| MPV value | 7.89±0.12 | 7.88±0.18 | 6.79±0.11 | 6.88±0.20 |
| PCT value | 0.92±0.06 | 0.91±0.06 | 0.64±0.04 | 0.73±0.06 |
| NEU % value | 74.63±2.82 | 77.42±2.26 | 83.13±1.78 | 81.35±3.31 |
| LYM % value | 5.22±0.78 | 4.69±0.23 | 7.86±1.06 | 8.31±1.63 |
| MONO % value | 19.43±3.20 | 16.97±1.68 | 8.86±1.05 | 10.06±2.09 |
| MONO count | 0.56±0.06 | 0.60±0.11 | 0.65±0.12 | 0.75±0.15 |
| MCV value | 49.72±1.33 | 49.33±0.51 | 50.23±0.46 | 52.22±2.16 |
| MCH value | 16.75±0.21 | 16.93±0.16 | 17.10±0.10 | 17.43±0.47 |
| MCHC value | 33.80±0.64 | 34.35±0.13 | 34.07±0.20 | 33.48±0.54 |
| PDW value | 18.12±0.19 | 18.42±0.30 | 18.90±0.57 | 18.18±0.30 |
Histological analysis of the SK-HEP-1 tumors
revealed that FD, LD, and SP94-LD induced apoptosis (Fig. 5D and E), with SP94-LD treatment
resulting in the greatest proportion of apoptotic cells (Fig. 5E). The CD31 (angiogenesis) index
(Fig. 5F and G) of SP94-LD treated
tumors was lower compared to that of tumors treated with FD, LD, or
saline.
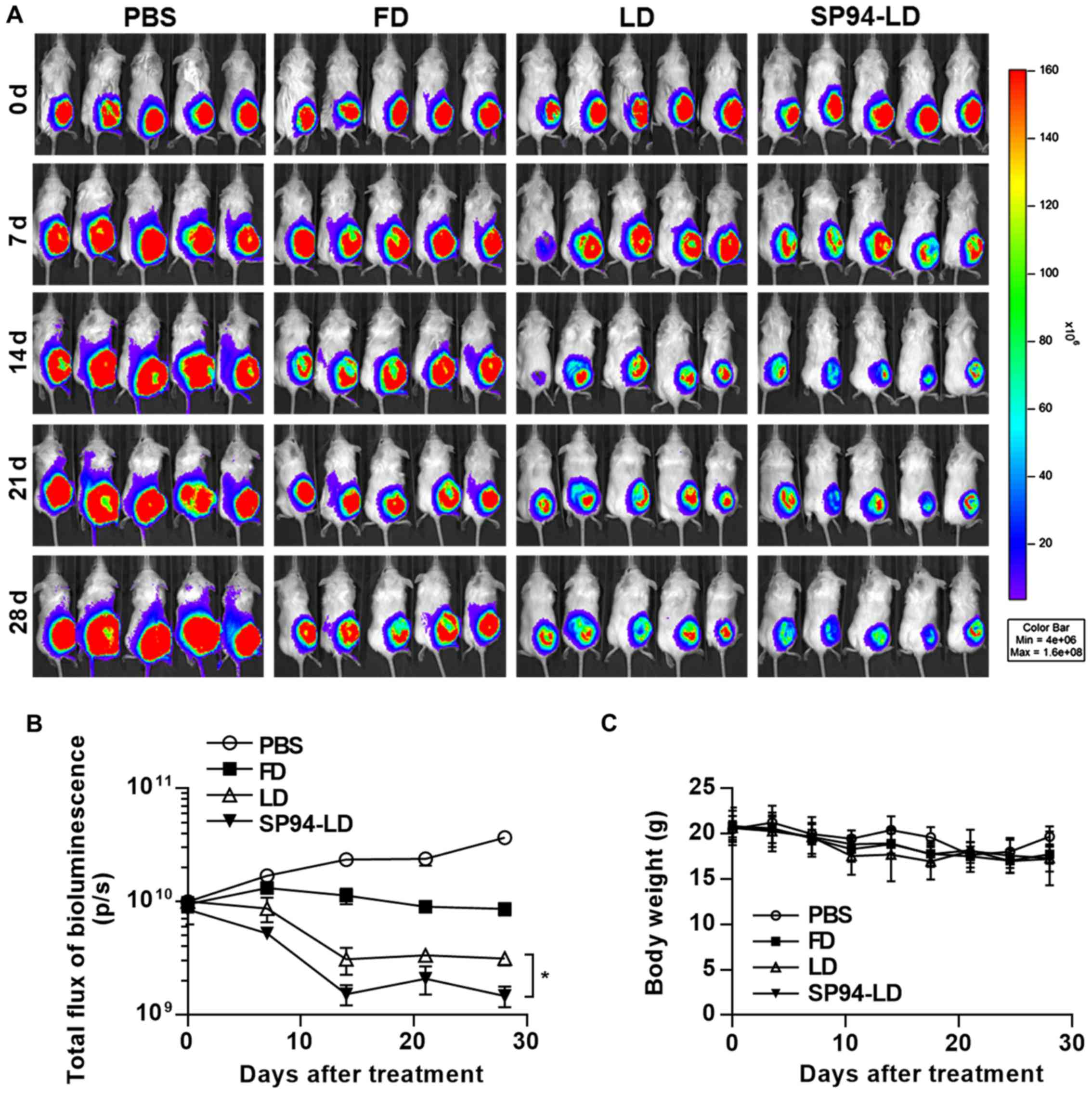
We have previously observed that
SP94-peptide-targeted liposomal doxorubicin (SP94-LD) inhibits the
proliferation of small tumors. Nevertheless, in many cancer cases
in humans, the tumors are detected when they are large. Thus, here
we examined the activities of FD, LD, and SP94-LD against large
tumors. Luciferase activity in live cells of mice bearing
hepatocellular carcinoma xenografts was measured after i.v.
injection with FD, LD, or SP94-LD (Fig. 6A). We observed a statistically
significant reduction of tumor growth after i.v. treatment of
hepatocellular carcinoma with SP94-LD, as compared with tumors in
untreated and FD or LD-treated mice (Fig. 6A and B). SP94-LD significantly
inhibited tumor growth as evidenced by a 91.47% reduction in
luminescence (P<0.01), whereas treatment with LD reduced
luminescence by 96.01% (P<0.01) compared with untreated
controls. SP94-LD inhibition of growth was more significant than
that by LD (P=0.011). Neither SP94-LD nor LD had a significant
effect on body weight during the treatment period (Fig. 6C).
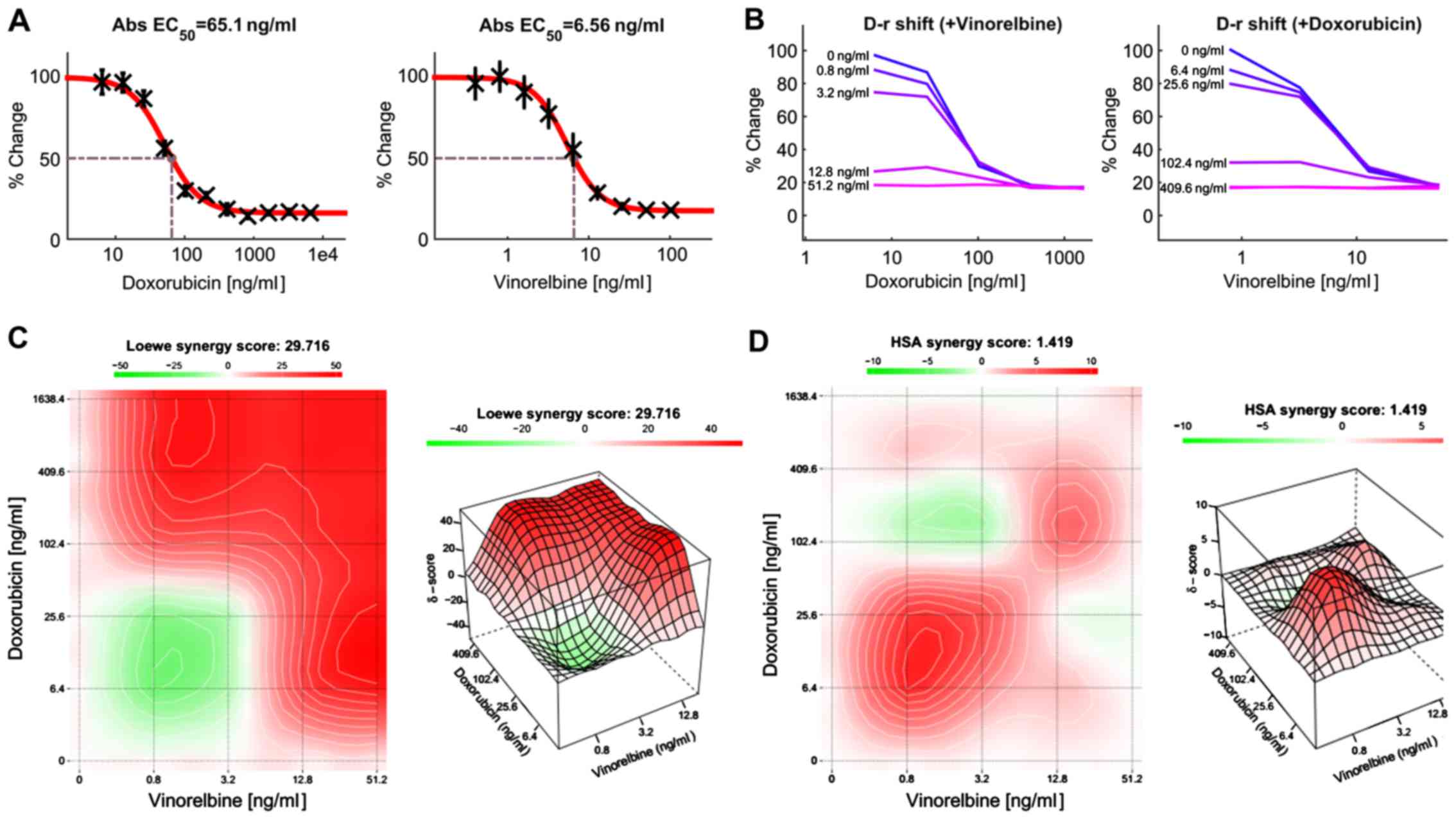
Effect of combination treatment with
doxorubicin and vinorelbine in HCC cells
Previous study has shown that doxorubicin can
inhibit HCC growth, but the clinical response was low. Combinations
of anticancer agents with different mechanisms of action may be
effective treatment regimens. For this reason, we attempted to
develop an effective treatment for HCC patients by combining two or
more anticancer drugs. In this study, HCC cells were treated with
doxorubicin or vinorelbine alone, or in combination using a
dose-matrix approach to evaluate the drug combination effects at
various drug ratios. To determine the combination response
(additivity, synergy, or antagonism), the Loewe Additivity score
(27) and the Highest Single Agent
(HAS) score (28) were calculated
for the drug combination using the software Combenefit (24) and SynergyFinder (25). Fig.
7A presents a dose-response matrix between 6 concentrations of
doxorubicin and 6 concentrations of vinorelbine in a 4-fold
dilution scheme. The cytotoxicity of doxorubicin or/and vinorelbine
over a wide range of molar ratios for HCC cells is shown in
Fig. 7B. Vinorelbine significantly
enhanced the effect of doxorubicin on the viability of HCC cells.
Similarly, doxorubicin also enhanced the cytotoxicity of
vinorelbine on HCC cells. The drug combination responses at various
dose levels were quantified by computational methods. The
dose-matrix combination showed the combination modeling between
doxorubicin and vinorelbine by using the Loewe Additivity model
(Fig. 7C) and HAS model (Fig. 7D). The synergy heatmaps showed that
doxorubicin and vinorelbine have additive/synergistic effects (red
areas in the model graph) on inhibiting cell proliferation at a
wide range of drug combination ratio. These results suggest that
the combined use of doxorubicin and vinorelbine may add an
advantage to the current therapeutic regimens in liver cancer.
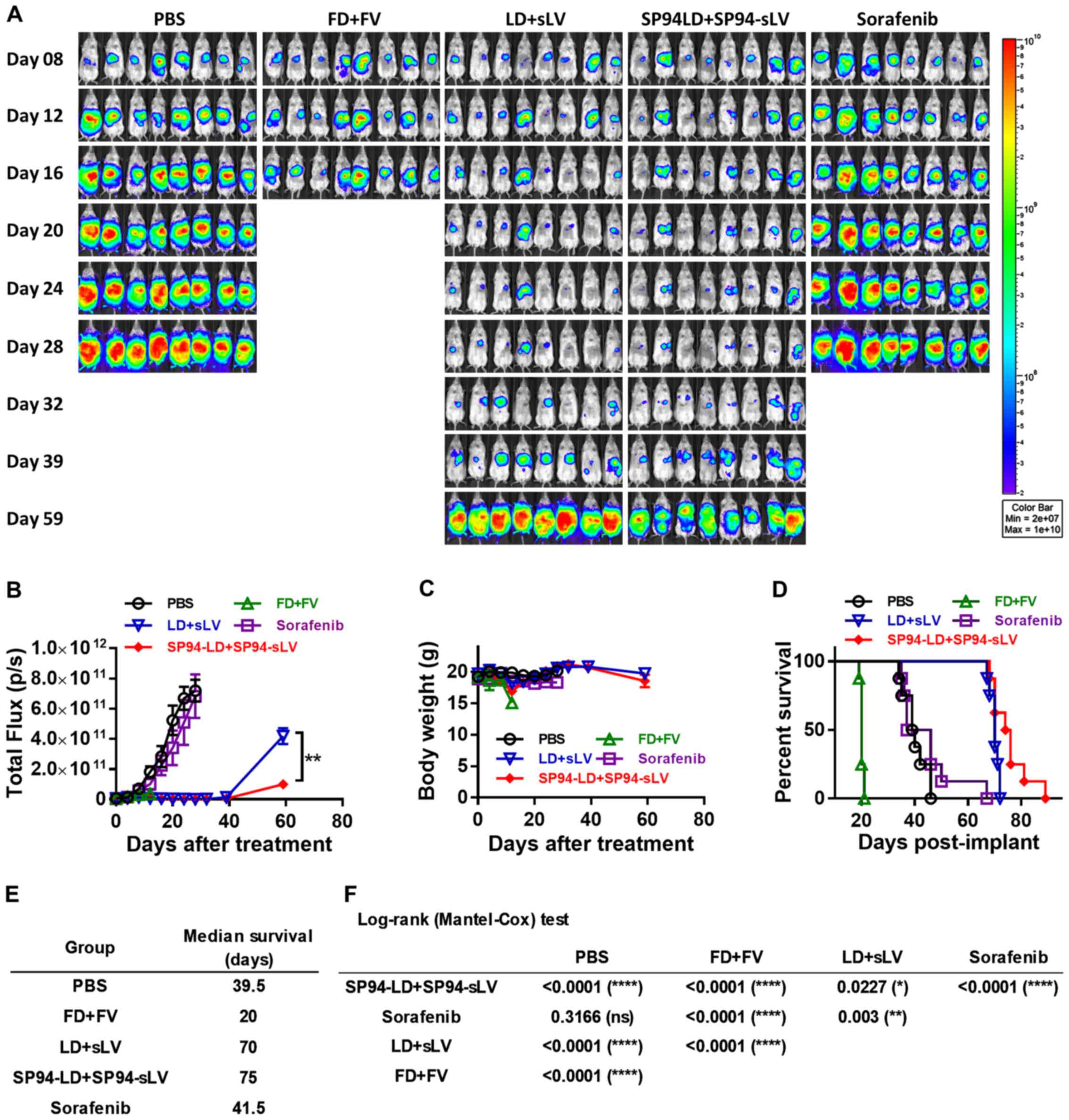
Therapeutic potential of combination
therapy in an orthotopic hepatocellular carcinoma model
Models based on the subcutaneous injection of cancer
cell lines may not accurately reproduce the biology of human HCC.
To study the influence of the liver microenvironment upon response
to therapy, we developed an orthotopic liver cancer model to
recapitulate the tumor growth pattern seen in liver cancer
patients. We investigated the antitumor potential of i.v.
administration of both SP94-targeted liposomal doxorubicin and
liposomal vinorelbine to SK-HEP-1-Luc tumors stably expressing
firefly luciferase. Orthotopic tumor growth was non-invasively
monitored by bioluminescence imaging. Prior to the first
therapeutic injection (4 days after tumor cell implantation),
growing orthotopic tumors were found to be localized mainly at the
liver (Fig. 8A).
Mice were treated with either vehicle alone (PBS),
FD (1 mg/kg) + free vinorelbine 11 (FV) (2 mg/kg), LD (1 mg/kg) +
stable liposomal vinorelbine (sLV) (2 mg/kg), SP94-LD (1 mg/kg) +
SP94-sLV (2 mg/kg), or sorafenib alone (30 mg/kg) every other day
for sixteen days. Bioluminescence was examined weekly to monitor
tumor burden. Bioluminescence images revealed significant
inhibition of tumor growth in the SP94-LD (1 mg/kg) + SP94-sLV (2
mg/kg)-treated group compared with the vehicle control group, the
FD (1 mg/kg) + FV (2 mg/kg) group, or the sorafenib (30 mg/kg)
group (Fig. 8A and B). Body weight
was not affected by any treatment regimen (Fig. 8C). Kaplan-Meier survival curves of
all groups are shown in Fig. 8D.
At the end of the study, the median survival times for the PBS, FD
+ FV, LD + sLV, SP94-LD + SP94-sLV, and sorafenib treatment groups
were 39.5, 20, 69, 75 and 41.5 days, respectively (Fig. 8D and E). A survival analysis with a
log-rank (Mantel-Cox) test revealed that SP94-LD + SP94-sLV
treatment significantly extended animal survival as compared with
PBS, FD + FV, LD + sLV, or sorafenib treatment (Fig. 8F).
Discussion
Lipid nanocarriers have been widely used to increase
the efficacy of chemotherapeutics, largely through passive
accumulation achieved by the enhanced permeability and retention
effect (29). Additionally, their
specificity and internalization by target tissues can be further
enhanced by surface conjugation with targeting moieties (29–31).
Nevertheless, insufficient differential affinity of targeting
moieties between tumor and normal tissues remains a critical
challenge for clinical application. This problem may arise because
the molecules recognized by targeting moieties are not only
expressed by tumor cells, but also by normal tissue. Therefore, it
is of great importance to study the biodistribution and targeting
potential of targeted nanocarriers.
Difficulties in developing specific imaging methods
for HCC are caused by the lack of specific molecular targets,
problems with drug delivery, and poor signal-to-noise ratios. It
was previously reported that near-infrared, fluorochrome-labeled
phage probes allow for cancer targeting and imaging in vivo
(22,32). In this study, we used near-infrared
fluorochrome-labeled PC94 phage and in vivo fluorescence
imaging techniques, which enabled us to study in vivo tumor
targeting and tissue distribution of SP94 peptide over time in the
living animal. The HiLyte Fluor 750-labeled PC94 phage was
able to bind to HCC and exhibited more rapid tumor localization
than the control, as evidenced by fluorescence images of the tumor
ex vivo and in living mice. Both HiLyte Fluor
750-labeled PC94 phage and control phage accumulated rapidly at
high concentrations in liver, spleen, and bone marrow, indicating
that the mononuclear phagocytes of the reticuloendothelial system
(RES) are involved in the clearance of some of the circulating
phages in mice. Although the tumor-to-background ratio was
satisfactory in this study, the uptake of near-infrared
fluorochrome-labeled phages by the RES is, nonetheless, quite
obvious.
To investigate the potential of SP94 peptide as a
target ligand for the delivery of therapeutics to tumors, we used
this peptide to modify a clinically-used PEGylated liposomal drug
through post-insertion technology, and examined the resulting
pharmacokinetic profile and biodistribution in vivo. This
post-insertion technique facilitates efficient insertion of
ligand-PEG-DSPE conjugates into preformed liposomes under the right
conditions (33). The plasma
pharmacokinetics of SP94-LD was found to be indistinguishable from
that of LD, indicating that the SP94 peptide conjugation did not
compromise circulation time or stability. Comparison of the
biodistribution of SP94-LD versus LD at 24 h post-treatment showed
that doxorubicin accumulation was greater in the tumor, and that
accumulation was 1.6-fold greater for SP94-LD-treated as compared
with LD-treated mice. The bioavailable drug concentrations of
SP94-LD reached maximum levels by 24 h after injection, and SP94
targeting increased drug delivery to intracellular tumors by
1.8-fold. While SP94-LD and LD have similar plasma circulation
profiles, the presence of the SP94-targeting moiety enhances drug
delivery to tumors and the bioavailability of drug in tumors.
However, SP94 targeting did not affect doxorubicin accumulation in
the brain, heart, lung, liver, or kidneys. These results indicate
that SP94-LD may improve the therapeutic index by increasing drug
accumulation in the tumor, but not in normal tissue.
The use of peptides as targeting ligands offers
several advantages, including low immunogenicity, small size, ready
diffusion, ease of manufacturing, and simple targeted formulation
assembly, when compared to larger biomolecules, such as antibodies.
Previous studies showed that using larger biomolecules as targeting
ligands may increase the clearance of antibody-modified
nanoparticles from the blood (34–36).
This may be due to non-specific binding and uptake of nanoparticles
by the RES (35). Our results
showed that the modification of liposomes with SP94 peptide does
not enhance immunogenicity, and has identical plasma
pharmacokinetics as the original formulation. In addition,
SP94-mediated targeting enhanced tumor delivery by increasing
overall tumor accumulation and cellular uptake, without affecting
delivery to noncancerous host tissues or enhancing host toxicity.
These findings were confirmed by in vivo near-infrared
fluorescence imaging and biodistribution analysis. Previous studies
with other targeted delivery systems have also suggested that
targeting moieties increase drug accumulation in tumor tissues
(37,38). However, targeted nanoparticles have
not always caused a significant increase in overall tumor
accumulation as compared to the non-targeted drugs. Previous
studies showed that the use of macromolecule targeting ligands,
such as antibodies (39) or
transferrin (40), have a
negligible impact on tumor accumulation and biodistribution. The
differential effects of nanoparticles modified with macromolecules
and those modified with small molecules may be due to differences
in molecular size, affinity, and penetrability of the targeting
ligand (41). It is possible that
targeting moieties with high affinity would be subject to greater
internalization and degradation by perivascular tumor cells,
thereby limiting their penetration of tumors and reducing their
tumor retention (42,43).
At the time of writing, only sorafenib has been
shown to exert a survival benefit in patients with hepatocellular
carcinoma (19,20). In our orthotopic liver cancer
model, we found that sorafenib had no effect on tumor growth. FD +
FV showed antitumor growth effects compared with PBS control group
at day 8, 12 and 16 (Fig. 8A).
However, the combination drugs induced the toxicity by markedly
decreasing their body weights, leading to the death of the mice
(Fig. 8C). LD + LV showed
significant antitumor growth effects and reduced drug toxicity.
SP94-LD + SP94-LV exhibited even higher therapeutic efficacy than
LD + LV in an orthotopic model of human HCC, by reducing tumor size
and prolonging the overall survival rate of the mice (Fig. 8). SP94-LD + SP94-sLV combination
therapy shows strong clinical potential for treatment of HCC.
In conclusion, SP94-modification of liposomal
doxorubicin significantly improves therapeutic efficacy in human
SK-HEP-1 tumor-bearing NOD.CB17-Prkdcscid/J mice,
and significantly inhibits tumor cell viability, resulting in
reduced tumor volumes and final average tumor weights compared with
control nonspecific treatments. The rapid, massive, and specific
accumulation of SP94-LD in tumor cells results in prominent tumor
growth regression. These therapeutic outcomes confirm the key role
of the tumor-specific binding and internalization of SP94-LD in
achieving elevated local concentrations of chemotherapeutic agents
inside tumors. These findings suggest a potential clinical benefit
from SP94-targeted liposomal doxorubicin and liposomal vinorelbine
combination therapy.
In conclusion, we have evaluated whether it is worth
considering SP94-LD for use in clinical trials against liver
cancer. SP94-LD caused significantly greater inhibition of human
hepatocellular carcinoma xenograft tumor growth when compared with
LD, without increasing toxicity. SP94-LD has similar plasma
pharmacokinetics as LD. Moreover, SP94-mediated targeting enhances
therapeutic efficacy in the hepatocellular carcinoma xenograft
mouse model by improving tumor pharmacokinetics and tissue
distribution, allowing large amounts of antitumor drug to
accumulate in tumors. These results indicate that SP94-mediated
targeting for cancer therapy has distinct advantages over standard
LD. Furthermore, it provides a promising opportunity to further
improve the therapeutic index of the original formulation, which
can be rapidly translated into the clinic given the availability of
LD. Our results should also encourage further research to expand
the application of this targeting ligand to various other
drug-delivery nanoparticles.
Acknowledgments
The authors thank the Core Facility of the Institute
of Cellular and Organismic Biology for their technical assistance.
This study was supported by grants from Academia Sinica and
Ministry of Science and Technology (MOST 104-0210-01-09-02, MOST
105-0210-01-13-01, MOST 106-2623-E-001-001 and MOST
106-0210-01-15-02), Taiwan (to H.-C. Wu).
References
|
1
|
Souto E: Lipid Nanocarriers in Cancer
Diagnosis and Therapy. iSmithers Rapra Publishing; Shrewsbury:
2011
|
|
2
|
Maeda H, Wu J, Sawa T, Matsumura Y and
Hori K: Tumor vascular permeability and the EPR effect in
macromolecular therapeutics: A review. J Controlled Release.
65:271–284. 2000. View Article : Google Scholar
|
|
3
|
Wu CH, Liu IJ, Lu RM and Wu HC:
Advancement and applications of peptide phage display technology in
biomedical science. J Biomed Sci. 23:82016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lopes de Menezes DE, Pilarski LM and Allen
TM: In vitro and in vivo targeting of immunoliposomal doxorubicin
to human B-cell lymphoma. Cancer Res. 58:3320–3330. 1998.PubMed/NCBI
|
|
5
|
Lu RM, Chang YL, Chen MS and Wu HC: Single
chain anti-c-Met antibody conjugated nanoparticles for in vivo
tumor-targeted imaging and drug delivery. Biomaterials.
32:3265–3274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farokhzad OC, Jon S, Khademhosseini A,
Tran TN, Lavan DA and Langer R: Nanoparticle-aptamer bioconjugates:
A new approach for targeting prostate cancer cells. Cancer Res.
64:7668–7672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gabizon A, Shmeeda H, Horowitz AT and
Zalipsky S: Tumor cell targeting of liposome-entrapped drugs with
phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv
Rev. 56:1177–1192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee RJ and Low PS: Folate-mediated tumor
cell targeting of liposome-entrapped doxorubicin in vitro. Biochim
Biophys Acta. 1233:134–144. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee TY, Wu HC, Tseng YL and Lin CT: A
novel peptide specifically binding to nasopharyngeal carcinoma for
targeted drug delivery. Cancer Res. 64:8002–8008. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lo A, Lin CT and Wu HC: Hepatocellular
carcinoma cell-specific peptide ligand for targeted drug delivery.
Mol Cancer Ther. 7:579–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu CH, Kuo YH, Hong RL and Wu HC:
α-Enolase-binding peptide enhances drug delivery efficiency and
therapeutic efficacy against colorectal cancer. Sci Transl Med.
7:290ra912015. View Article : Google Scholar
|
|
12
|
Yeh CY, Hsiao JK, Wang YP, Lan CH and Wu
HC: Peptide-conjugated nanoparticles for targeted imaging and
therapy of prostate cancer. Biomaterials. 99:1–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen WC, Completo GC, Sigal DS, Crocker
PR, Saven A and Paulson JC: In vivo targeting of B-cell lymphoma
with glycan ligands of CD22. Blood. 115:4778–4786. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashley CE, Carnes EC, Phillips GK, Padilla
D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, et
al: The targeted delivery of multicomponent cargos to cancer cells
by nanoporous particle-supported lipid bilayers. Nat Mater.
10:389–397. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ashley CE, Carnes EC, Epler KE, Padilla
DP, Phillips GK, Castillo RE, Wilkinson DC, Wilkinson BS, Burgard
CA, Kalinich RM, et al: Delivery of small interfering RNA by
peptide-targeted mesoporous silica nanoparticle-supported lipid
bilayers. ACS Nano. 6:2174–2188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashley CE, Carnes EC, Phillips GK, Durfee
PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman
CL, et al: Cell-specific delivery of diverse cargos by
bacteriophage MS2 virus-like particles. ACS Nano. 5:5729–5745.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Hu Y, Xiao J, Liu G, Li X, Zhao Y,
Tan H, Shi H and Cheng D: Investigation of SP94 peptide as a
specific probe for hepatocellular carcinoma imaging and therapy.
Sci Rep. 6:335112016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Ng HLH, Lu A, Lin C, Zhou L, Lin
G, Zhang Y, Yang Z and Zhang H: Drug delivery system targeting
advanced hepatocellular carcinoma: Current and future. Nanomedicine
(Lond). 12:853–869. 2016. View Article : Google Scholar
|
|
19
|
Villanueva A, Hernandez-Gea V and Llovet
JM: Medical therapies for hepatocellular carcinoma: A critical view
of the evidence. Nat Rev Gastroenterol Hepatol. 10:34–42. 2013.
View Article : Google Scholar
|
|
20
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jaye DL, Geigerman CM, Fuller RE, Akyildiz
A and Parkos CA: Direct fluorochrome labeling of phage display
library clones for studying binding specificities: Applications in
flow cytometry and fluorescence microscopy. J Immunol Methods.
295:119–127. 2004. View Article : Google Scholar
|
|
22
|
Kelly KA, Waterman P and Weissleder R: In
vivo imaging of molecularly targeted phage. Neoplasia. 8:1011–1018.
2006. View Article : Google Scholar
|
|
23
|
Laginha KM, Verwoert S, Charrois GJ and
Allen TM: Determination of doxorubicin levels in whole tumor and
tumor nuclei in murine breast cancer tumors. Clin Cancer Res.
11:6944–6949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Di Veroli GY, Fornari C, Wang D, Mollard
S, Bramhall JL, Richards FM and Jodrell DI: Combenefit: An
interactive platform for the analysis and visualization of drug
combinations. Bioinformatics. 32:2866–2868. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ianevski A, He L, Aittokallio T and Tang
J: SynergyFinder: A web application for analyzing drug combination
dose-response matrix data. Bioinformatics. 33:2413–2415. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woodcock J and Woosley R: The FDA critical
path initiative and its influence on new drug development. Annu Rev
Med. 59:1–12. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loewe S: The problem of synergism and
antagonism of combined drugs. Arzneimittelforschung. 3:285–290.
1953.PubMed/NCBI
|
|
28
|
Berenbaum MC: What is synergy? Pharmacol
Rev. 41:93–141. 1989.PubMed/NCBI
|
|
29
|
Torchilin VP: Passive and active drug
targeting: Drug delivery to tumors as an example. Handb Exp
Pharmacol. 197:3–53. 2010. View Article : Google Scholar
|
|
30
|
Mohanty C, Das M, Kanwar JR and Sahoo SK:
Receptor mediated tumor targeting: An emerging approach for cancer
therapy. Curr Drug Deliv. 8:45–58. 2011. View Article : Google Scholar
|
|
31
|
Brown KC: Peptidic tumor targeting agents:
The road from phage display peptide selections to clinical
applications. Curr Pharm Des. 16:1040–1054. 2010. View Article : Google Scholar
|
|
32
|
Hilderbrand SA, Kelly KA, Niedre M and
Weissleder R: Near infrared fluorescence-based bacteriophage
particles for ratio-metric pH imaging. Bioconjug Chem.
19:1635–1639. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moreira JN, Ishida T, Gaspar R and Allen
TM: Use of the post-insertion technique to insert peptide ligands
into pre-formed stealth liposomes with retention of binding
activity and cytotoxicity. Pharm Res. 19:265–269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harding JA, Engbers CM, Newman MS,
Goldstein NI and Zalipsky S: Immunogenicity and pharmacokinetic
attributes of poly(ethylene glycol)-grafted immunoliposomes.
Biochim Biophys Acta. 1327:181–192. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koning GA, Kamps JA and Scherphof GL:
Interference of macrophages with immunotargeting of liposomes. J
Liposome Res. 12:107–119. 2002. View Article : Google Scholar
|
|
36
|
ElBayoumi TA and Torchilin VP:
Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo
of doxorubicin-loaded, long-circulating liposomes modified with
cancer-specific monoclonal antibody. Clin Cancer Res. 15:1973–1980.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu Z, Cai W, He L, Nakayama N, Chen K,
Sun X, Chen X and Dai H: In vivo biodistribution and highly
efficient tumour targeting of carbon nanotubes in mice. Nat
Nanotechnol. 2:47–52. 2007. View Article : Google Scholar
|
|
38
|
Qian X, Peng XH, Ansari DO, Yin-Goen Q,
Chen GZ, Shin DM, Yang L, Young AN, Wang MD and Nie S: In vivo
tumor targeting and spectroscopic detection with surface-enhanced
Raman nanoparticle tags. Nat Biotechnol. 26:83–90. 2008. View Article : Google Scholar
|
|
39
|
Kirpotin DB, Drummond DC, Shao Y, Shalaby
MR, Hong K, Nielsen UB, Marks JD, Benz CC and Park JW: Antibody
targeting of long-circulating lipidic nanoparticles does not
increase tumor localization but does increase internalization in
animal models. Cancer Res. 66:6732–6740. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bartlett DW, Su H, Hildebrandt IJ, Weber
WA and Davis ME: Impact of tumor-specific targeting on the
biodistribution and efficacy of siRNA nanoparticles measured by
multimodality in vivo imaging. Proc Natl Acad Sci USA.
104:15549–15554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Moghimi SM, Hunter AC and Andresen TL:
Factors controlling nanoparticle pharmacokinetics: An integrated
analysis and perspective. Annu Rev Pharmacol Toxicol. 52:481–503.
2012. View Article : Google Scholar
|
|
42
|
Adams GP, Schier R, McCall AM, Simmons HH,
Horak EM, Alpaugh RK, Marks JD and Weiner LM: High affinity
restricts the localization and tumor penetration of single-chain fv
antibody molecules. Cancer Res. 61:4750–4755. 2001.PubMed/NCBI
|
|
43
|
Rudnick SI, Lou J, Shaller CC, Tang Y,
Klein-Szanto AJ, Weiner LM, Marks JD and Adams GP: Influence of
affinity and antigen internalization on the uptake and penetration
of Anti-HER2 antibodies in solid tumors. Cancer Res. 71:2250–2259.
2011. View Article : Google Scholar : PubMed/NCBI
|